
A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia

 ,
,  and
and
Abstract
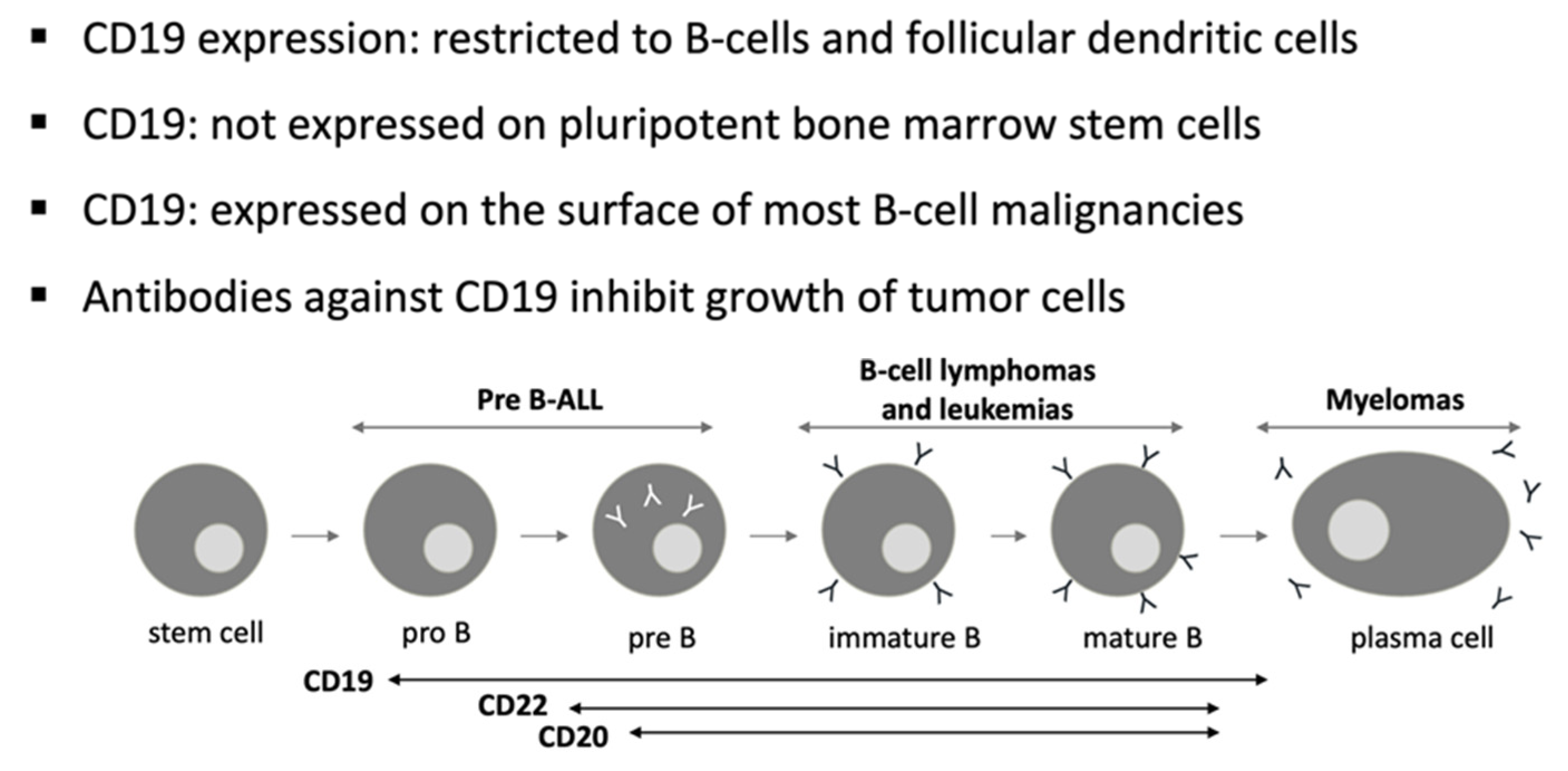
:1. Introduction
2. CAR-T Constructs
3. Anti-CD19 CAR T-Cell Studies
4. Relapse after CAR-T
5. Future Developments
5.1. CD19 CAR-T: Ongoing Trials
5.2. CD22 CAR T-Cell
5.3. Dual-Target CARs
6. Allogeneic CAR-T
7. Allo-SCT after CAR-T
8. Expert Opinion
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Cancer Stat Facts: Leukemia—Acute Lymphocytic Leukemia (ALL). Available online: https://seer.cancer.gov/statfacts/html/alyl.html (accessed on 1 January 2021).
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J. Clin. Oncol. 1999, 17, 3835–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [Green Version]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction therapy for adults with acute lymphoblastic leukemia: Results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Thomas, D.; O’Brien, S.; Cortes, J.; Giles, F.; Jeha, S.; Bueso-Ramos, C.E.; Pierce, S.; Shan, J.; Koller, C.; et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 2004, 101, 2788–2801. [Google Scholar] [CrossRef]
- Linker, C.; Damon, L.; Ries, C.; Navarro, W. Intensified and shortened cyclical chemotherapy for adult acute lymphoblastic leukemia. J. Clin. Oncol. 2002, 20, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.K.; Richards, S.M.; Chopra, R.; Lazarus, H.M.; Litzow, M.R.; Buck, G.; Durrant, I.J.; Luger, S.M.; Marks, D.I.; Franklin, I.M.; et al. Medical Research Council of the United Kingdom Adult ALL Working Party; Eastern Cooperative Oncology Group. The outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007, 109, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Tavernier, E.; Boiron, J.M.; Huguet, F.; Bradstock, K.; Vey, N.; Kovacsovics, T.; Delannoy, A.; Fegueux, N.; Fenaux, P.; Stamatoullas, A.; et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia 2007, 21, 1907–1914. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; De Angelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Ribrag, V.; Koscielny, S.; Bosq, J.; Leguay, T.; Casasnovas, O.; Fornecker, L.M.; Recher, C.; Ghesquieres, H.; Morschhauser, F.; Girault, S.; et al. Rituximab and dose-dense chemotherapy for adults with burkitt’s lymphoma: A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 2402–2411. [Google Scholar] [CrossRef]
- Dai, H.; Wang, Y.; Lu, X.; Han, W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. Natl. Cancer Inst. 2016, 108, PII djv439. [Google Scholar] [CrossRef] [Green Version]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCR zeta/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Tedder, T.F.; Zhou, L.J.; Engel, P. The CD19/CD21 signal transduction complex of B lymphocytes. Immunol. Today 1994, 15, 437–442. [Google Scholar] [CrossRef]
- Matsuo, Y.; Drexler, H.G. Establishment and characterization of human B cell precursor-leukemia cell lines. Leuk. Res. 1998, 22, 567–579. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des. Devel. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD41:CD81 composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22- targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19- targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained remissions with CD19- specific chimeric antigen receptor (CAR)- modified T cells in children with relapsed/refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Grupp, S.A.; Maude, S.L.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Callahan, C.; Lacey, S.F.; Levine, B.L.; Melenhorst, J.J.; Motley, L.; et al. Durable remissions in children with re- lapsed/refractory ALL treated with T cells engineered with a CD19-targeted chimeric antigen receptor (CTL019). Blood 2015, 126, 681. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Maude, S.L.; Rives, S.; Baruchel, A.; Boyer, M.W.; Bittencourt, H.; Bader, P.; Büchner, J.; Laetsch, T.W.; Stefanski, H.; et al. Updated Analysis of the Efficacy and Safety of Tisagenlecleucel in Pediatric and Young Adult Patients with Relapsed/Refractory (r/r) Acute Lymphoblastic Leukemia. Blood 2018, 132 (Suppl. 1), 895. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.; Baer, M.R.; Donnellan, W.B.; Carr-O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. End of phase I results of ZUMA-3, a phase 1/2 study of KTE-X19, anti-CD19 chimeric antigen receptor (CAR) T cell therapy, in adult patients (pts) with relapsed/refractory (R/R) acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 2019, 37 (Suppl. 15), 7006. [Google Scholar] [CrossRef]
- Schultz, L.M.; Baggott, C.; Prabhu, S.; Pacenta, H.; Phillips, C.L.; Rossoff, J.; Stefanski, H.; Talano, J.A.; Moskop, A.; Margossian, S.P.; et al. Disease Burden Impacts Outcomes in Pediatric and Young Adult B-Cell Acute Lymphoblastic Leukemia after Commercial Tisagenlecleucel: Results from Pediatric Real World CAR Consortium (PRWCC). Blood 2020, 136, 14–15. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Hu, Z.H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef]
- Anagnostou, T.; Riaz, I.B.; Hashmi, S.K.; Murad, M.H.; Kenderian, S.S. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: A systematic review and meta-analysis. Lancet Haematol. 2020, 7, e816–e826. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.A.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL- rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Riviere, I.; Wang, X.; Bernal, Y.B.; Yoo, S.; Purdon, T.; Halton, E.; Quintanilla, H.; Curran, K.J.; Sauter, C.S.; et al. CD19-Targeted 19–28z CAR-modified autologous T cells induce high rates of complete remission and durable responses in adult patients with relapsed, refractory B-cell ALL. Blood 2014, 124, 382. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisagenlecleucel Versus Blinatumomab or Inotuzumab for Adult Patients with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia: A Randomized Open Label, Multicenter, Phase III Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT03628053 (accessed on 15 December 2020).
- A Phase II Trial of Tisagenlecleucel in First-Line High-Risk (HR) Pediatric and Young Adult Patients With B-Cell Acute Lymphoblastic Leukemia (B-ALL) Who Are Minimal Residual Disease (MRD) Positive at the End of Consolidation (EOC) Therapy. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03876769 (accessed on 1 January 2021).
- Roddie, C.; O’Reilly, M.A.; Marzolini, M.A.V.; Wood, L.; Dias, J.; Cadinanos Garai, A.; Bosshard, L.; Abbasian, M.; Lowdell, M.W.; Wheeler, G.; et al. ALLCAR19: Updated Data Using AUTO1, a Novel Fast-Off Rate CD19 CAR in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukaemia and Other B-Cell Malignancies. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. Abstract 160. [Google Scholar]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; FitzGerald, D.J.; Barrett, D.M.; et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef] [Green Version]
- Martyniszyn, A.; Krahl, A.C.; André, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 Bispecific CAR T Cells for the Treatment of B-Cell Malignancies. Hum. Gene. Ther. 2017, 28, 1147–1157. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Schultz, L.M.; Muffly, L.S.; Spiegel, J.Y.; Ramakrishna, S.; Hossain, N.; Baggott, C.; Sahaf, B.; Patel, S.; Craig, J.; Yoon, J.; et al. Phase I Trial Using CD19/CD22 Bispecific CAR T Cells in Pediatric and Adult Acute Lymphoblastic Leukemia (ALL). In Proceedings of the 61nd ASH Annual Meeting and Exposition, Orlando, FL, USA, 7–10 December 2019. Abstract 744. [Google Scholar]
- Yang, J.; Jiang, P.; Zhang, X.; Li, J.; Wu, Y.; Xu, L.; Su, Y.; Hu, X.; Zhao, X.; Dong, Q.; et al. Successful 24-Hours Manufacture of Anti-CD19/CD22 Dual Chimeric Antigen Receptor (CAR) T Cell Therapy for B-Cell Acute Lymphoblastic Leukemia (B-ALL) Clinically Relevant Abstract. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. Abstract 159. [Google Scholar]
- Shah, N.; Maatman, T.; Hari, P.N.; Johnson, B. Multi-targeted CAR- T cell therapies for B-cell malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.E.; Zhang, H.; Wada, M.; Fang, L.; Feng, J.; Zhang, W.; Chen, Q.; Cao, Y.; Pinz, K.G.; Chen, K.H.; et al. Targeting Two Antigens Associated with B-ALL with CD19-CD123 Compound Car T Cell Therapy. Stem. Cell. Rev. Rep. 2020, 16, 385–396. [Google Scholar] [CrossRef]
- Hassanein, N.M.; Alcancia, F.; Perkinson, K.R.; Buckley, P.J.; Lagoo, A.S. Distinct expression patterns of CD123 and CD34 on normal bone marrow B-cell precursors (“hematogones”) and B lymphoblastic leukemia blasts. Am. J. Clin. Pathol. 2009, 132, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Fousek, K.; Watanabe, J.; George, A.; An, X.; Samaha, H.S.; Navai, S.A.; Byrd, T.T.; Jang, A.; Kim, H.; Sujith, J.; et al. Targeting CD19-negative relapsed B-acute lymphoblastic leukemia using trivalent CAR T cells. J. Clin. Oncol. 2018, 36, 121. [Google Scholar] [CrossRef]
- Harrer, D.C.; Schuler, G.; Dörrie, J.; Schaft, N. CSPG4-specific CAR T cells for high-risk childhood B cell precursor leukemia. Int. J. Mol. Sci. 2019, 20, 2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, C.F.; Mezzanotte, C.; Cappuzzello, C.; Bardini, M.; Tettamanti, S.; Fazio, G.; Cooper, L.J.N.; Dastoli, G.; Cazzaniga, G.; Biondi, A.; et al. Preclinical Efficacy and Safety of CD19CAR Cytokine-Induced Killer Cells Transfected with Sleeping Beauty Transposon for the Treatment of Acute Lymphoblastic Leukemia. Hum. Gene. Ther. 2018, 29, 602–613. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Magnani, C.F.; Tettamanti, S.; Gaipa, G.; Biagi, E. Redirecting T cells with Chimeric Antigen Receptor (CAR) for the treatment of childhood acute lymphoblastic leukemia. J. Autoimmun. 2017, 85, 141–152. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Buracchi, C.; Borleri, G.M.; Zaninelli, S.; Rizzuto, G.; et al. Donor-Derived CAR T Cells Engineered with Sleeping Beauty Achieve Anti-Leukemic Activity without Severe Toxicity. In Proceedings of the 62nd ASH Annual Meeting and Exposition, San Diego, CA, USA, 5–8 December 2020. Abstract 741. [Google Scholar]
- Zhang, X.; Yang, J.; Li, W.; Zhang, G.; Su, Y.; Shi, Y.; Song, D.; Zhang, M.; He, J.; Xu, L.; et al. Feasibility, and Efficacy of Donor-Derived cd19-Targeted Car t-Cell Therapy in Refractory/Relapsed(r/r)b-Cell Acute Lymphoblastic Leukemia (b-all) Patients. Blood 2020, 136 (Suppl. 1), 4–6. [Google Scholar] [CrossRef]
- Jain, N.; Roboz, G.J.; Konopleva, M.; Liu, H.; Jabbour, E.; Poirot, C.; Schiffer-Manniou, C.; Gouble, A.; Haider, A.; Zernovak, O.; et al. Preliminary Results of Balli-01: A Phase I Study of UCART22 (allogeneic engineered T-cells expressing anti-CD22 Chimeric Antigen Receptor) in Adult Patients with Relapsed or Refractory (R/R) CD22+ B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2020, 136 (Suppl. 1), 7–8. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Herrera, A.F.; Budde, L.E.; DeAngelo, D.J.; Heery, C.; Stein, A.; Jain, M.D.; Bijal Shah, B. Initial Findings of the Phase 1 Trial of PBCAR0191, a CD19 Targeted Allogeneic CAR-T Cell Therapy. Blood 2019, 134 (Suppl. 1), 4107. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, Y.; Mei, H. Consolidative allogeneic hematopoietic stem cell transplantation after chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia: Who? When? Why? Biomark. Res. 2020, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Jain, T.; Santomasso, B.D.; Mead, E.; Wudhikarn, K.; Silverberg, M.L.; Batlevi, Y.; Shouval, R.; Devlin, S.M.; Batlevi, C.; et al. Comparing CAR T-cell toxicity grading systems: Application of the ASTCT grading system and implications for management. Blood Adv. 2020, 4, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy— assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Mahadeo, K.M.; Khazal, S.J.; Abdel-Azim, H.; Fitzgerald, J.C.; Taraseviciute, A.; Bollard, C.M.; Tewari, P.; Duncan, C.; Traube, C.; McCall, D.; et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 45–63. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Hari, P.; Hu, Y.; Mei, H. Biomarkers in individualized management of chimeric antigen receptor T cell therapy. Biomark. Res. 2020, 8, 13. [Google Scholar] [CrossRef]
- Yang, J.; He, J.; Zhang, X.; Wang, Z.; Zhang, Y.; Cai, S.; Sun, Z.; Ye, X.; He, Y.; Shen, L.; et al. A Feasibility and Safety Study of a New CD19-Directed Fast CAR-T Therapy for Refractory and Relapsed B Cell Acute Lymphoblastic Leukemia. Blood 2019, 134 (Suppl. 1), 825. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Ciocarlie, O.; NJain, N.; Jabbour, E.J.; Maus, M.V.; Frigault, M.; Boissel, N.; et al. Preliminary Data on Safety, Cellular Kinetics and Anti-Leukemic Activity of UCART19, an Allogeneic Anti-CD19 CAR T-Cell Product, in a Pool of Adult and Pediatric Patients with High-Risk CD19+ Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132 (Suppl. 1), 896. [Google Scholar] [CrossRef]
- Qasim, W. Allogeneic CAR T cell therapies for leukemia. Am. J. Hematol. 2019, 94, S50–S54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Antigen Target | CAR Structure |
|---|---|
| CD19 | CD3ζ and CD28 or CD3ζ and 4-1BB |
| CD22 | CD3ζ and CD28 |
| CD20 | CD3ζ or and CD3ζ and 4-1BB |
| Author (Years) | Population | T-Cell Product | Age, Med (Range) | NO. | Priorhsct | CR | Post-Carhsct | Survival |
|---|---|---|---|---|---|---|---|---|
| Park (2018) | Adults | 19–28z | 44 (23–74) | 53 | 36% | 83% | 39% | 39% relapse (24% CD19-) Median OS/EFS: 12.9/6.1 months |
| Hay (2019) | Adults | 19–41BBz | 39 (20–76) | 53 | 43% | 85% | 40% | 49% relapse (27% CD19-) |
| Buechner (2017) | Pediatrics | 19–41BBz | 12 (3–23) | 68 | 61% | 83% | 12% | 36% relapse (68% CD19-) 6 months EFS: 73% |
| Lee (2015) | Pediatrics | 19–28z | 14 (5–27) | 20 | 38% | 70% | 71% | 10 months OS: 51.6% |
| Gardner (2017) | Pediatrics | 19–41BBz | 12 (1–25) | 45 | 62% | 89% | 24% | 12 months EFS: 51% (45% relapse) 12 months OS: 70% |
| ELIANA | MSKCC | ZUMA-3 | |
|---|---|---|---|
| CAR T-cell agent | Tisagenleucel | JCAR015 | Brexucabtagene autoleucel |
| Author (Years) | Maude (2018) | Park (2018) | Shah (2019) |
| Study phase | II | I | I/II |
| Study population | Pediatric/young adults | Adults | Adults |
| No. Patients | 75 | 53 | 45 |
| CR % | MRD negative: 81 | Overall: 83 | Overall: 68 RP2D: 84 |
| Median OS, mos | 19.1 | 12.9 | -- |
| Median EFS, mos | NR | 6.1 | -- |
| Median DoR, mos | NR | -- | RP2D: 12.9 |
| Median follow-up, mos | 13.1 | 29 | 16 |
| Trial | Phase | Treatment | Population | Endpoints |
|---|---|---|---|---|
| OBERON (NCT03628053) | III | Tisagenlecleucel vs. blinatumomab or inotuzumabozogamicin | Adults with B-cell precursor ALL; R/R after 1–2 lines of therapy or ASCT | OS |
| CASSIOPEIA (NCT03876769) | II | Tisagenlecleucel | Pediatric/young adult high-risk B-cell ALL; MRD positive after first-line therapy | DFS |
| ZUMA-4 (NCT02625480) | I/II | Axicabtageneciloleucel | Pediatric/adolescent pts with R/R B-precursor ALL or R/R B-cell NHL | AEs, CRR, ORR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martino, M.; Alati, C.; Canale, F.A.; Musuraca, G.; Martinelli, G.; Cerchione, C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042150
Martino M, Alati C, Canale FA, Musuraca G, Martinelli G, Cerchione C. A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2021; 22(4):2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042150
Chicago/Turabian StyleMartino, Massimo, Caterina Alati, Filippo Antonio Canale, Gerardo Musuraca, Giovanni Martinelli, and Claudio Cerchione. 2021. "A Review of Clinical Outcomes of CAR T-Cell Therapies for B-Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 22, no. 4: 2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042150