
Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway

 , , and
, , and 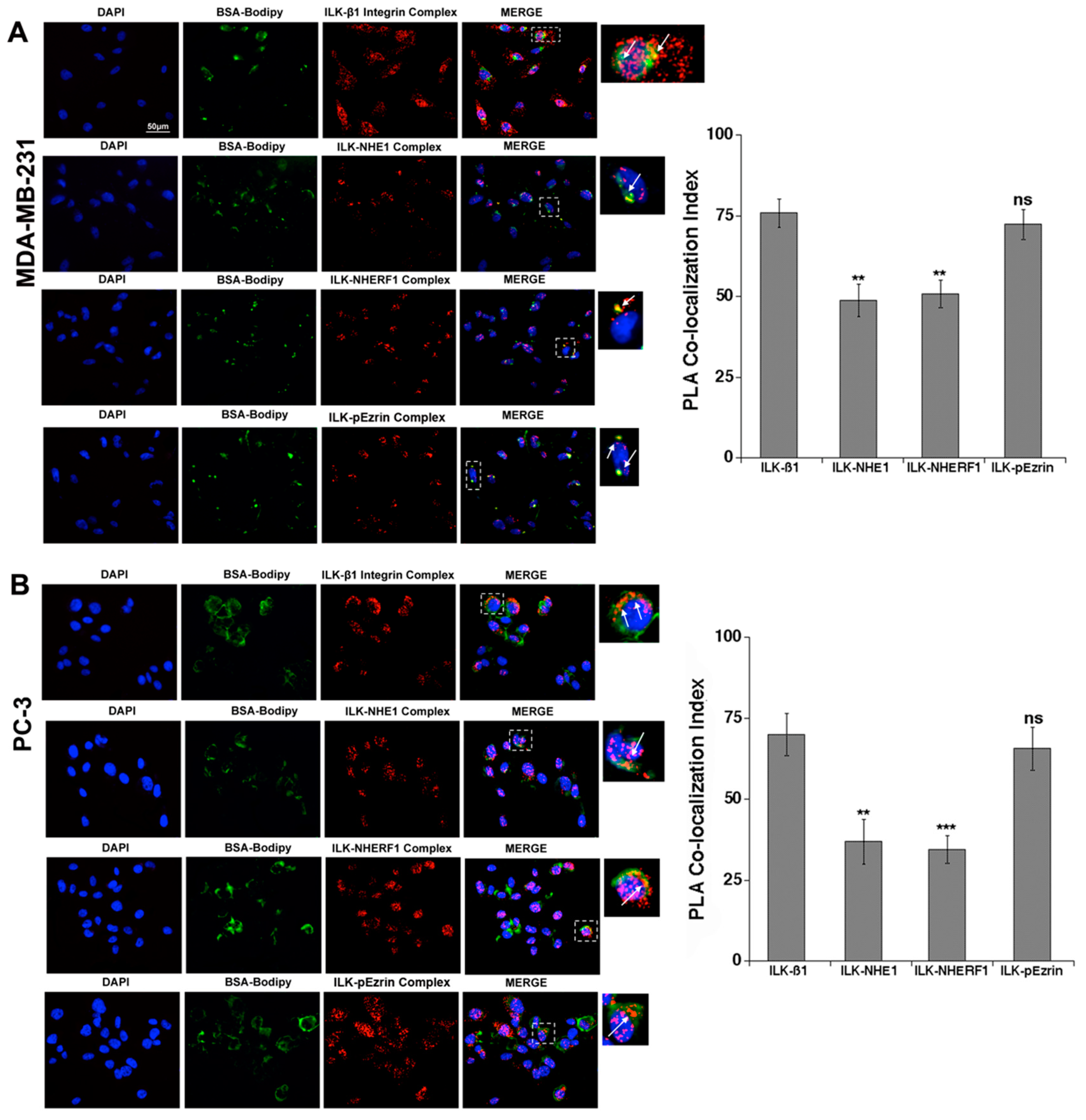{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
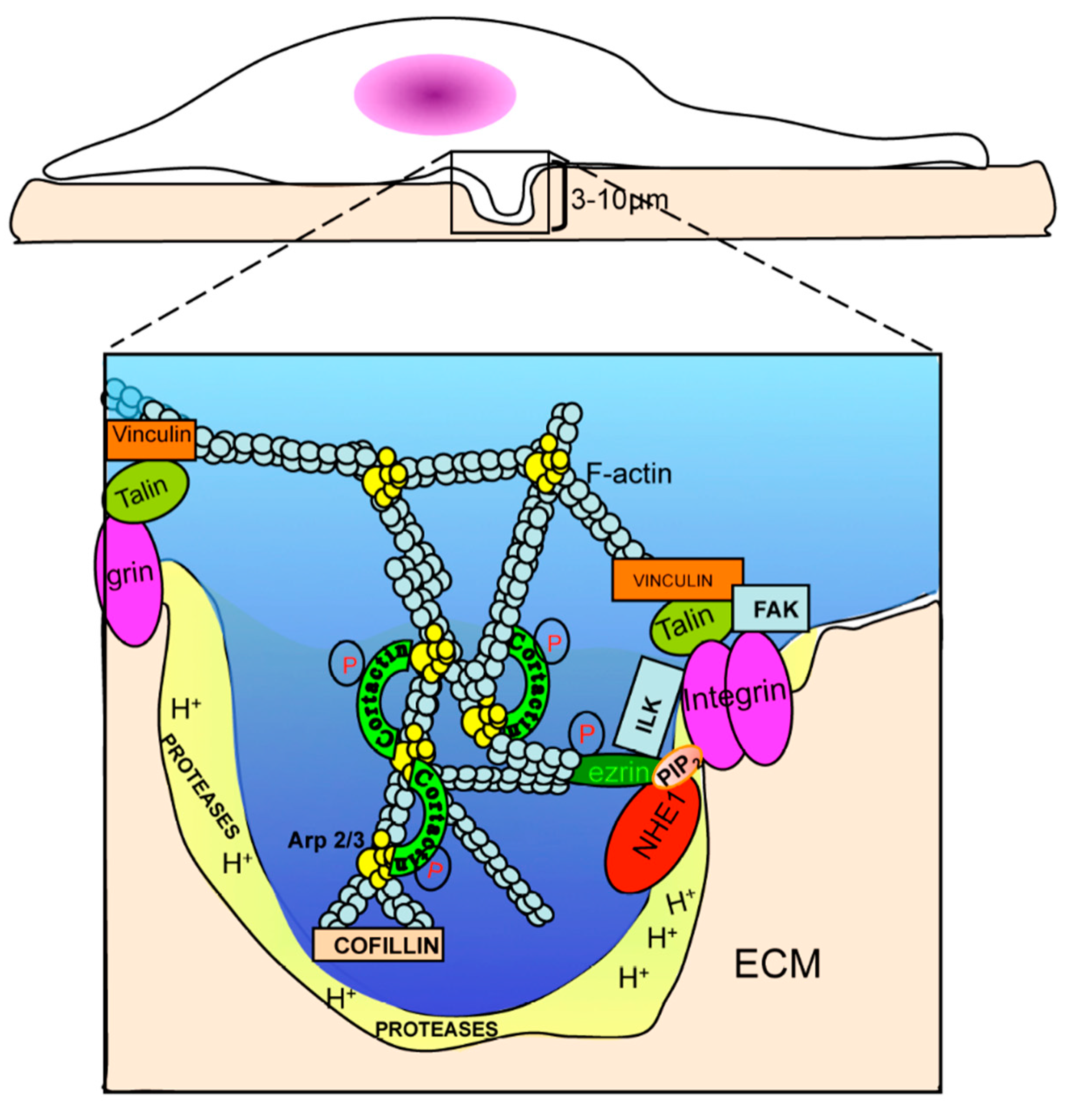
Abstract
:1. Introduction
2. Results
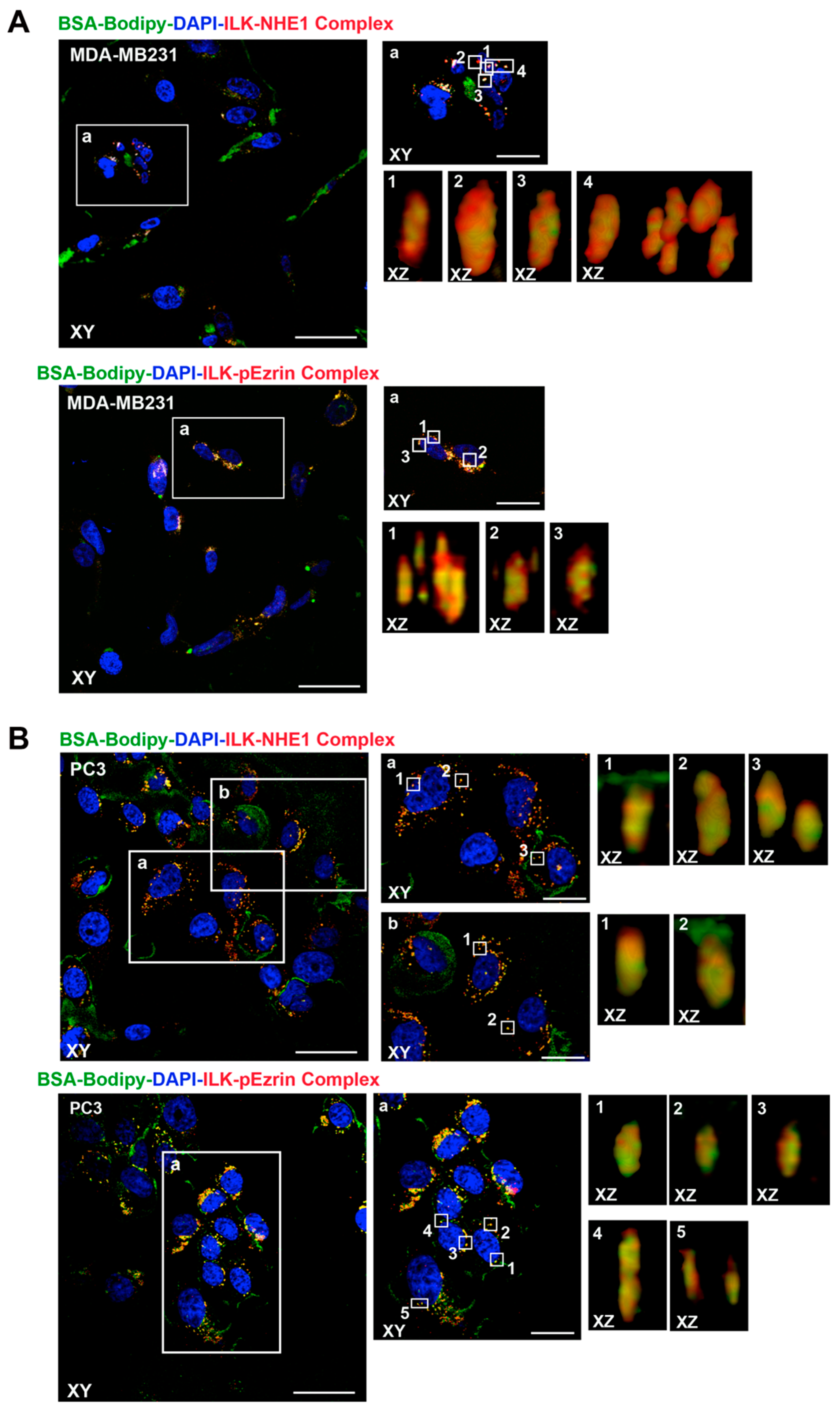
2.1. ILK Co-Localizes with β1-Integrin, NHERF1, p-Ezrin and NHE1 at Sites of Focal ECM Proteolysis
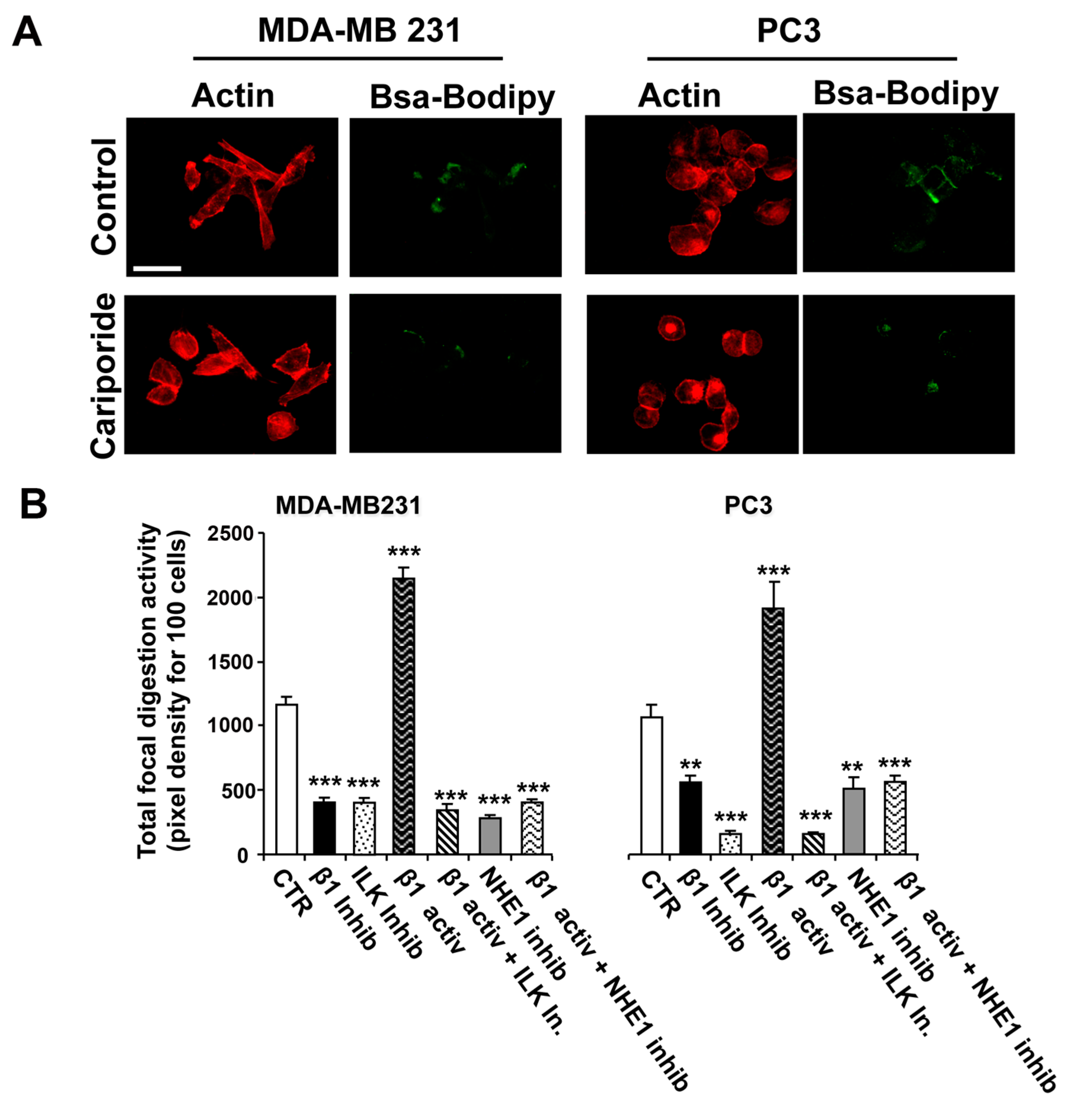
2.2. Role of ILK and NHE1 in Regulating β1-Integrin-Driven Invadopodia Proteolytic Activity
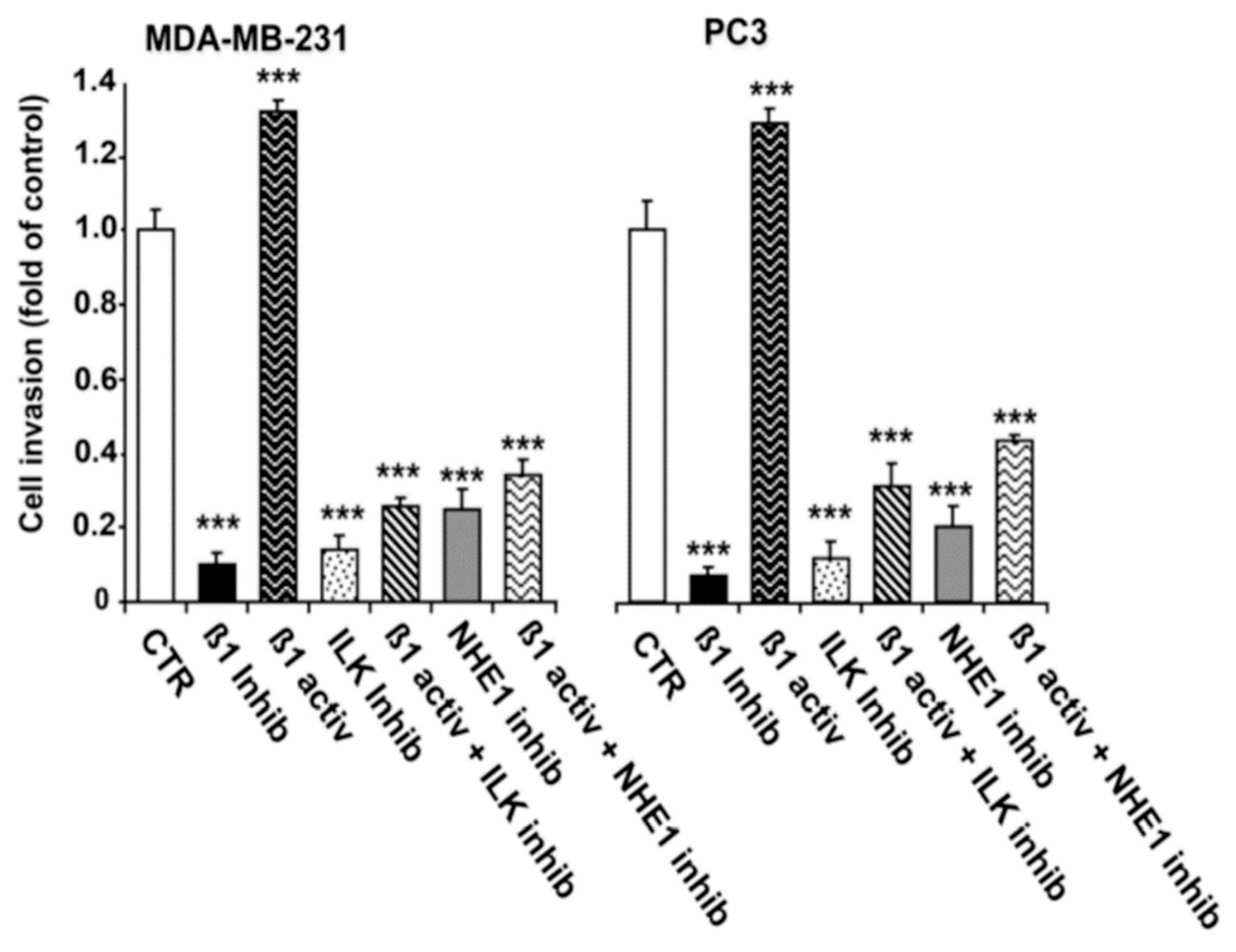
2.3. ILK and NHE1 Mediate β1-Integrin-Driven Invasion
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection of Constructs
4.2. Matrigel Layer Preparation and Invadopodia Activity Assay Using In Situ Zymography
4.3. Invasion across Matrigel Layer in Boyden Chambers
4.4. Proximity Ligation Assay
4.5. Image Analysis
4.6. Statistical Procedures
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Lehembre, F.; Regenass, U. Metastatic disease: A drug discovery perspective. Semin. Cancer Biol. 2012, 22, 261–271. [Google Scholar] [CrossRef]
- Fidler, I.J.; Kripke, M.L. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015, 34, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Linder, S.; Wiesner, C.; Himmel, M. Degrading devices: Invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 2011, 27, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H. Pathological roles of invadopodia in cancer invasion and metastasis. Eur. J. Cell Biol. 2012, 91, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Brisson, L.; Reshkin, S.J.; Gore, J.; Roger, S. pH regulators in invadosomal functioning: Proton delivery for matrix tasting. Eur. J. Cell Biol. 2012, 91, 847–860. [Google Scholar] [CrossRef]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [Green Version]
- Havrylov, S.; Park, M. MS/MS-based strategies for proteomic profiling of invasive cell structures. Proteomics 2015, 15, 272–286. [Google Scholar] [CrossRef]
- Seano, G.; Primo, L. Podosomes and invadopodia: Tools to breach vascular basement membrane. Cell Cycle 2015, 14, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Pelaez, R.; Pariente, A.; Perez-Sala, A.; Larrayoz, I.M. Integrins: Moonlighting Proteins in Invadosome Formation. Cancers 2019, 11, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Yang, J. Targeting invadopodia to block breast cancer metastasis. Oncotarget 2011, 2, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, E.K.; Courtneidge, S.A. Invadosomes are coming: New insights into function and disease relevance. FEBS J. 2018, 285, 8–27. [Google Scholar] [CrossRef]
- Ji, K.; Mayernik, L.; Moin, K.; Sloane, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Gil-Henn, H. Targeting invadopodia for blocking breast cancer metastasis. Drug Resist. Updates 2018, 39, 1–17. [Google Scholar] [CrossRef]
- Nakahara, H.; Mueller, S.C.; Nomizu, M.; Yamada, Y.; Yeh, Y.; Chen, W.T. Activation of beta1 integrin signaling stimulates tyrosine phosphorylation of p190RhoGAP and membrane-protrusive activities at invadopodia. J. Biol. Chem. 1998, 273, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, H.; Nomizu, M.; Akiyama, S.K.; Yamada, Y.; Yeh, Y.; Chen, W.T. A mechanism for regulation of melanoma invasion. Ligation of alpha6beta1 integrin by laminin G peptides. J. Biol. Chem. 1996, 271, 27221–27224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artym, V.V. Dense fibrillar collagen is a master activator of invadopodia. Mol. Cell. Oncol. 2016, 3, e1035476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artym, V.V.; Swatkoski, S.; Matsumoto, K.; Campbell, C.B.; Petrie, R.J.; Dimitriadis, E.K.; Li, X.; Mueller, S.C.; Bugge, T.H.; Gucek, M.; et al. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J. Cell Biol. 2015, 208, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Beaty, B.T.; Sharma, V.P.; Bravo-Cordero, J.J.; Simpson, M.A.; Eddy, R.J.; Koleske, A.J.; Condeelis, J. β1 integrin regulates Arg to promote invadopodial maturation and matrix degradation. Mol. Biol. Cell 2013, 24, 1661–1675. [Google Scholar] [CrossRef]
- Antelmi, E.; Cardone, R.A.; Greco, M.R.; Rubino, R.; di Sole, F.; Martino, N.A.; Casavola, V.; Carcangiu, M.; Moro, L.; Reshkin, S.J. β1 integrin binding phosphorylates ezrin at T567 to activate a lipid raft signalsome driving invadopodia activity and invasion. PLoS ONE 2013, 8, e75113. [Google Scholar] [CrossRef] [Green Version]
- Bellizzi, A.; Mangia, A.; Malfettone, A.; Cardone, R.A.; Simone, G.; Reshkin, S.J.; Paradiso, A. Na+/H+ exchanger regulatory factor 1 expression levels in blood and tissue predict breast tumour clinical behaviour. Histopathology 2011, 58, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.R.; Antelmi, E.; Busco, G.; Guerra, L.; Rubino, R.; Casavola, V.; Reshkin, S.J.; Cardone, R.A. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol. Rep. 2014, 31, 940–946. [Google Scholar] [CrossRef]
- Greco, M.R.; Bon, E.; Rubino, R.; Guerra, L.; Bernabe-Garcia, M.; Cannone, S.; Cayuela, M.L.; Ciaccia, L.; Marionneau-Lambot, S.; Oullier, T.; et al. Phosphorylation of NHERF1 S279 and S301 differentially regulates breast cancer cell phenotype and metastatic organotropism. Biochimica et biophysica acta. Mol. Basis Dis. 2019, 1865, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Beaty, B.T.; Condeelis, J. Digging a little deeper: The stages of invadopodium formation and maturation. Eur. J. Cell Biol. 2014, 93, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, M.A.; Larson, D.R.; Mader, C.C.; Bravo-Cordero, J.J.; Gil-Henn, H.; Oser, M.; Chen, X.; Koleske, A.J.; Condeelis, J. Cortactin phosphorylation regulates cell invasion through a pH-dependent pathway. J. Cell Biol. 2011, 195, 903–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, P.; Schwab, A.; et al. A Novel NHE1-Centered Signaling Cassette Drives Epidermal Growth Factor Receptor–Dependent Pancreatic Tumor Metastasis and Is a Target for Combination Therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Bellizzi, A.; Greco, M.R.; Rubino, R.; Paradiso, A.; Forciniti, S.; Zeeberg, K.; Cardone, R.A.; Reshkin, S.J. The scaffolding protein NHERF1 sensitizes EGFR-dependent tumor growth, motility and invadopodia function to gefitinib treatment in breast cancer cells. Int. J. Oncol. 2015, 46, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Lucien, F.; Brochu-Gaudreau, K.; Arsenault, D.; Harper, K.; Dubois, C.M. Hypoxia-induced invadopodia formation involves activation of NHE-1 by the p90 ribosomal S6 kinase (p90RSK). PLoS ONE 2011, 6, e28851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, S. The matrix corroded: Podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- Zellweger, M.J. Looking at the whole picture. J. Nucl. Cardiol. 2015, 22, 901–902. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.J.; Ortega, F.G.; Alvarez-Cubero, M.J.; Nadal, R.; Sanchez-Rovira, P.; Salido, M.; Rodriguez, M.; Garcia-Puche, J.L.; Delgado-Rodriguez, M.; Sole, F.; et al. EMT and EGFR in CTCs cytokeratin negative non-metastatic breast cancer. Oncotarget 2014, 5, 7486–7497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kharbili, M.; Robert, C.; Witkowski, T.; Danty-Berger, E.; Barbollat-Boutrand, L.; Masse, I.; Gadot, N.; de la Fouchardiere, A.; McDonald, P.C.; Dedhar, S.; et al. Tetraspanin 8 is a novel regulator of ILK-driven beta1 integrin adhesion and signaling in invasive melanoma cells. Oncotarget 2017, 8, 17140–17155. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.C.; Hu, H.F.; Hong, P.; Zhang, Q.H.; Xu, W.W.; He, Q.Y.; Li, B. Significance of integrin-linked kinase (ILK) in tumorigenesis and its potential implication as a biomarker and therapeutic target for human cancer. Am. J. Cancer Res. 2019, 9, 186–197. [Google Scholar]
- Branch, K.M.; Hoshino, D.; Weaver, A.M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 2012, 1, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013, 5, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- LaFlamme, S.E.; Mathew-Steiner, S.; Singh, N.; Colello-Borges, D.; Nieves, B. Integrin and microtubule crosstalk in the regulation of cellular processes. Cell. Mol. Life Sci. 2018, 75, 4177–4185. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Greco, M.R.; Capulli, M.; Weinman, E.J.; Busco, G.; Bellizzi, A.; Casavola, V.; Antelmi, E.; Ambruosi, B.; Dell’Aquila, M.E.; et al. NHERF1 acts as a molecular switch to program metastatic behavior and organotropism via its PDZ domains. Mol. Biol. Cell 2012, 23, 2028–2040. [Google Scholar] [CrossRef]
- Cardone, R.A.; Bellizzi, A.; Busco, G.; Weinman, E.J.; Dell’Aquila, M.E.; Casavola, V.; Azzariti, A.; Mangia, A.; Paradiso, A.; Reshkin, S.J. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol. Biol. Cell 2007, 18, 1768–1780. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, G.; Ray, R.; Misra, U.K.; Pizzo, S.V. Modulation of the unfolded protein response by GRP78 in prostate cancer. Methods Enzymol. 2011, 489, 245–257. [Google Scholar]
- Cardone, R.A.; Bagorda, A.; Bellizzi, A.; Busco, G.; Guerra, L.; Paradiso, A.; Casavola, V.; Zaccolo, M.; Reshkin, S.J. Protein kinase A gating of a pseudopodial-located RhoA/ROCK/p38/NHE1 signal module regulates invasion in breast cancer cell lines. Mol. Biol. Cell 2005, 16, 3117–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderberg, O.; Leuchowius, K.J.; Gullberg, M.; Jarvius, M.; Weibrecht, I.; Larsson, L.G.; Landegren, U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 2008, 45, 227–232. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, M.R.; Moro, L.; Forciniti, S.; Alfarouk, K.; Cannone, S.; Cardone, R.A.; Reshkin, S.J. Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway. Int. J. Mol. Sci. 2021, 22, 2162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042162
Greco MR, Moro L, Forciniti S, Alfarouk K, Cannone S, Cardone RA, Reshkin SJ. Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway. International Journal of Molecular Sciences. 2021; 22(4):2162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042162
Chicago/Turabian StyleGreco, Maria Raffaella, Loredana Moro, Stefania Forciniti, Khalid Alfarouk, Stefania Cannone, Rosa Angela Cardone, and Stephan Joel Reshkin. 2021. "Integrin-Linked Kinase Links Integrin Activation to Invadopodia Function and Invasion via the p(T567)-Ezrin/NHERF1/NHE1 Pathway" International Journal of Molecular Sciences 22, no. 4: 2162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042162