
Exploring Molecular Contacts of MUC1 at CIN85 Binding Interface to Address Future Drug Design Efforts

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
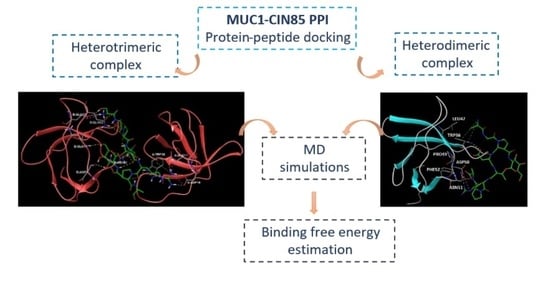
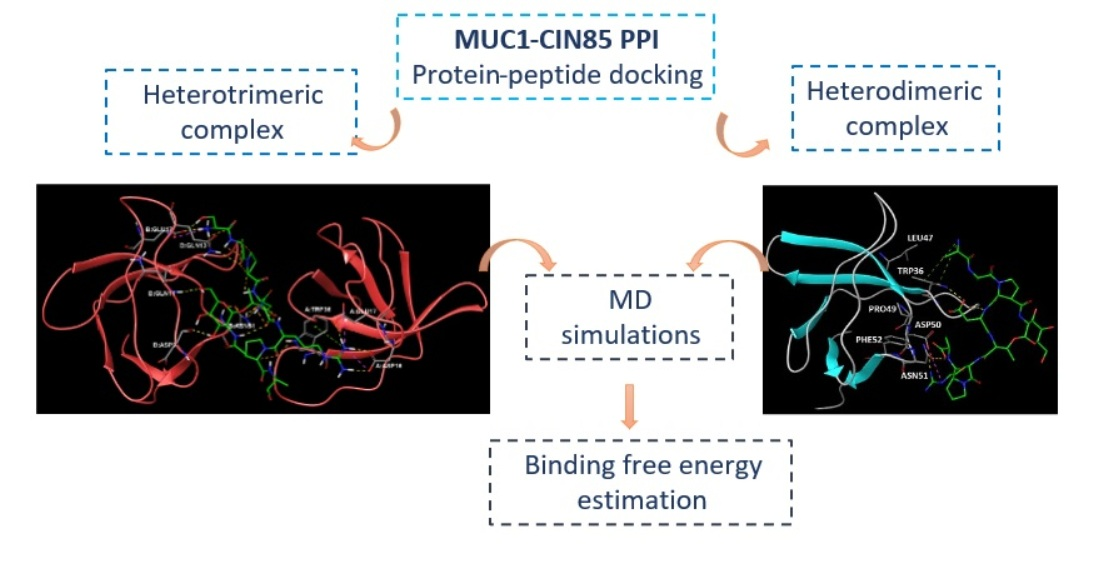
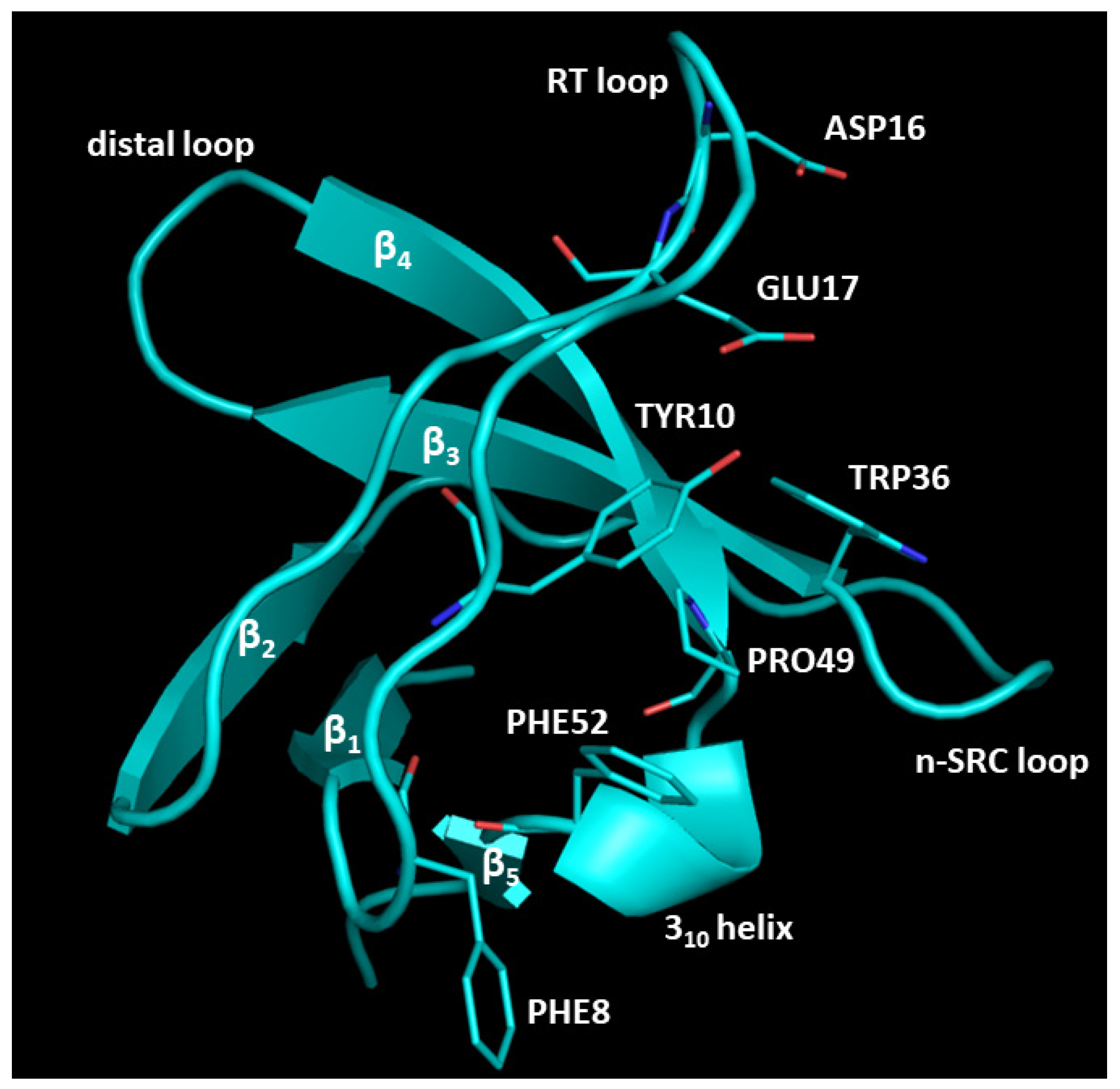
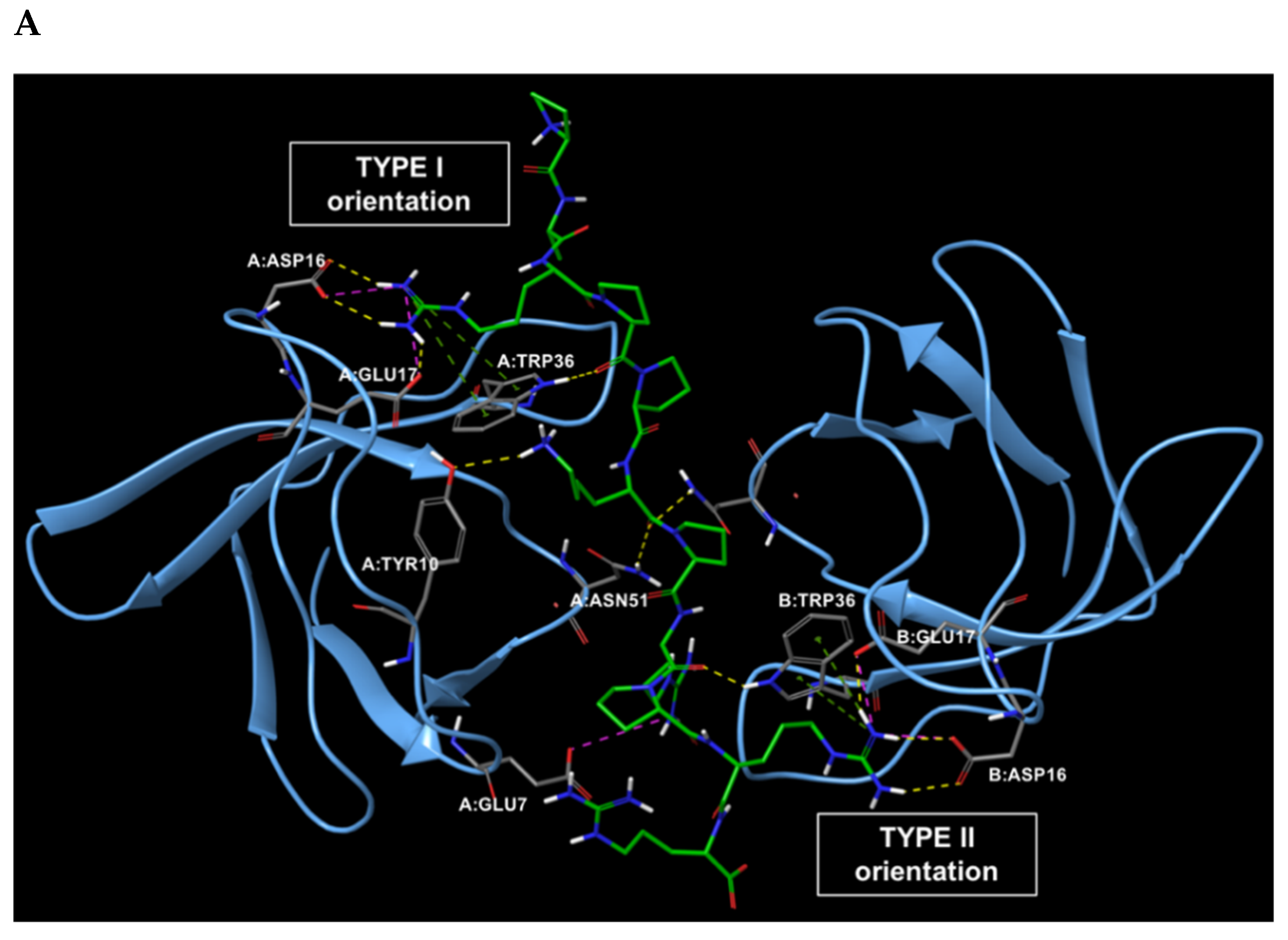
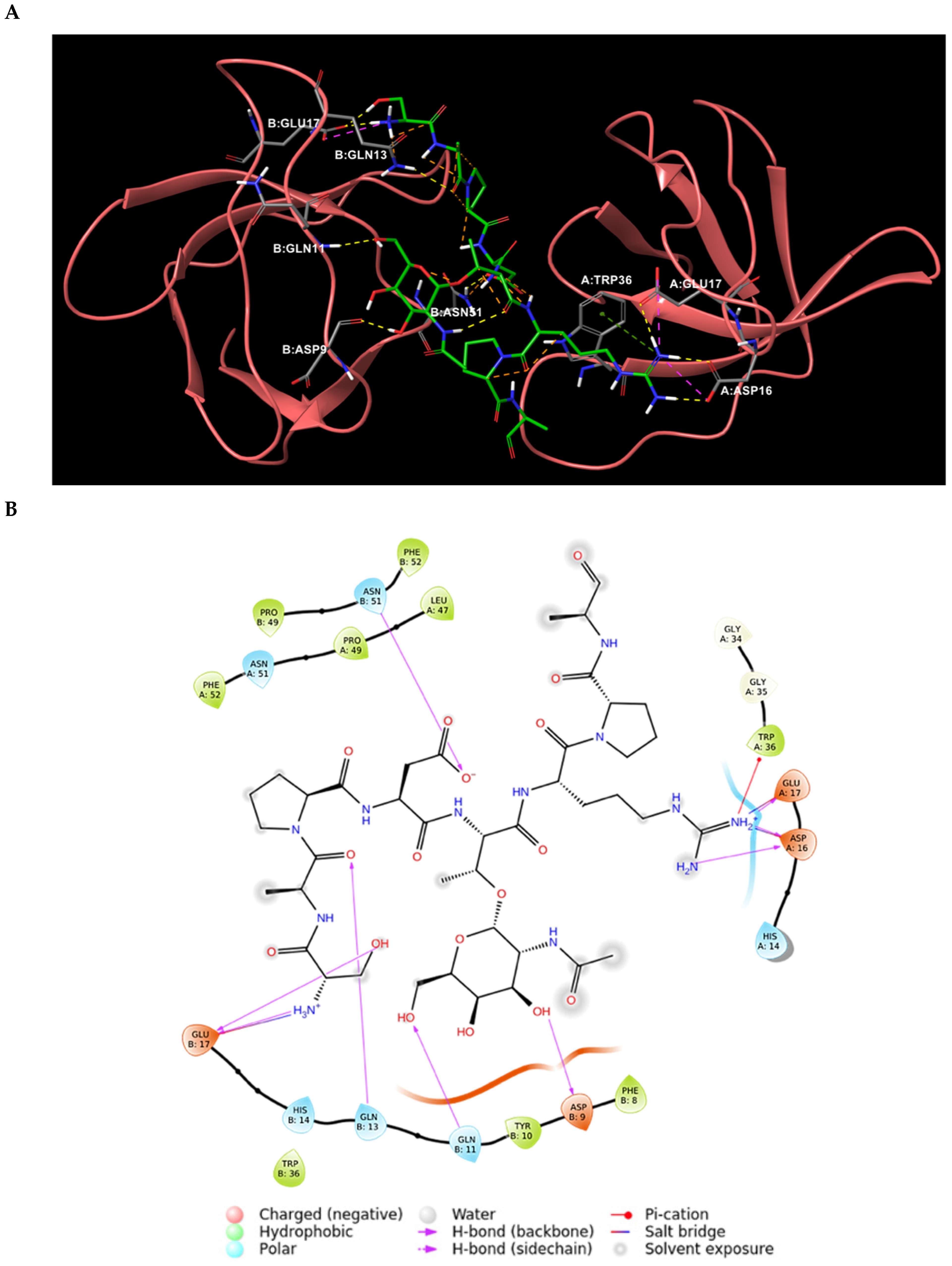
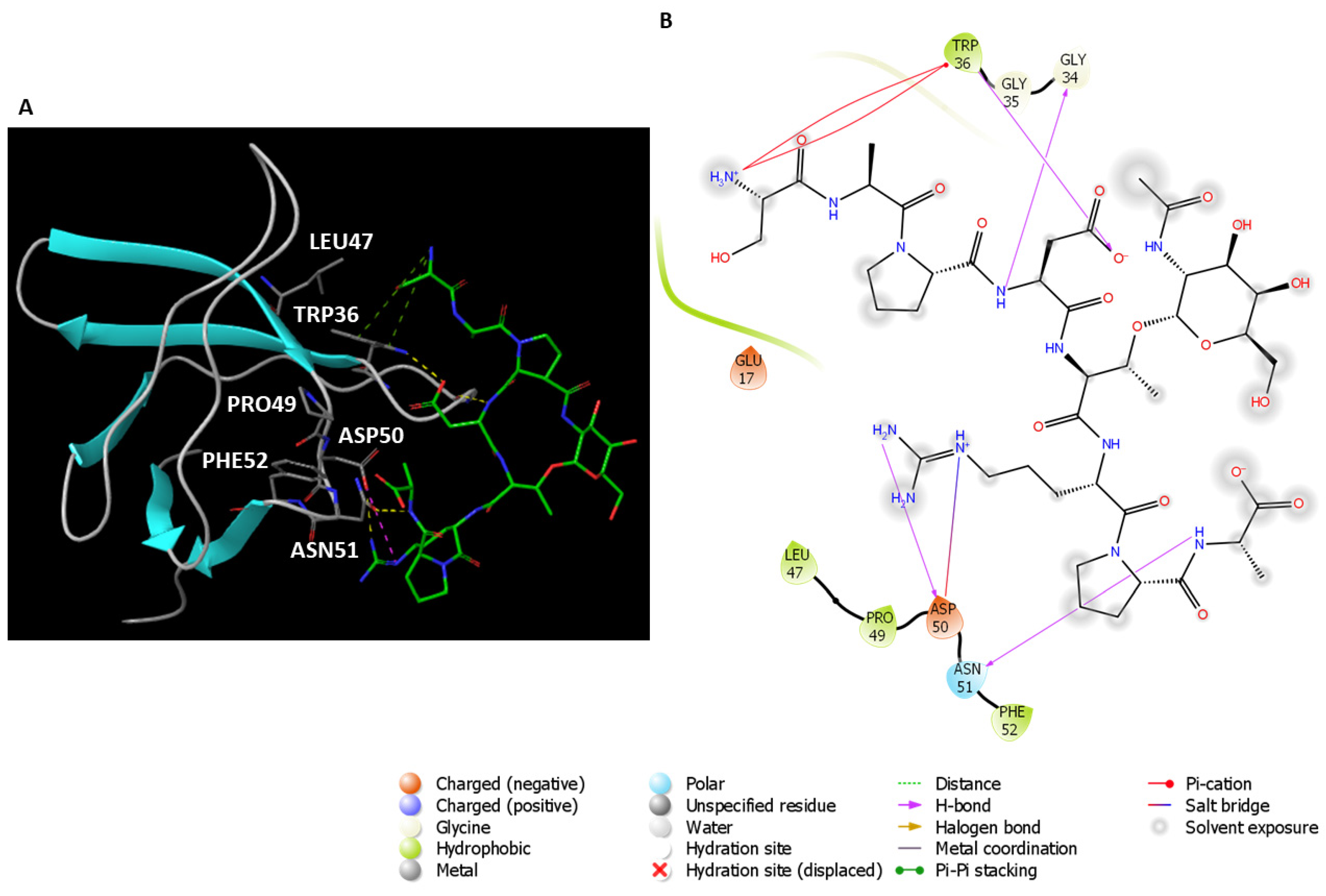
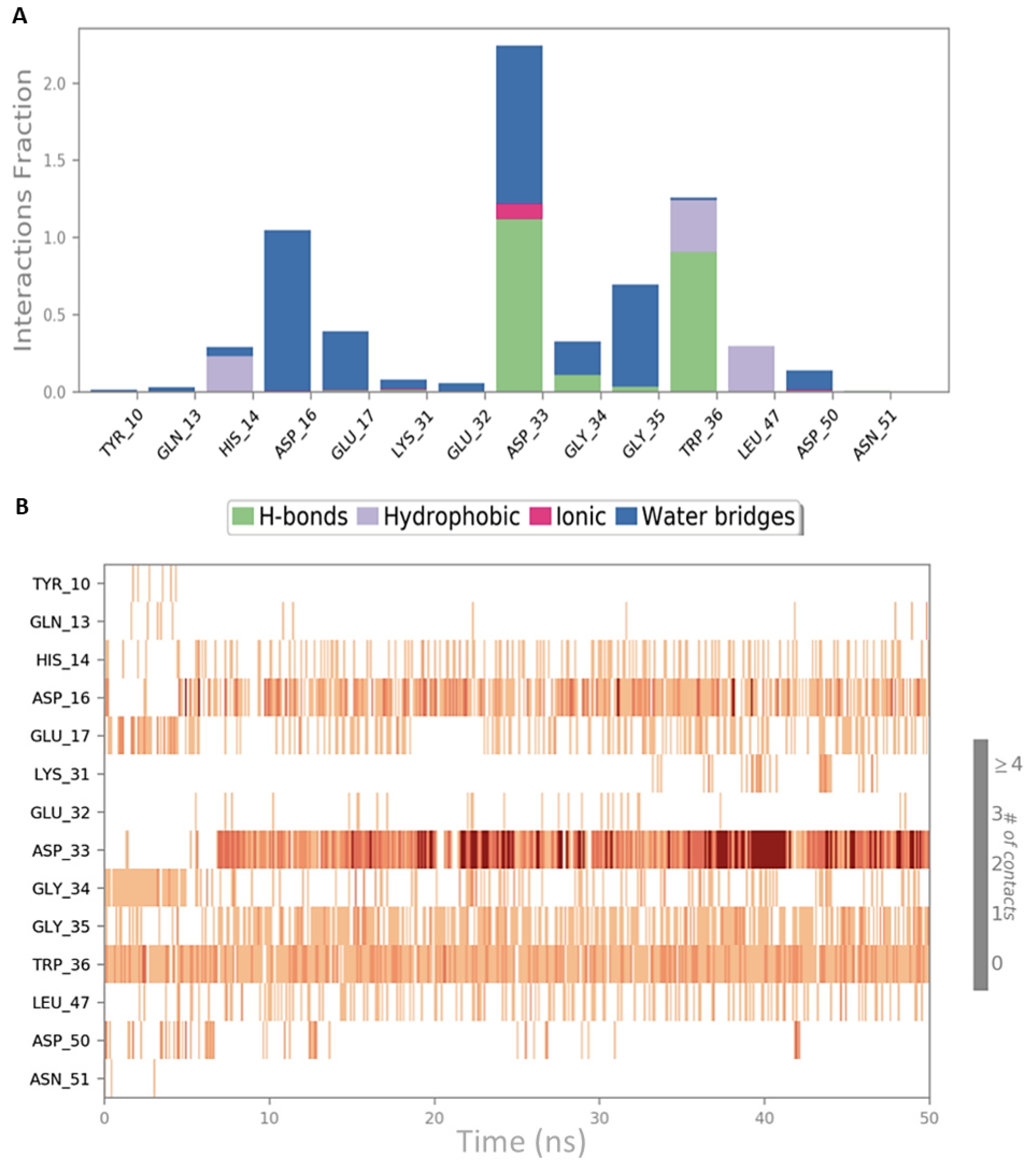
2.1. Analysis of the Experimentally Solved CIN85-Cbl-b Heterotrimeric Complex and MD Exploration
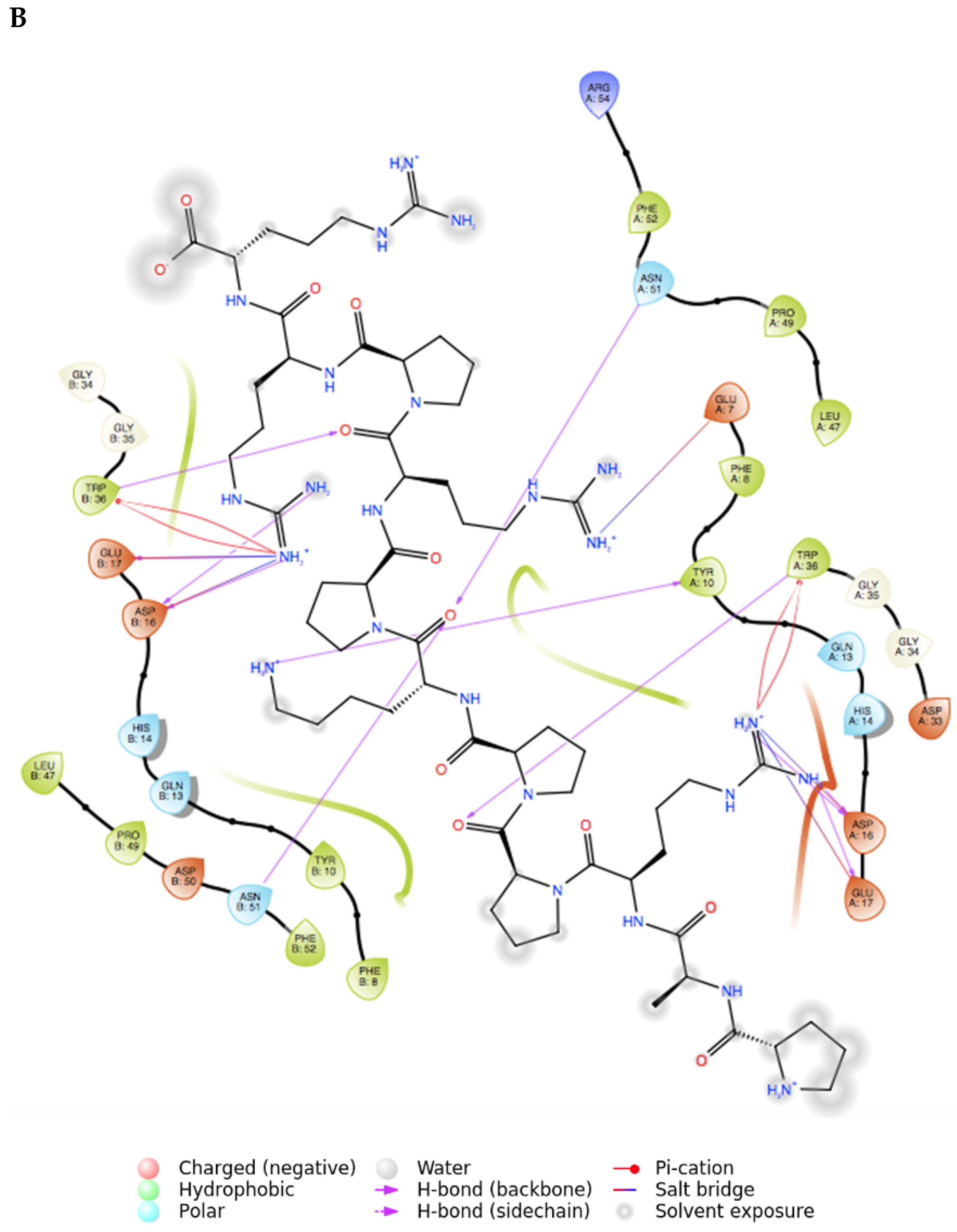
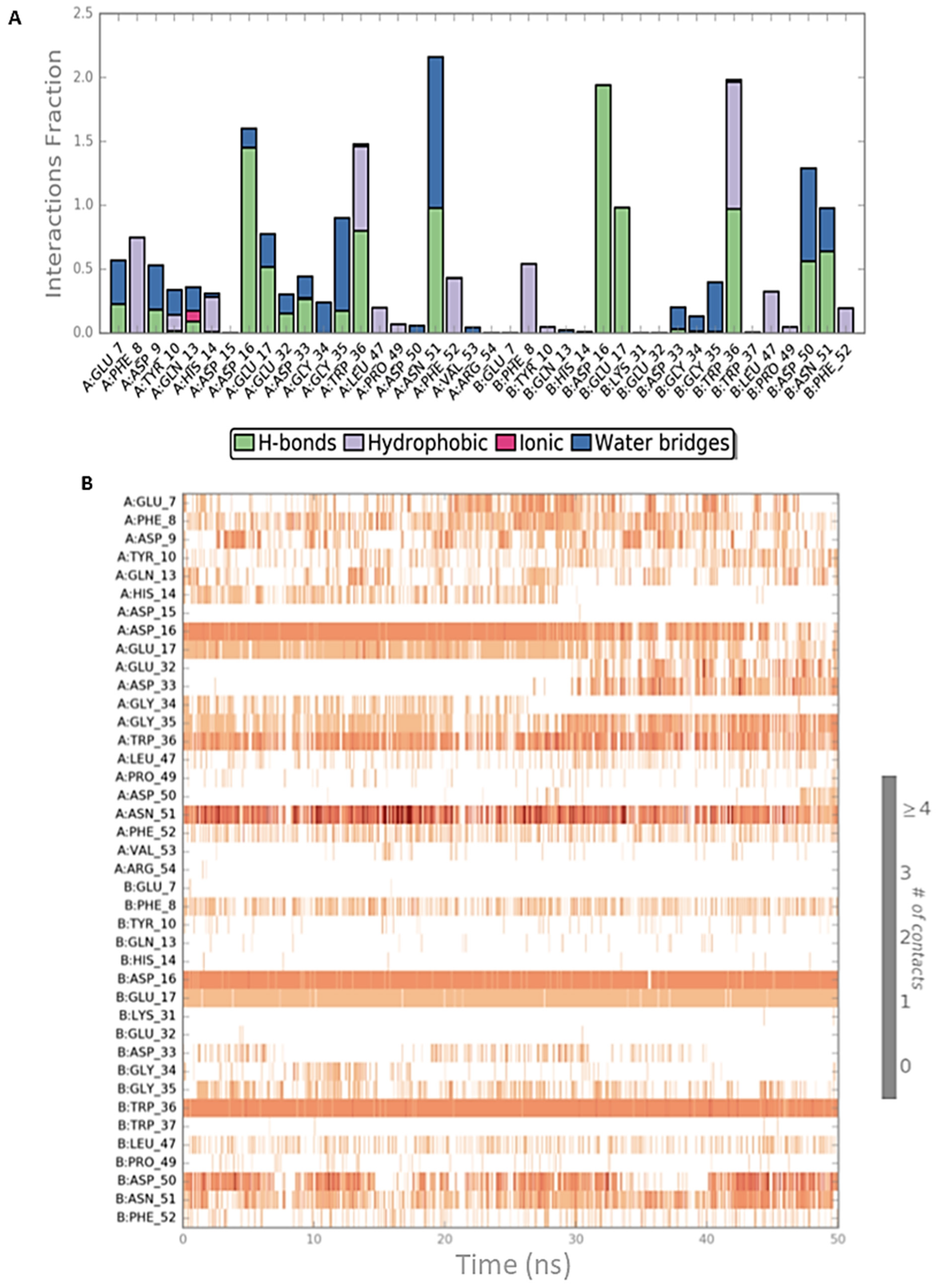
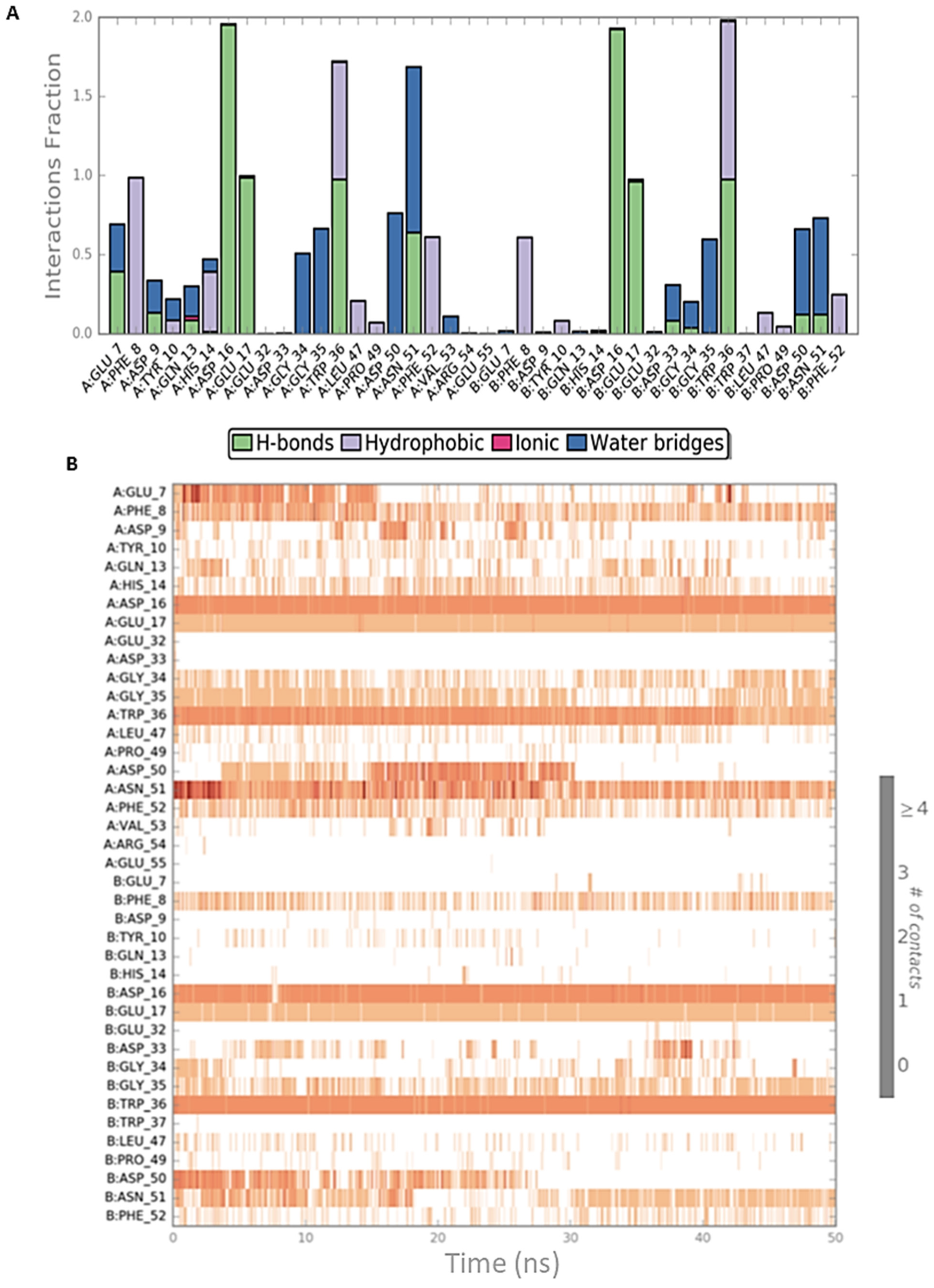
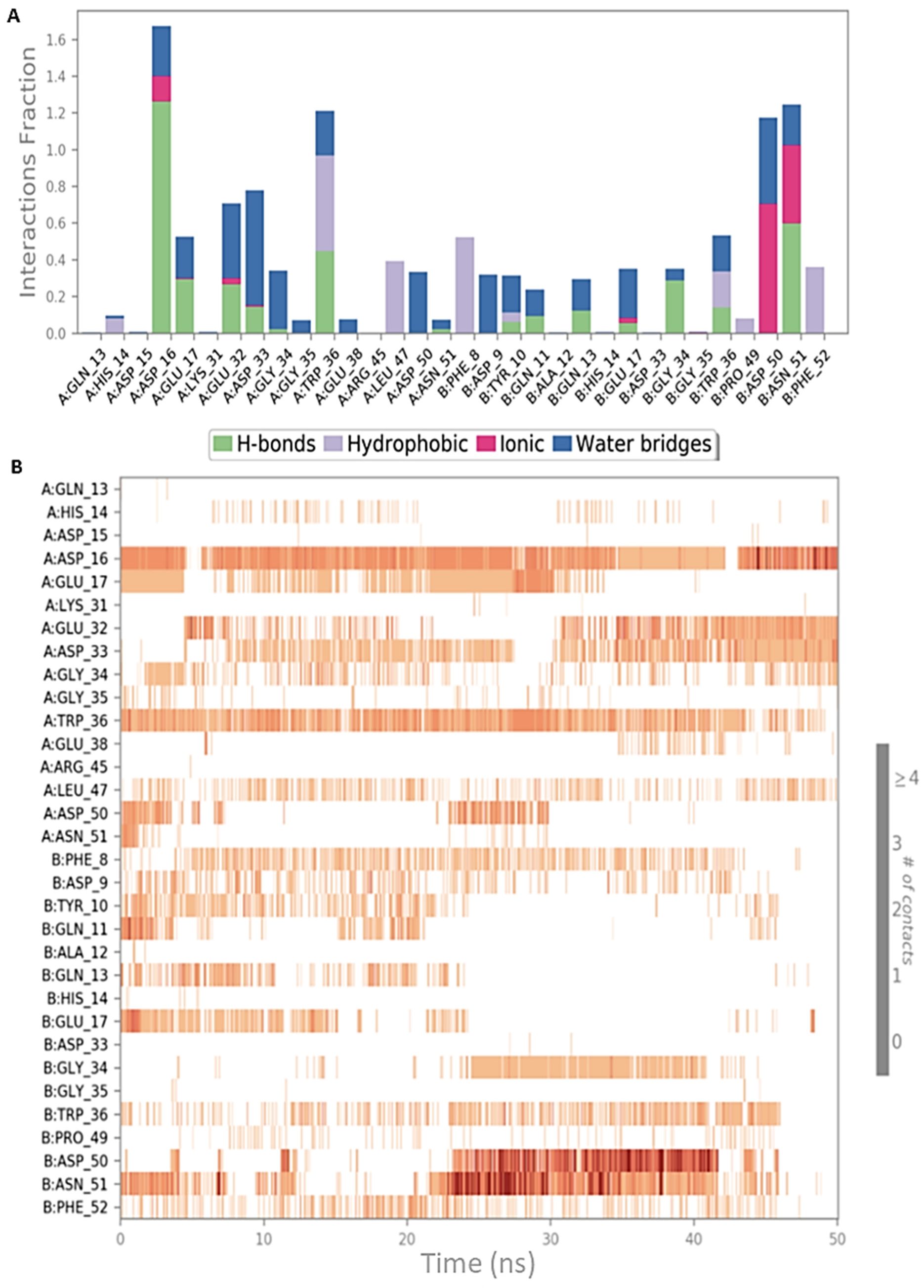
2.2. Peptide Docking and MD Simulations of CIN85 Dimer and MUC1 Peptide
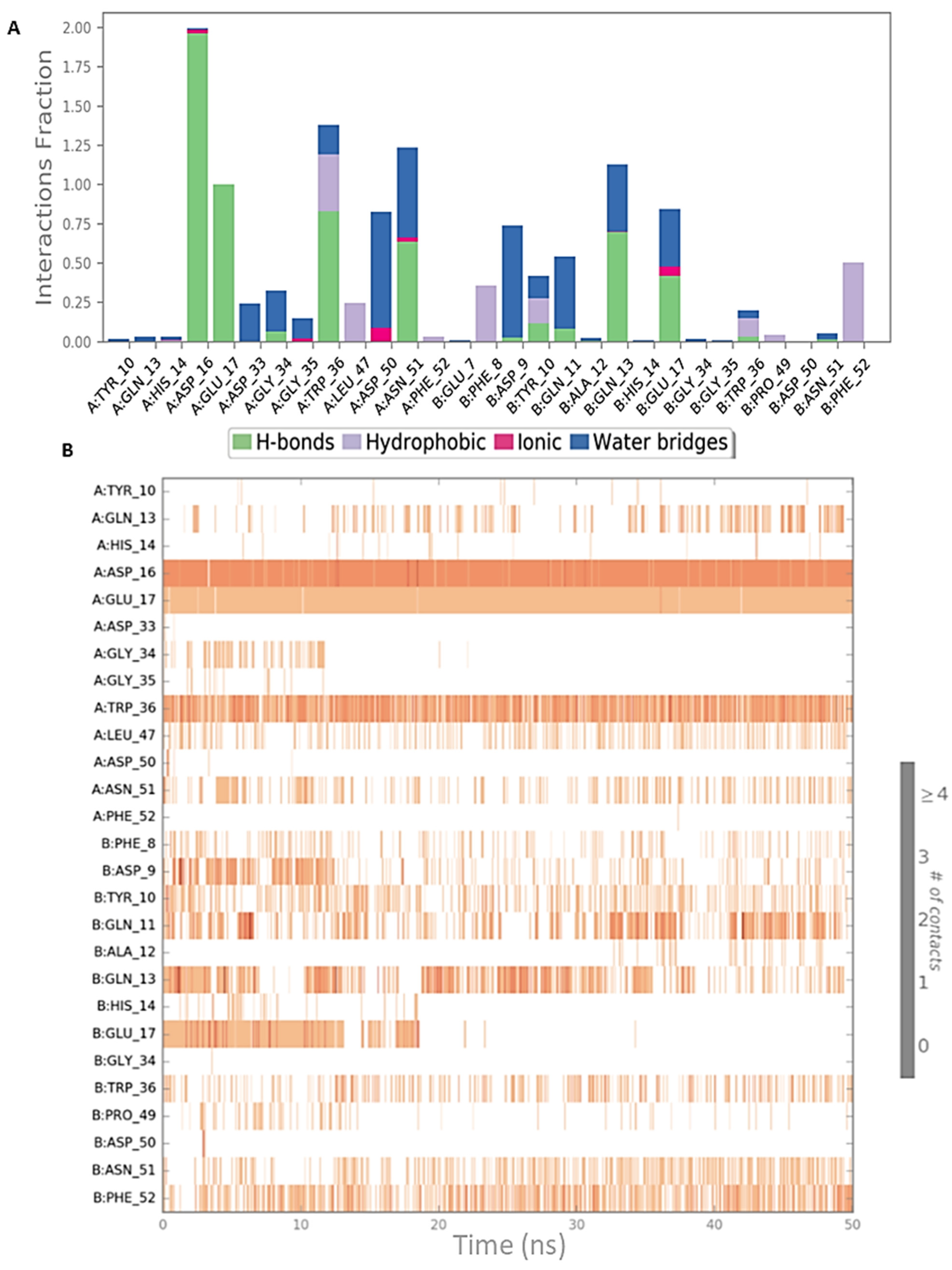
2.3. Peptide Docking and MD Simulation of CIN85 Monomer and MUC1 Peptide
2.4. MM-GBSA Calculations of MD Simulations
3. Materials and Methods
3.1. Preparation of PDB Structures
3.2. Receptor Grids Generation of CIN85 Dimer and Monomer and Peptide Docking
3.3. MD Simulations of CIN85 in Complex with MUC1 and Cbl-b and MM-GBSA Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shin, W.-H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein–protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef]
- Perricone, U.; Gulotta, M.R.; Lombino, J.; Parrino, B.; Cascioferro, S.; Diana, P.; Cirrincione, G.; Padova, A. An overview of recent molecular dynamics applications as medicinal chemistry tools for the undruggable site challenge. Medchemcomm 2018, 9, 920–936. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Li, X.; He, X.; Pu, J.; Zhang, J.; Lu, S. Computational methods-guided design of modulators targeting protein-protein interactions (PPIs). Eur. J. Med. Chem. 2020, 207, 112764. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-J.; Lei, P.-M.; Liu, H.; Wu, C.; Leung, C.-H.; Ma, D.-L. Mimicking Strategy for Protein–Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules 2019, 24, 4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, S.; Finn, O. Complex of MUC1, CIN85 and Cbl in Colon Cancer Progression and Metastasis. Cancers 2015, 7, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascio, S.; Farkas, A.M.; Hughey, R.P.; Finn, O.J. Altered glycosylation of MUC1 influences its association with CIN85: The role of this novel complex in cancer cell invasion and migration. Oncotarget 2013, 4, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Cen, Q.; Lei, H. A review on development of MUC1-based cancer vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, D.M.; Cudic, M. Tumor-associated O-glycans of MUC1: Carriers of the glyco-code and targets for cancer vaccine design. Semin. Immunol. 2020, 47, 101389. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. CIN85/CMS family of adaptor molecules. FEBS Lett. 2002, 529, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein–protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saksela, K.; Permi, P. SH3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Deloia, M.A.; Kang, Y.; Litchke, C.; Zhang, N.; Titus, M.A.; Walters, K.J. The SH3 domain of a M7 interacts with its C-terminal proline-rich region. Protein Sci. 2007, 16, 189–196. [Google Scholar] [CrossRef]
- Jozic, D.; Cárdenes, N.; Deribe, Y.L.; Moncalián, G.; Hoeller, D.; Groemping, Y.; Dikic, I.; Rittinger, K.; Bravo, J. Cbl promotes clustering of endocytic adaptor proteins. Nat. Struct. Mol. Biol. 2005, 12, 972–979. [Google Scholar] [CrossRef]
- Büchse, T.; Horras, N.; Lenfert, E.; Krystal, G.; Körbel, S.; Schümann, M.; Krause, E.; Mikkat, S.; Tiedge, M. CIN85 Interacting Proteins in B Cells-Specific Role for SHIP-1. Mol. Cell. Proteom. 2011, 10, M110.006239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidos, G.; Soni, S.; Oswald, D.J.; Toselli, P.A.; Kirsch, K.H. Structure and function analysis of the CMS/CIN85 protein family identifies actin-bundling properties and heterotypic-complex formation. J. Cell Sci. 2007, 120, 2366–2377. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, W.; Wang, Y.; Liu, W. Biochemical and Structural Studies of the Interaction between ARAP1 and CIN85. Biochemistry 2018, 57, 2132–2139. [Google Scholar] [CrossRef]
- Cascio, S.; Sciurba, J.; Hughey, R.; Camacho, C.; Finn, O. Abstract 3151: Muc1/Cin85 complex is a new molecular target for control of cancer invasion and metastasis. In Proceedings of the Tumor Biology, AACR Annual Meeting 2014, San Diego, CA, USA, 5–9 April 2014; American Association for Cancer Research: Philadelphia, PA, USA, 2014; p. 3151. [Google Scholar]
- Vittorio, S.; Seidel, T.; Garon, A.; Gitto, R.; Langer, T.; De Luca, L. In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain. Int. J. Mol. Sci. 2021, 22, 534. [Google Scholar] [CrossRef] [PubMed]
- Ceregido, M.A.; Garcia-Pino, A.; Ortega-Roldan, J.L.; Casares, S.; López Mayorga, O.; Bravo, J.; van Nuland, N.A.J.; Azuaga, A.I. Multimeric and differential binding of CIN85/CD2AP with two atypical proline-rich sequences from CD2 and Cbl-b*. FEBS J. 2013, 280, 3399–3415. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Association for Computing Machinery: New York, NY, USA, 2006. [Google Scholar]
- Somovilla, V.J.; Bermejo, I.A.; Albuquerque, I.S.; Martínez-Sáez, N.; Castro-López, J.; García-Martín, F.; Compañón, I.; Hinou, H.; Nishimura, S.-I.; Jiménez-Barbero, J.; et al. The Use of Fluoroproline in MUC1 Antigen Enables Efficient Detection of Antibodies in Patients with Prostate Cancer. J. Am. Chem. Soc. 2017, 139, 18255–18261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protein Data Bank. Available online: https://pdb101.rcsb.org (accessed on 21 May 2020).
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Wakui, H.; Tanaka, Y.; Ose, T.; Matsumoto, I.; Kato, K.; Min, Y.; Tachibana, T.; Sato, M.; Naruchi, K.; Martin, F.G.; et al. A straightforward approach to antibodies recognising cancer specific glycopeptidic neoepitopes. Chem. Sci. 2020, 11, 4999–5006. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction Type | Cbl-b Peptide | CIN85 SH3 Domains |
|---|---|---|
| H-Bond | Arg904, Arg911, Lys907, Arg909 | Asp16, Glu17, Asn51, Trp36 |
| Salt bridge | Arg904, Arg911 | Asp16, Glu17 |
| π-Cation | Arg904, Arg911 | Trp36 |
| Hydrophobic | Pro906, Pro908, Pro910 | Trp36, Phe52 |
| MD Simulations of the Complexes | ||||
|---|---|---|---|---|
| CIN85 Dimer—Cbl-b Peptide | CIN85 Dimer—MUC1 Peptide | CIN85 Monomer—MUC1 Peptide | ||
| First MD | Average ΔGbinding (kcal/mol) | −141.449 | −54.624 | −36.009 |
| ΔGbinding range (kcal/mol) | −164.158 to −112.656 | −101.514 to −18.227 | −55.595 to −10.065 | |
| Second MD | Average ΔGbinding (kcal/mol) | −136.904 | −62.681 | −26.516 |
| ΔGbinding range (kcal/mol) | −163.5629 to −116.8524 | −109.318 to −34.366 | −47.012 to −6.81 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulotta, M.R.; Vittorio, S.; Gitto, R.; Perricone, U.; De Luca, L. Exploring Molecular Contacts of MUC1 at CIN85 Binding Interface to Address Future Drug Design Efforts. Int. J. Mol. Sci. 2021, 22, 2208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042208
Gulotta MR, Vittorio S, Gitto R, Perricone U, De Luca L. Exploring Molecular Contacts of MUC1 at CIN85 Binding Interface to Address Future Drug Design Efforts. International Journal of Molecular Sciences. 2021; 22(4):2208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042208
Chicago/Turabian StyleGulotta, Maria Rita, Serena Vittorio, Rosaria Gitto, Ugo Perricone, and Laura De Luca. 2021. "Exploring Molecular Contacts of MUC1 at CIN85 Binding Interface to Address Future Drug Design Efforts" International Journal of Molecular Sciences 22, no. 4: 2208. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042208