
GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
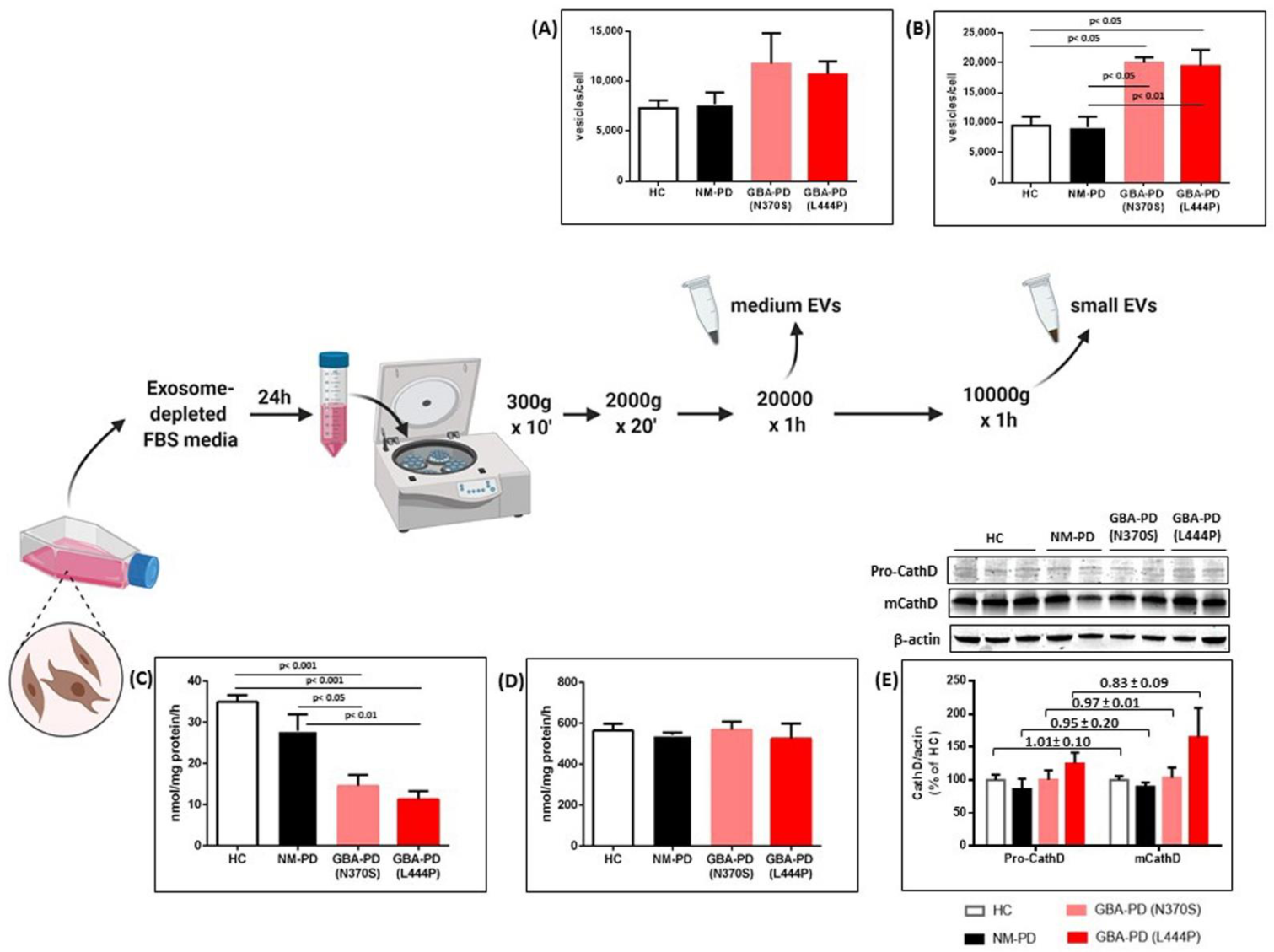
2.1. GCase Deficiency Promotes the Release of Small Extracellular Vesicles
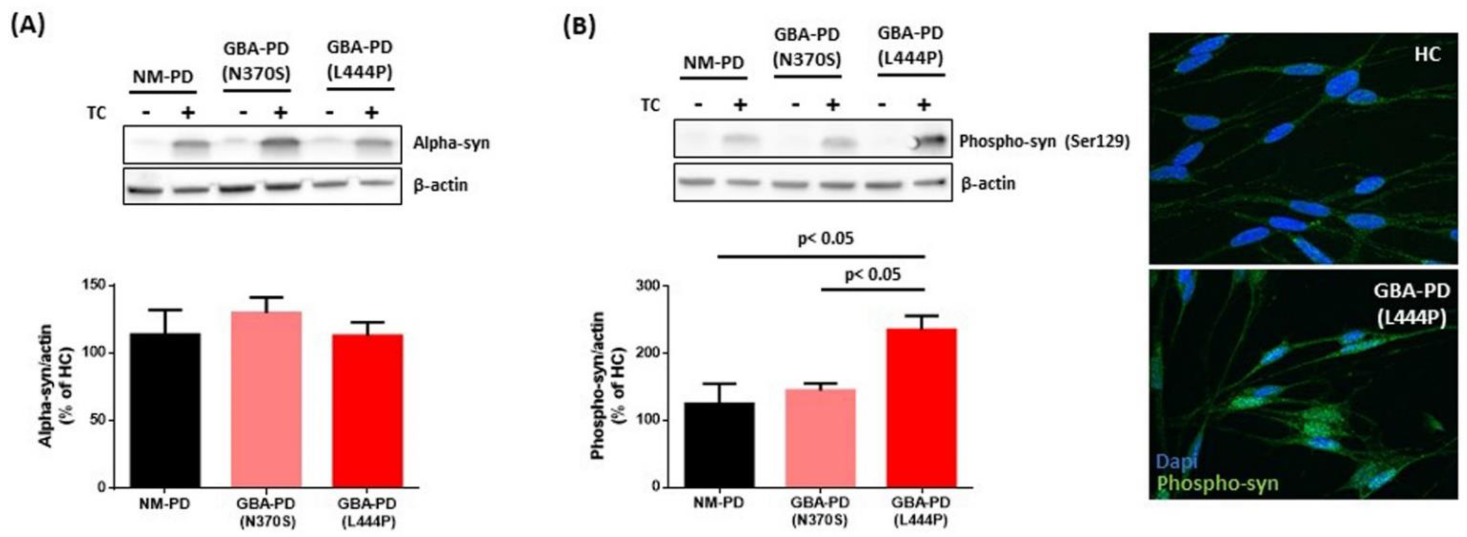
2.2. Small EVs Derived from PD Fibroblasts Carrying Severe GBA Mutation Increase the Levels of Phospho-Ser129 α-Syn in Recipient Cells
3. Discussion
4. Materials and Methods
4.1. Fibroblast Cell Culture
4.2. Enzyme Activity Assay
4.3. EV Isolation and Quantification
4.4. Transmission Electron Microscopy (TEM)
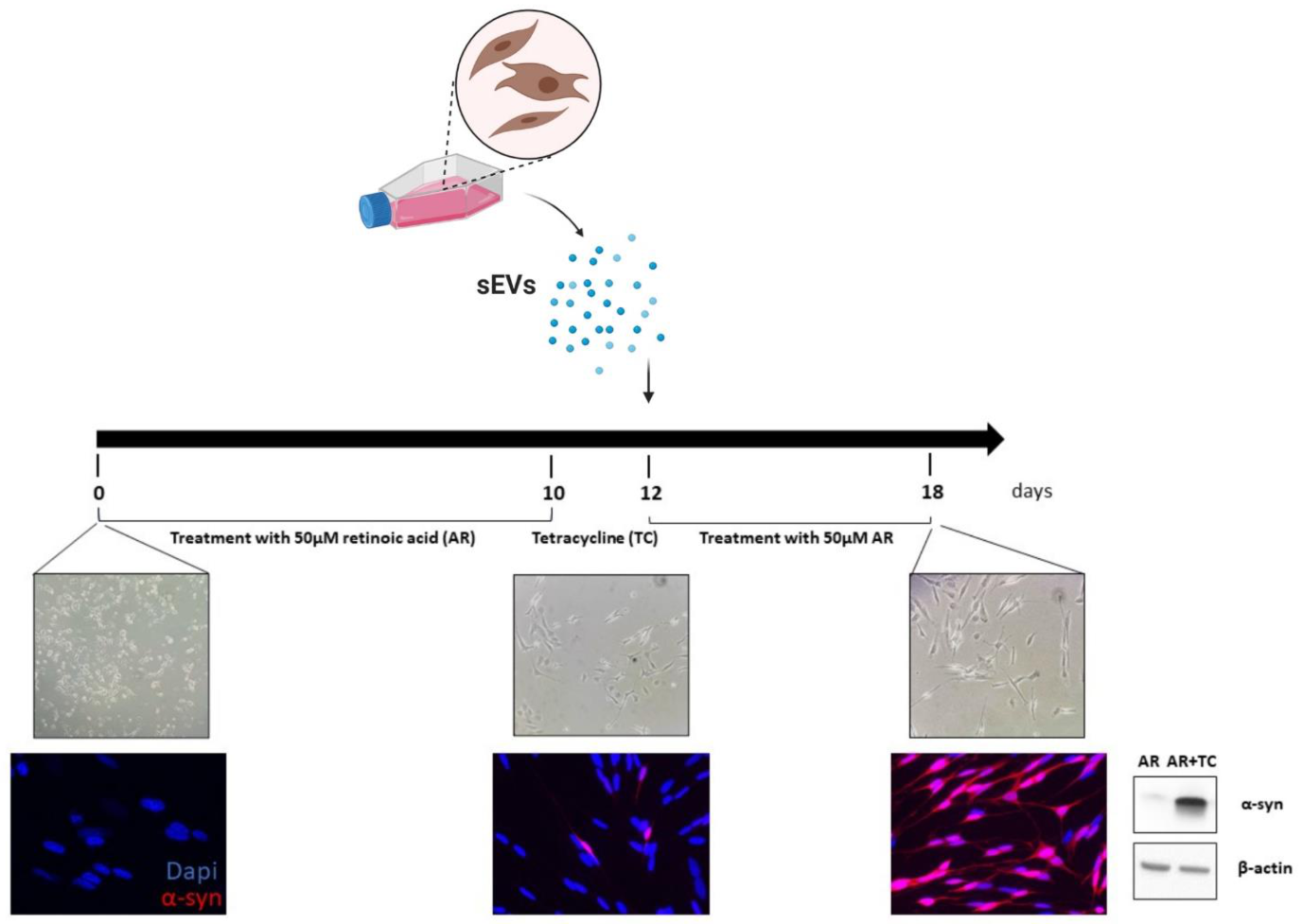
4.5. SH-SY5Y Cell Culture and sEV Treatment
4.6. Western Blot Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avenali, M.; Blandini, F.; Cerri, S. Glucocerebrosidase Defects as a Major Risk Factor for Parkinson’s Disease. Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Barkhuizen, M.; Anderson, D.G.; Grobler, A.F. Advances in GBA-associated Parkinson’s disease—Pathology, presentation and therapies. Neurochem. Int. 2016, 93, 6–25. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA—Associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217. [Google Scholar] [CrossRef] [Green Version]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA--Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, A.; Bregman, N.; Gurevich, T.; Shiner, T.; Dror, Y.; Zmira, O.; Gan-Or, Z.; Bar-Shira, A.; Gana-Weisz, M.; Orr-Urtreger, A.; et al. Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat. Disord. 2018, 55, 45–49. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef]
- Alecu, I.; Bennett, S.A.L. Dysregulated Lipid Metabolism and Its Role in α-Synucleinopathy in Parkinson’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, M.; Mi, N.; Yang, W.; Li, X.; Wang, P.; Yin, N.; Li, Y.; Yue, F.; Chan, P.; et al. Increased oligomerization and phosphorylation of α-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol. Aging 2015, 36, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Miranda, A.M.; Lasiecka, Z.M.; Xu, Y.; Neufeld, J.; Shahriar, S.; Simoes, S.; Chan, R.B.; Oliveira, T.G.; Small, S.A.; Di Paolo, G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 2018, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateescu, B.; Kowal, E.J.K.; van Balkom, B.W.M.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.N.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.F.; Teo, H.S.; Park, J.E.; Dutta, B.; Tse, S.W.; Leow, M.K.-S.; Wahli, W.; Sze, S.K. Exploring Extracellular Vesicles Biogenesis in Hypothalamic Cells through a Heavy Isotope Pulse/Trace Proteomic Approach. Cells 2020, 9, 1320. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [Green Version]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Cicalini, I.; Lanuti, P.; Ercolino, E.; di Ioia, M.; Zucchelli, M.; Zappacosta, R.; Miscia, S.; Marchisio, M.; Sacchetta, P.; et al. Enhanced release of acid sphingomyelinase-enriched exosomes generates a lipidomics signature in CSF of Multiple Sclerosis patients. Sci. Rep. 2018, 8, 3071. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Minakaki, G.; Menges, S.; Kittel, A.; Emmanouilidou, E.; Schaeffner, I.; Barkovits, K.; Bergmann, A.; Rockenstein, E.; Adame, A.; Marxreiter, F.; et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018, 14, 98–119. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, V.E.; Nikolopoulou, G.; Antoniadou, I.; Karachaliou, A.; Arianoglou, G.; Emmanouilidou, E.; Sardi, S.P.; Stefanis, L.; Vekrellis, K. Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.E.; Vincow, E.S.; Merrihew, G.E.; MacCoss, M.J.; Davis, M.Y.; Pallanck, L.J. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLOS Genet. 2018, 14, e1007694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, F.N.; Pampliega, O.; Bourdenx, M.; Meissner, W.G.; Bezard, E.; Dehay, B. Exosomes, an Unmasked Culprit in Neurodegenerative Diseases. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced -Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107.e10. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Sango, K.; Wada, K.; Nagai, Y. Pathological role of lipid interaction with α-synuclein in Parkinson’s disease. Neurochem. Int. 2018, 119, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Cerri, S.; Schapira, A.H.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. BioRxiv 2020. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-Synuclein Aggregation by Exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, S.E.; Grijalva, A.; Xu, X.; Ables, E.; Nomani, A.; Ferrante, A.W. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science 2019, 363, 989–993. [Google Scholar] [CrossRef]
- Garcia, N.A.; González-King, H.; Grueso, E.; Sánchez, R.; Martinez-Romero, A.; Jávega, B.; O’Connor, J.E.; Simons, P.J.; Handberg, A.; Sepúlveda, P. Circulating exosomes deliver free fatty acids from the bloodstream to cardiac cells: Possible role of CD36. PLoS ONE 2019, 14, e0217546. [Google Scholar] [CrossRef]
- Canerina-Amaro, A.; Pereda, D.; Diaz, M.; Rodriguez-Barreto, D.; Casañas-Sánchez, V.; Heffer, M.; Garcia-Esparcia, P.; Ferrer, I.; Puertas-Avendaño, R.; Marin, R. Differential Aggregation and Phosphorylation of Alpha Synuclein in Membrane Compartments Associated With Parkinson Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.; Wickel, M.; Winoto-Morbach, S.; Schneider-Brachert, W.; Weber, T.; Brunner, J.; Saftig, P.; Peters, C.; Krönke, M.; Schütze, S. Ceramide as an Activator Lipid of Cathepsin D. In Cellular Peptidases in Immune Functions and Diseases 2; Kluwer Academic Publishers: Dordrecht, Netherlands, 2000; pp. 305–315. [Google Scholar]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| small EVs | medium EVs | |||
|---|---|---|---|---|
| Mode (nm) | Mean (nm) | Mode (nm) | Mean (nm) | |
| HC fibroblasts | 98.2±4.7 | 155.6±10.0 | 239.2±1.8 | 311.7±13.3 |
| NM-PD fibroblasts | 99.7±3.0 | 155.9±14.6 | 237.2±2.6 | 303.5±11.5 |
| GBA-PD (N370S) fibroblasts | 101.7±3.3 | 153.8±11.1 | 242.2±16.4 | 285.6±15.2 |
| GBA-PD (L444P) fibroblasts | 103.5±1.9 | 151.4±6.3 | 239.2±6.2 | 300.2±14.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerri, S.; Ghezzi, C.; Ongari, G.; Croce, S.; Avenali, M.; Zangaglia, R.; Di Monte, D.A.; Valente, E.M.; Blandini, F. GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 2215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042215
Cerri S, Ghezzi C, Ongari G, Croce S, Avenali M, Zangaglia R, Di Monte DA, Valente EM, Blandini F. GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(4):2215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042215
Chicago/Turabian StyleCerri, Silvia, Cristina Ghezzi, Gerardo Ongari, Stefania Croce, Micol Avenali, Roberta Zangaglia, Donato A. Di Monte, Enza Maria Valente, and Fabio Blandini. 2021. "GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 4: 2215. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042215