
Neural Stem Cell-Based Therapies and Glioblastoma Management: Current Evidence and Clinical Challenges

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
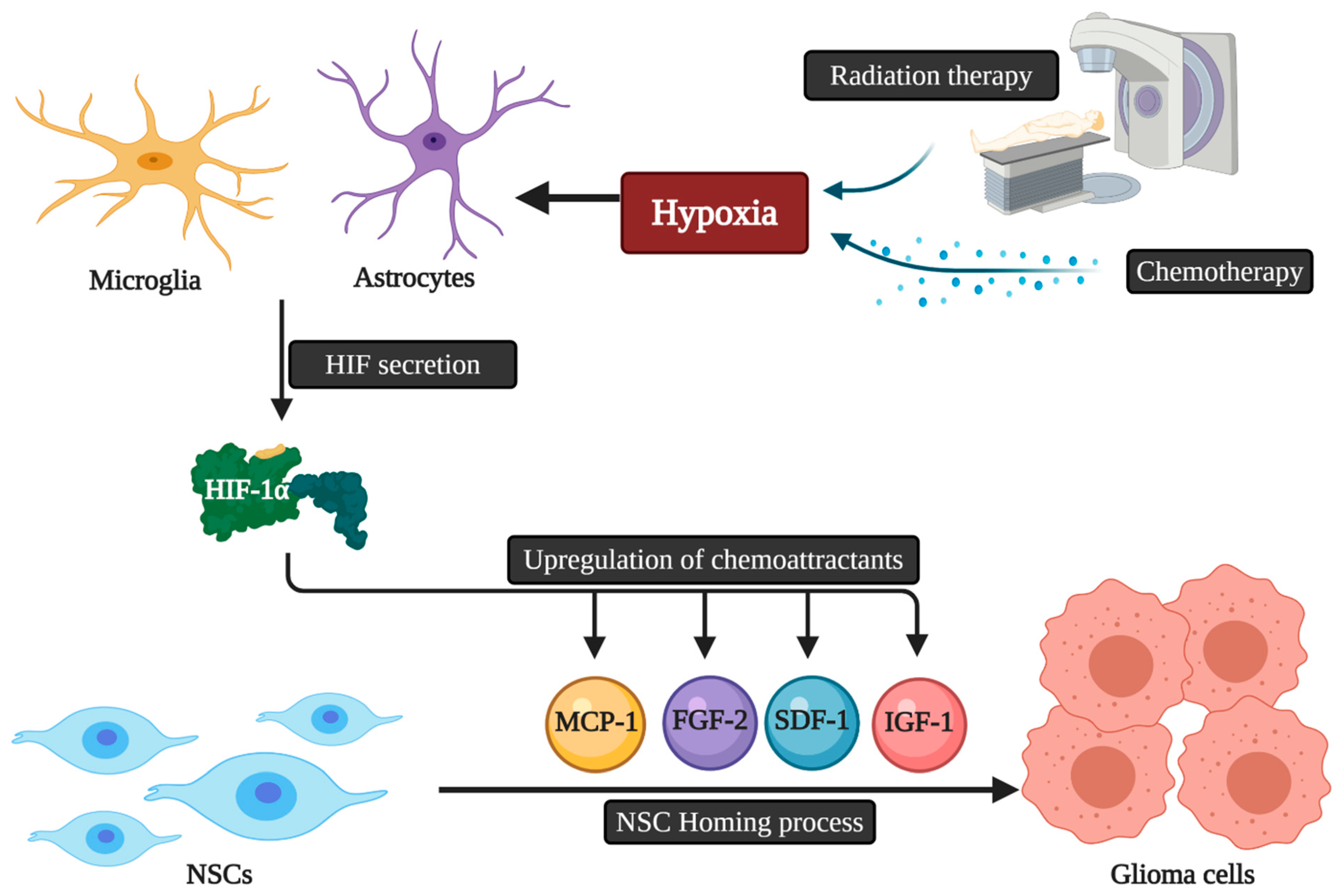
2. Tropism, Migration, and Tumor Homing Properties of Neural Stem Cells
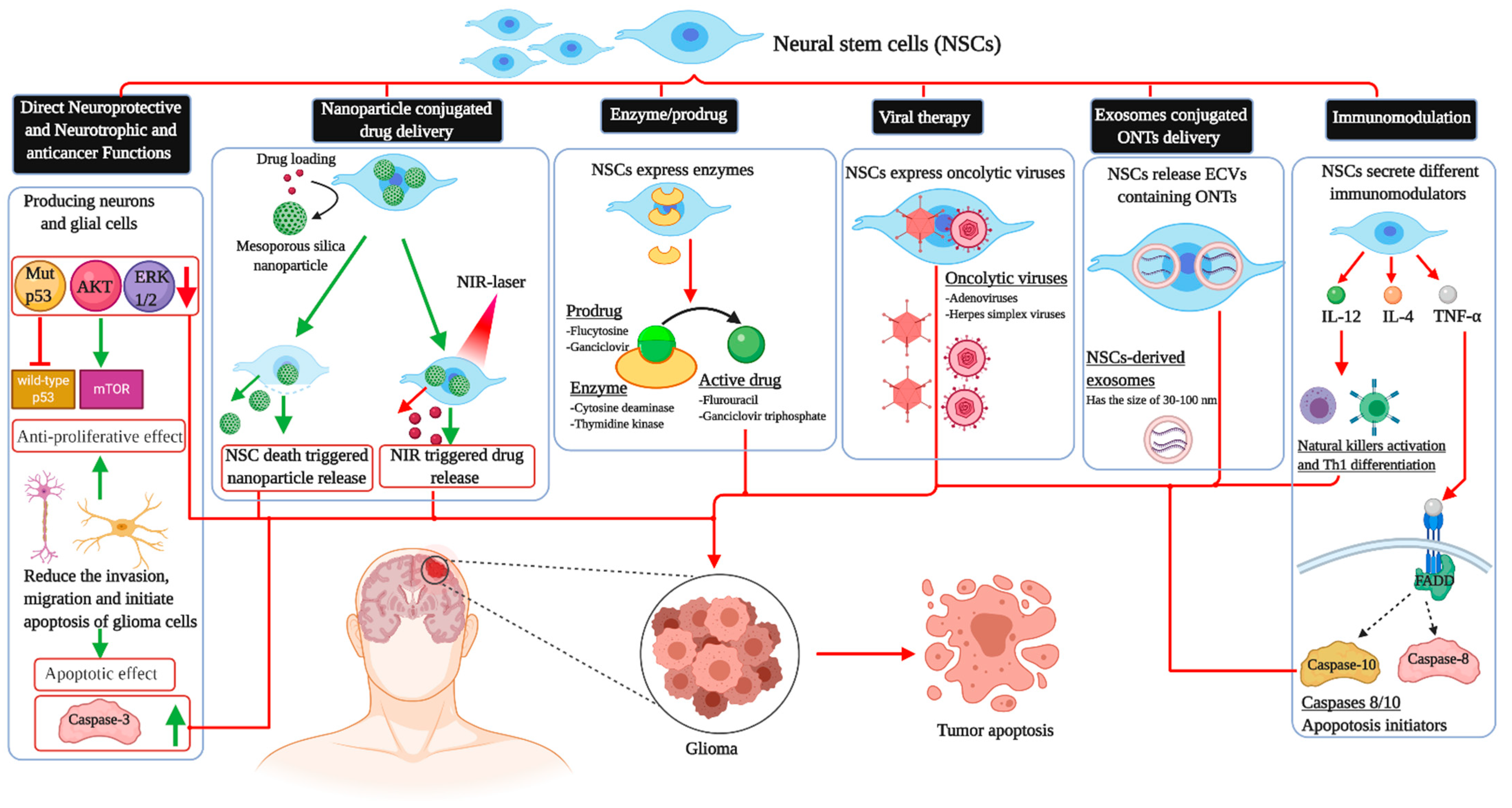
3. Neuroprotective and Neurotrophic Functions of Neural Stem Cells Therapy
4. Effects of Neural Stem Cells in Glioma
5. Immunomodulation
6. Enzyme-Prodrug System
7. Viral Vectors
8. Other Potential Approaches and Considerations
9. Clinical Trials and Administration of Therapy
10. Limitations of Therapy
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro. Oncol. 2019, 21, V1–V100. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Guideline Alliance (UK). Brain Tumours (Primary) and Brain Metastases in Adults; National Institute for Health and Care Excellence: London, UK, 2018. [Google Scholar]
- Sepúlveda-Sánchez, J.M.; Langa, J.M.; Arráez, M.; Fuster, J.; Laín, A.H.; Reynés, G.; González, V.R.; Vicente, E.; Denis, M.V. Gallego SEOM clinical guideline of diagnosis and management of low-grade glioma (2017). Clin. Transl. Oncol. 2018, 20, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Shah, K. Stem cell-based therapies for tumors in the brain: Are we there yet? Neuro. Oncol. 2016, 18, 1066–1078. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, H.D.; Vidane, A.S.; Gonçalves, N.N.; Ambrósio, C.E. Mesenchymal and induced pluripotent stem cells: General insights and clinical perspectives. Stem Cells Cloning Adv. Appl. 2015, 8, 125–134. [Google Scholar] [CrossRef]
- Gage, F.H. Mammalian Neural Stem Cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Carbajal, K.S.; Schaumburg, C.; Strieter, R.; Kane, J.; Lane, T.E. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2010, 107, 11068–11073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.A.J.M.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namba, H.; Kawaji, H.; Yamasaki, T. Use of genetically engineered stem cells for Glioma therapy (Review). Oncol. Lett. 2016, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaeth, E.; Klopp, A.; Dembinski, J.; Andreeff, M.; Marini, F. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008, 15, 730–738. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, J.H.; Lee, J.K.; Choi, S.J.; Kim, J.S.; Jeun, S.S.; Oh, W.; Yang, Y.S.; Chang, J.W. Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev. 2009, 18, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Egea, V.; Von Baumgarten, L.; Schichor, C.; Berninger, B.; Popp, T.; Neth, P.; Goldbrunner, R.; Kienast, Y.; Winkler, F.; Jochum, M.; et al. TNF-α respecifies human mesenchymal stem cells to a neural fate and promotes migration toward experimental glioma. Cell Death Differ. 2011, 18, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Menon, L.G.; Picinich, S.; Koneru, R.; Gao, H.; Lin, S.Y.; Koneru, M.; Mayer-Kuckuk, P.; Glod, J.; Banerjee, D. Differential Gene Expression Associated with Migration of Mesenchymal Stem Cells to Conditioned Medium from Tumor Cells or Bone Marrow Cells. Stem Cells 2007, 25, 520–528. [Google Scholar] [CrossRef]
- Young, J.S.; Morshed, R.A.; Kim, J.W.; Balyasnikova, I.V.; Ahmed, A.U.; Lesniak, M.S. Advances in stem cells, induced pluripotent stem cells, and engineered cells: Delivery vehicles for anti-glioma therapy. Expert Opin. Drug Deliv. 2014, 11, 1733–1746. [Google Scholar] [CrossRef]
- Jeung, H.A.; Soo, Y.L.; Jeong, Y.J.; Kyung, G.C.; Kim, S.U.; Myung, A.L. Identification of gliotropic factors that induce human stem cell migration to malignant tumor. J. Proteome Res. 2009, 8, 2873–2881. [Google Scholar]
- Ziu, M.; Schmidt, N.O.; Cargioli, T.G.; Aboody, K.S.; Black, P.M.L.; Carroll, R.S. Glioma-produced extracellular matrix influences brain tumor tropism of human neural stem cells. J. Neurooncol. 2006, 79, 125–133. [Google Scholar] [CrossRef]
- Pincus, D.W.; Keyoung, H.M.; Harrison-Restelli, C.; Goodman, R.R.; Fraser, R.A.R.; Edgar, M.; Sakakibara, S.; Okano, H.; Nedergaard, M.; Goldman, S.A. Fibroblast growth factor-2/brain-derived neurotrophic factor—associated maturation of new neurons generated from adult human subependymal cells. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1998, 43, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Wang, S.; Jiang, L.; Kang, J.; Benraiss, A.; Harrison-Restelli, C.; Fraser, R.A.R.; Couldwell, W.T.; Kawaguchi, A.; Okano, H. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat. Med. 2000, 6, 271–277. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Alexiades, N.G.; Lesniak, M.S. The use of neural stem cells in cancer gene therapy: Predicting the path to the clinic. Curr. Opin. Mol. Ther. 2010, 12, 546–552. [Google Scholar] [PubMed]
- Kim, S.-K.; Kim, S.U.; Park, I.H.; Bang, J.H.; Aboody, K.S.; Wang, K.-C.; Cho, B.-K.; Kim, M.; Menon, L.G.; Black, P.M. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clin. Cancer Res. 2006, 12, 5550–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.U.; Thaci, B.; Alexiades, N.G.; Han, Y.; Qian, S.; Liu, F.; Balyasnikova, I.V.; Ulasov, I.Y.; Aboody, K.S.; Lesniak, M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 2011, 19, 1714–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, M.Z.; Gutova, M.; Lacey, S.F.; Abramyants, Y.; Vo, T.; Gilchrist, M.; Tirughana, R.; Ghoda, L.Y.; Barish, M.E.; Brown, C.E. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: Implications for clinical use. Stem Cells Transl. Med. 2013, 2, 983–992. [Google Scholar] [CrossRef]
- Mooney, R.; Weng, Y.; Tirughana-Sambandan, R.; Valenzuela, V.; Aramburo, S.; Garcia, E.; Li, Z.; Gutova, M.; Annala, A.J.; Berlin, J.M. Neural stem cells improve intracranial nanoparticle retention and tumor-selective distribution. Futur. Oncol. 2014, 10, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboody, K.S.; Brown, A.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboody, K.S.; Bush, R.A.; Garcia, E.; Metz, M.Z.; Najbauer, J.; Justus, K.A.; Phelps, D.A.; Remack, J.S.; Yoon, K.J.; Gillespie, S.; et al. Development of a tumor-selective approach to treat metastatic cancer. PLoS ONE 2006, 1, e23. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.M.; Park, I.H.; Shin, J.Y.; Jin, J.; Kang, B.G.; Kim, M.H.; Lee, S.J.; Jo, M.; Kim, S.U.; Nam, D.-H. Human neural stem cells can target and deliver therapeutic genes to breast cancer brain metastases. Mol. Ther. 2009, 17, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lee, J.-E.; Kim, S.U.; Cho, K.-G. Stereological analysis on migration of human neural stem cells in the brain of rats bearing glioma. Neurosurgery 2010, 66, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Barish, M.E.; Herrmann, K.; Tang, Y.; Argalian Herculian, S.; Metz, M.; Aramburo, S.; Tirughana, R.; Gutova, M.; Annala, A.; Moats, R.A.; et al. Human Neural Stem Cell Biodistribution and Predicted Tumor Coverage by a Diffusible Therapeutic in a Mouse Glioma Model. Stem Cells Transl. Med. 2017, 6, 1522–1532. [Google Scholar] [CrossRef]
- Carey-Ewend, A.G.; Hagler, S.B.; Bomba, H.N.; Goetz, M.J.; Bago, J.R.; Hingtgen, S.D. Developing Bio-Inspired 3D Models of Brain Cancer to Evaluate Tumor-Homing Neural Stem Cell Therapy. Tissue Eng. 2020, 22, 417–429. [Google Scholar]
- Müller, F.-J.; Snyder, E.Y.; Loring, J.F. Gene therapy: Can neural stem cells deliver? Nat. Rev. Neurosci. 2006, 7, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Park, K.I.; Hack, M.A.; Ourednik, J.; Yandava, B.; Flax, J.D.; Stieg, P.E.; Gullans, S.; Jensen, F.E.; Sidman, R.L.; Ourednik, V.; et al. Acute injury directs the migration, proliferation, and differentiation of solid organ stem cells: Evidence from the effect of hypoxia-ischemia in the CNS on clonal “reporter” neural stem cells. Exp. Neurol. 2006, 199, 156–178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Najbauer, J.; Garcia, E.; Metz, M.Z.; Gutova, M.; Glackin, C.A.; Kim, S.U.; Aboody, K.S. Neural stem cell tropism to glioma: Critical role of tumor hypoxia. Mol. Cancer Res. 2008, 6, 1819–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Oh, J.H.; Park, S.A.; Ryu, C.H.; Lim, J.Y.; Kim, D.-S.; Chang, J.W.; Oh, W.; Jeun, S.-S. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells 2010, 28, 2217–2228. [Google Scholar] [CrossRef]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Thored, P.; Arvidsson, A.; Cacci, E.; Ahlenius, H.; Kallur, T.; Darsalia, V.; Ekdahl, C.T.; Kokaia, Z.; Lindvall, O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 2006, 24, 739–747. [Google Scholar] [CrossRef]
- Yamashita, T.; Ninomiya, M.; Acosta, P.H.; García-Verdugo, J.M.; Sunabori, T.; Sakaguchi, M.; Adachi, K.; Kojima, T.; Hirota, Y.; Kawase, T.; et al. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006, 26, 6627–6636. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.D.; Liao, W.-L.; Choi, H.; Konya, D.; Sabharwal, S.; Langer, R.; Sidman, R.L.; Snyder, E.Y.; Frontera, W.R. Physical activity-mediated functional recovery after spinal cord injury: Potential roles of neural stem cells. Regen. Med. 2006, 1, 763–776. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, Y.-W.; Chen, W.-G.; Liu, J. Neuroregeneration and functional recovery after stroke: Advancing neural stem cell therapy toward clinical application. Neural Regen. Res. 2021, 16, 80–92. [Google Scholar]
- Ross, H.H.; Ambrosio, F.; Trumbower, R.D.; Reier, P.J.; Behrman, A.L.; Wolf, S.L. Neural Stem Cell Therapy and Rehabilitation in the Central Nervous System: Emerging Partnerships. Phys. Ther. 2016, 96, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annese, V.; Navarro-Guerrero, E.; Rodríguez-Prieto, I.; Pardal, R. Physiological plasticity of neural-crest-derived stem cells in the adult mammalian carotid body. Cell Rep. 2017, 19, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.G.; Buller, B.; Chopp, M. Exosomes—beyond stem cells for restorative therapy in stroke and neurological injury. Nat. Rev. Neurol. 2019, 15, 193–203. [Google Scholar] [CrossRef]
- Baker, E.W.; Kinder, H.A.; West, F.D. Neural stem cell therapy for stroke: A multimechanistic approach to restoring neurological function. Brain Behav. 2019, 9, e01214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taupin, P. Adult neurogenesis, neuroinflammation and therapeutic potential of adult neural stem cells. Int. J. Med. Sci. 2008, 5, 127. [Google Scholar] [CrossRef] [Green Version]
- Reekmans, K.P.; Praet, J.; De Vocht, N.; Tambuyzer, B.R.; Bergwerf, I.; Daans, J.; Baekelandt, V.; Vanhoutte, G.; Goossens, H.; Jorens, P.G. Clinical potential of intravenous neural stem cell delivery for treatment of neuroinflammatory disease in mice? Cell Transplant. 2011, 20, 851–870. [Google Scholar] [CrossRef]
- Rong, Y.; Liu, W.; Wang, J.; Fan, J.; Luo, Y.; Li, L.; Kong, F.; Chen, J.; Tang, P.; Cai, W. Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, K.S.; Park, I.H.; Kim, S.U. Human Neural Stem Cells Over-Expressing VEGF Provide Neuroprotection, Angiogenesis and Functional Recovery in Mouse Stroke Model. PLoS ONE 2007, 2, e156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Lee, S.-H.; Kim, S.U.; Yoon, B.-W. Human neural stem cells promote proliferation of endogenous neural stem cells and enhance angiogenesis in ischemic rat brain. Neural Regen. Res. 2016, 11, 298–304. [Google Scholar]
- Aboody, K.S.; Najbauer, J.; Metz, M.Z.; D’Apuzzo, M.; Gutova, M.; Annala, A.J.; Synold, T.W.; Couture, L.A.; Blanchard, S.; Moats, R.A.; et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: Preclinical studies. Sci. Transl. Med. 2013, 5, 184ra59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Shah, K.; Messerli, S.M.; Snyder, E.; Breakefield, X.; Weissleder, R. In vivo tracking of neural progenitor cell migration to glioblastomas. Hum. Gene Ther. 2003, 14, 1247–1254. [Google Scholar] [CrossRef]
- Einstein, O.; Ben-Hur, T. The changing face of neural stem cell therapy in neurologic diseases. Arch. Neurol. 2008, 65, 452–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluchino, S.; Quattrini, A.; Brambilla, E.; Gritti, A.; Salani, G.; Dina, G.; Galli, R.; Del Carro, U.; Amadio, S.; Bergami, A.; et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 2003, 422, 688–694. [Google Scholar] [CrossRef]
- Díaz-Coránguez, M.; Segovia, J.; López-Ornelas, A.; Puerta-Guardo, H.; Ludert, J.; Chávez, B.; Meraz-Cruz, N.; González-Mariscal, L. Transmigration of Neural Stem Cells across the Blood Brain Barrier Induced by Glioma Cells. PLoS ONE 2013, 8, e60665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.-H.; Eve, D.J.; Sanberg, P.R.; Musso, J., 3rd; Bachstetter, A.D.; Wolfson, A.; Schlunk, A.; Baradez, M.-O.; Sinden, J.D.; Gemma, C. Increased neuronal proliferation in the dentate gyrus of aged rats following neural stem cell implantation. Stem Cells Dev. 2010, 19, 175–180. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Yan, H.; Li, X.; Tan, R.; Chen, X.; Zhang, Z.; Liu, Y.; Zhang, P.; Lu, H.; Liu, Y. The inhibiting effect of neural stem cells on proliferation and invasion of glioma cells. Oncotarget 2017, 8, 76949–76960. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tan, R.; Hu, X.; Jiao, Q.; Rahman, M.S.; Chen, X.; Zhang, P.; An, J.; Lu, H.; Liu, Y. Neural stem cell-derived factors inhibit the growth and invasion of U87 stem-like cells in vitro. J. Cell. Biochem. 2019, 120, 5472–5479. [Google Scholar] [CrossRef]
- Glass, R.; Synowitz, M.; Kronenberg, G.; Walzlein, J.; Markovic, D.S.; Wang, L.; Gast, D.; Kempermann, G.; Kettenmann, H. Glioblastoma-Induced Attraction of Endogenous Neural Precursor Cells Is Associated with Improved Survival. J. Neurosci. 2005, 25, 2637–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginisty, A.; Oliver, L.; Arnault, P.; Vallette, F.; Benzakour, O.; Coronas, V. The vitamin K-dependent factor, protein S, regulates brain neural stem cell migration and phagocytic activities towards glioma cells. Eur. J. Pharmacol. 2019, 855, 30–39. [Google Scholar] [CrossRef]
- Benedetti, S.; Pirola, B.; Pollo, B.; Magrassi, L.; Bruzzone, M.G.; Rigamonti, D.; Galli, R.; Selleri, S.; Di Meco, F.; De Fraja, C.; et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 2000, 6, 447–450. [Google Scholar] [CrossRef]
- Ehtesham, M.; Kabos, P.; Kabosova, A.; Neuman, T.; Black, K.L.; Yu, J.S. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002, 62, 5657–5663. [Google Scholar] [PubMed]
- Wojno, E.D.T.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Faber, C.; Terao, E.; Morga, E.; Heuschling, P. Interleukin-4 Enhances the In Vitro Precursor Cell Recruitment for Tumor-Specific T Lymphocytes in Patients With Glioblastoma. J. Immunother. 2000, 23, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, P.V.; Hamner, J.B.; Burger, R.A.; Garcia, E.; Ouma, A.A.; Kim, S.U.; Ng, C.Y.C.; Gray, J.T.; Aboody, K.S.; Danks, M.K.; et al. Intravascular administration of tumor tropic neural progenitor cells permits targeted delivery of interferon-beta and restricts tumor growth in a murine model of disseminated neuroblastoma. J. Pediatr. Surg. 2007, 42, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Hu, J.; Belladonna, M.L.; Black, K.L.; Yu, J.S. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer Res. 2006, 66, 2630–2638. [Google Scholar] [CrossRef] [Green Version]
- Hingtgen, S.; Kasmieh, R.; Elbayly, E.; Nesterenko, I.; Figueiredo, J.-L.; Dash, R.; Sarkar, D.; Hall, D.; Kozakov, D.; Vajda, S.; et al. A first-generation multi-functional cytokine for simultaneous optical tracking and tumor therapy. PLoS ONE 2012, 7, e40234. [Google Scholar] [CrossRef] [Green Version]
- Dent, P.; Yacoub, A.; Hamed, H.A.; Park, M.A.; Dash, R.; Bhutia, S.K.; Sarkar, D.; Gupta, P.; Emdad, L.; Lebedeva, I.V.; et al. MDA-7/IL-24 as a cancer therapeutic: From bench to bedside. Anticancer. Drugs 2010, 21, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-E.; Liu, L.-F.; Dusting, G.J.; Weng, W.-T.; Chen, S.-C.; Kung, M.-L.; Tee, R.; Liu, G.-S.; Tai, M.-H. Pro-opiomelanocortin gene delivery suppresses the growth of established Lewis lung carcinoma through a melanocortin-1 receptor-independent pathway. J. Gene Med. 2012, 14, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Abdulghani, J.; El-Deiry, W.S. TRAIL receptor signaling and therapeutics. Expert Opin. Ther. Targets 2010, 14, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.B.; Salvesen, G.S.; Green, D.R. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef] [Green Version]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Activation and Function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [PubMed]
- Hingtgen, S.; Ren, X.; Terwilliger, E.; Classon, M.; Weissleder, R.; Shah, K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and temozolomide. Mol. Cancer Ther. 2008, 7, 3575–3585. [Google Scholar] [CrossRef] [Green Version]
- Kock, N.; Kasmieh, R.; Weissledery, R.; Shah, K. Tumor therapy mediated by lentiviral expression of shBcl-2 and S-TRAIL1. Neoplasia 2007, 9, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balyasnikova, I.V.; Ferguson, S.D.; Han, Y.; Liu, F.; Lesniak, M.S. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011, 310, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhao, Y.P.; Zhou, L.; Zhang, T.P.; Chen, G. Bcl-2 upregulation induced by miR-21 via a direct interaction is associated with apoptosis and chemoresistance in MIA PaCa-2 pancreatic cancer cells. Arch. Med. Res. 2011, 42, 8–14. [Google Scholar] [CrossRef]
- Chen, J.-J.; Chou, C.-W.; Chang, Y.-F.; Chen, C.-C. Proteasome Inhibitors Enhance TRAIL-Induced Apoptosis through the Intronic Regulation of DR5: Involvement of NF-κB and Reactive Oxygen Species-Mediated p53 Activation. J. Immunol. 2008, 180, 8030–8039. [Google Scholar] [CrossRef] [Green Version]
- Kohlhaas, S.L.; Craxton, A.; Sun, X.M.; Pinkoski, M.J.; Cohen, G.M. Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J. Biol. Chem. 2007, 282, 12831–12841. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Jiang, X.; Kim, D.; Guan, T.; Nicolls, M.R.; Rockson, S.G. Leukotrienes in Tumor-Associated Inflammation. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Lim, J.Y.; Jeun, S.S. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. 2012, 72, 4807–4817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barresi, V.; Belluardo, N.; Sipione, S.; Mudó, G.; Cattaneo, E.; Condorelli, D.F. Transplantation of prodrug-converting neural progenitor cells for brain tumor therapy. Cancer Gene Ther. 2003, 10, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tokuyama, T.; Yamamoto, J.; Koide, M.; Yokota, N.; Namba, H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005, 12, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonabend, A.M.; Ulasov, I.V.; Tyler, M.A.; Rivera, A.A.; Mathis, J.M.; Lesniak, M.S. Mesenchymal Stem Cells Effectively Deliver an Oncolytic Adenovirus to Intracranial Glioma. Stem Cells 2008, 26, 831–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Δ24-RGD to human gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef] [Green Version]
- MacLean, A.R.; Ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpont neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the “a” sequence. J. Gen. Virol. 1991, 72, 631–639. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interactions, V.; Peters, C.; Paget, M.; Tshilenge, K.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; et al. Crossm Restriction of Replication of Oncolytic Herpes Simplex Virus. J. Virol. 2018, 92, 1–17. [Google Scholar]
- Kanai, R.; Zaupa, C.; Sgubin, D.; Antoszczyk, S.J.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Effect of γ34.5 Deletions on Oncolytic Herpes Simplex Virus Activity in Brain Tumors. J. Virol. 2012, 86, 4420–4431. [Google Scholar] [CrossRef] [Green Version]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Morshed, R.; Cheng, S.; Tobias, A.; Auffinger, B.; Wainwright, D.A.; Zhang, L.; Yunis, C.; Han, Y.; Chen, C. Nanoparticle-Programmed Self-Destructive Neural Stem Cells for Glioblastoma Targeting and Therapy. Small 2013, 9, 4123–4129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, R.; Roma, L.; Zhao, D.; Van Haute, D.; Garcia, E.; Kim, S.U.; Annala, A.J.; Aboody, K.S.; Berlin, J.M. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano 2014, 8, 12450–12460. [Google Scholar] [CrossRef]
- Hwang, J.; Fitzgerald, D.J.; Adhya, S.; Pastan, I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell 1987, 48, 129–136. [Google Scholar] [CrossRef]
- Husain, S.R.; Joshi, B.H.; Puri, R.K. Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. Int. J. Cancer 2001, 92, 168–175. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Immunotoxins in the treatment of hematologic malignancies. Curr. Drug Targets 2006, 7, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Hingtgen, S.D.; Karakas, N.; Rich, B.E.; Shah, K. Engineering toxin-resistant therapeutic stem cells to treat brain tumors. Stem Cells 2015, 33, 589–600. [Google Scholar] [CrossRef] [Green Version]
- van Eekelen, M.; Sasportas, L.S.; Kasmieh, R.; Yip, S.; Figueiredo, J.-L.; Louis, D.N.; Weissleder, R.; Shah, K. Human stem cells expressing novel TSP-1 variant have anti-angiogenic effect on brain tumors. Oncogene 2010, 29, 3185–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krichevsky, A.M.; Uhlmann, E.J. Oligonucleotide Therapeutics as a New Class of Drugs for Malignant Brain Tumors: Targeting mRNAs, Regulatory RNAs, Mutations, Combinations, and Beyond. Neurotherapeutics 2019, 16, 319–347. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Sun, X.; Liu, L.; Jiang, H.; Shen, Y.; Xu, X.; Li, J.; Zhang, G.; Huang, J.; Lin, Z.; et al. Exosomes and Their Therapeutic Potentials of Stem Cells. Stem Cells Int. 2016, 2016, 7653489. [Google Scholar] [CrossRef] [Green Version]
- Tobias, A.L.; Thaci, B.; Auffinger, B.; Rincón, E.; Balyasnikova, I.V.; Kim, C.K.; Han, Y.; Zhang, L.; Aboody, K.S.; Ahmed, A.U.; et al. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl. Med. 2013, 2, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Portnow, J.; Synold, T.W.; Badie, B.; Tirughana, R.; Lacey, S.F.; D’Apuzzo, M.; Metz, M.Z.; Najbauer, J.; Bedell, V.; Vo, T.; et al. Neural stem cell-based anticancer gene therapy: A first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 2017, 23, 2951–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Pilot Feasibility Study of Oral 5-Fluorocytosine and Genetically-Modified Neural Stem Cells Expressing, E.Coli Cytosine Deaminase for Treatment of Recurrent High Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT01172964 (accessed on 25 September 2020).
- Genetically Modified Neural Stem Cells, Flucytosine, and Leucovorin for Treating Patients With Recurrent High-Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT02015819 (accessed on 25 September 2020).
- Neural Stem Cell Based Virotherapy of Newly Diagnosed Malignant Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT03072134 (accessed on 25 September 2020).
- Oncolytic Adenovirus DNX-2401 in Treating Patients With Recurrent High-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT03896568 (accessed on 25 September 2020).
- Bexell, D.; Gunnarsson, S.; Svensson, A.; Tormin, A.; Henriques-Oliveira, C.; Siesjö, P.; Paul, G.; Salford, L.G.; Scheding, S.; Bengzon, J. Rat multipotent mesenchymal stromal cells lack long-distance tropism to 3 different rat glioma models. Neurosurgery 2012, 70, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Gutova, M.; Flores, L.; Adhikarla, V.; Tsaturyan, L.; Tirughana, R.; Aramburo, S.; Metz, M.; Gonzaga, J.; Annala, A.; Synold, T.W.; et al. Quantitative evaluation of intraventricular delivery of therapeutic neural stem cells to orthotopic glioma. Front. Oncol. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panciani, P.P.; Fontanella, M.; Tamagno, I.; Battaglia, L.; Garbossa, D.; Inghirami, G.; Fagioli, F.; Pagano, M.; Ducati, A.; Lanotte, M. Stem cells based therapy in high grade glioma: Why the intraventricular route should be preferred? J. Neurosurg. Sci. 2012, 56, 221–229. [Google Scholar]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, L.; Baker, A.-M.; Graham, T.A. Tumour Cell Heterogeneity. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Registration Number | Stem Cell Type | Approach | Phase | Anti-tumor Agent | Route of Administration | Study Status |
|---|---|---|---|---|---|---|
| NCT01172964 | NSC | Enzyme-prodrug | Phase I | 5-fluorouracil | Intracerebral | Completed |
| NCT02015819 | NSC | Enzyme-prodrug | Phase I | 5-fluorouracil | Intracerebral | Active, not recruiting |
| NCT03072134 | NSC | Oncolytic virus | Phase I | Oncolytic Adenovirus | Intracerebral | Active, not recruiting |
| NCT03896568 | NSC | Oncolytic virus | Phase I | Oncolytic Adenovirus Ad5-DNX-2401 | Intracerebral | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benmelouka, A.Y.; Munir, M.; Sayed, A.; Attia, M.S.; Ali, M.M.; Negida, A.; Alghamdi, B.S.; Kamal, M.A.; Barreto, G.E.; Ashraf, G.M.; et al. Neural Stem Cell-Based Therapies and Glioblastoma Management: Current Evidence and Clinical Challenges. Int. J. Mol. Sci. 2021, 22, 2258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052258
Benmelouka AY, Munir M, Sayed A, Attia MS, Ali MM, Negida A, Alghamdi BS, Kamal MA, Barreto GE, Ashraf GM, et al. Neural Stem Cell-Based Therapies and Glioblastoma Management: Current Evidence and Clinical Challenges. International Journal of Molecular Sciences. 2021; 22(5):2258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052258
Chicago/Turabian StyleBenmelouka, Amira Yasmine, Malak Munir, Ahmed Sayed, Mohamed Salah Attia, Mohamad M. Ali, Ahmed Negida, Badrah S. Alghamdi, Mohammad Amjad Kamal, George E. Barreto, Ghulam Md Ashraf, and et al. 2021. "Neural Stem Cell-Based Therapies and Glioblastoma Management: Current Evidence and Clinical Challenges" International Journal of Molecular Sciences 22, no. 5: 2258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052258