
New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
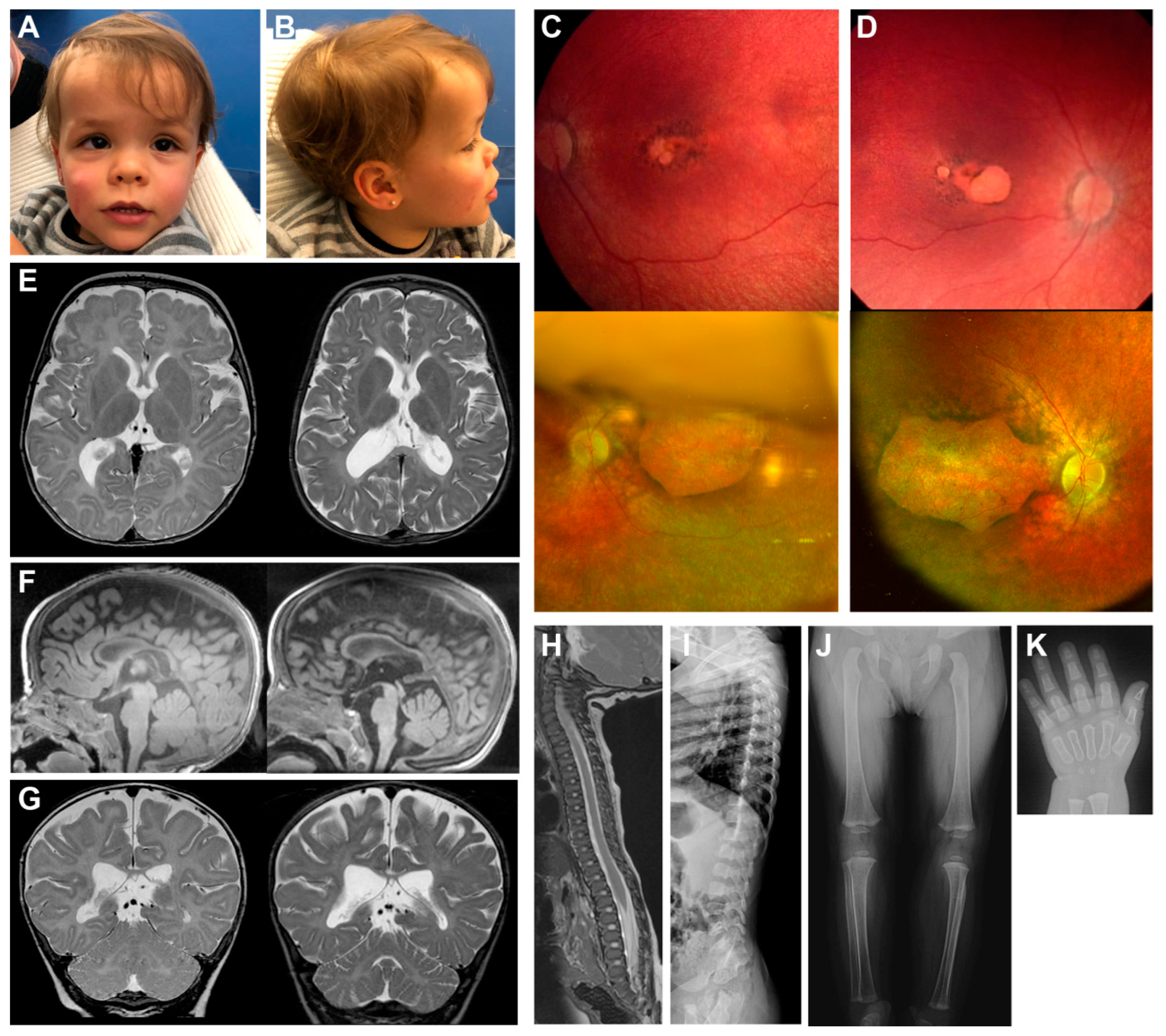
2.1. Clinical Presentation
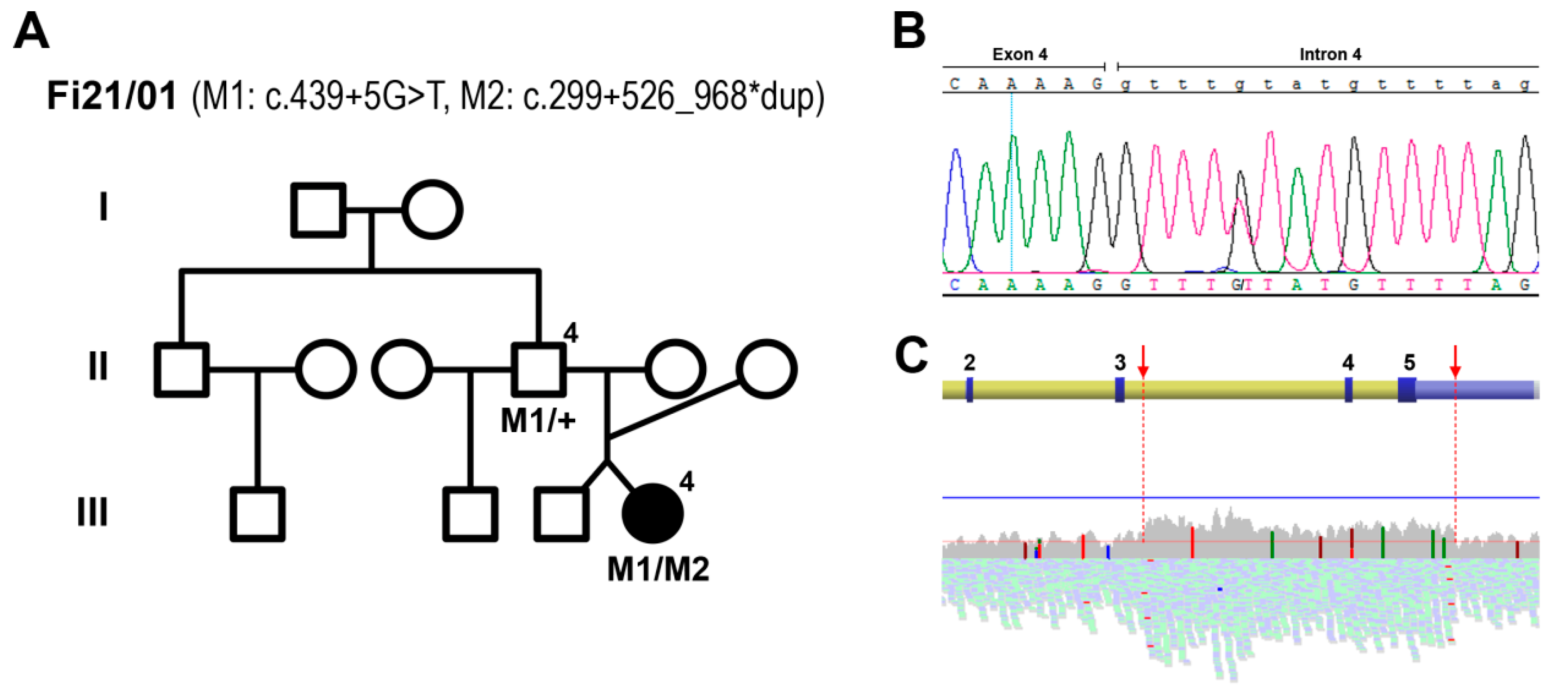
2.2. Genetic Analyses
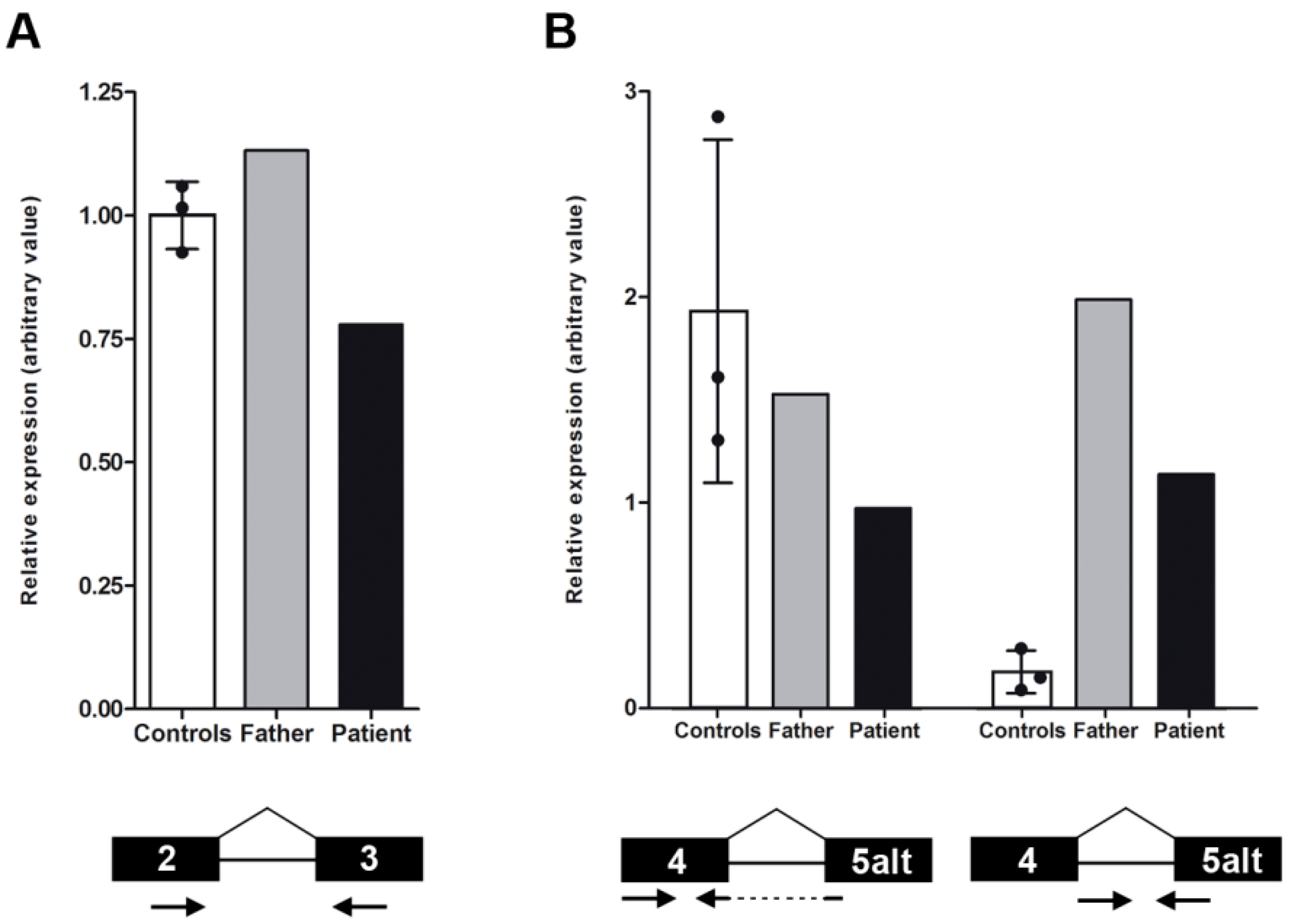
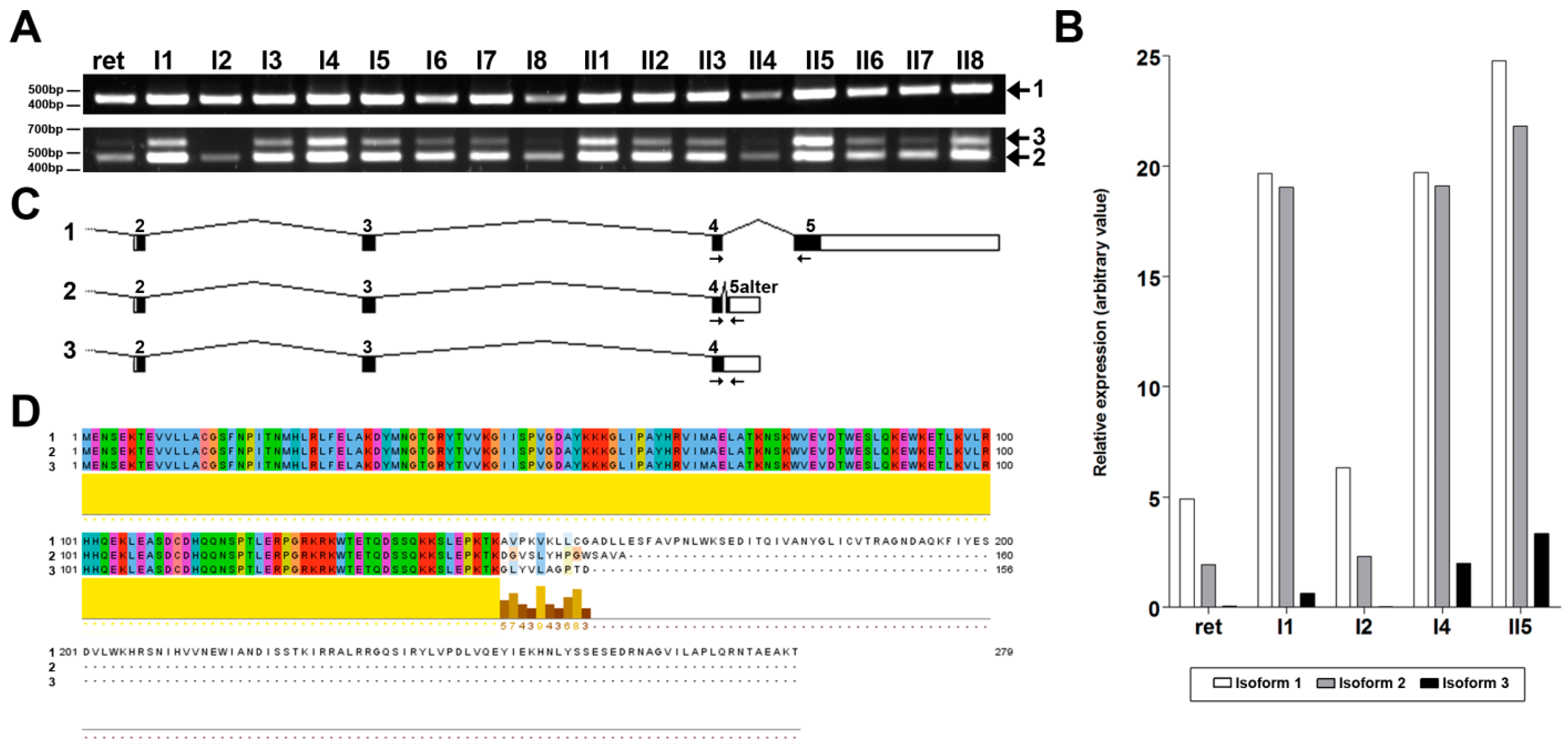
2.3. NMNAT1 Expression
3. Discussion
4. Materials and Methods
4.1. Clinical Examinations
4.2. Genetic Screenings
4.3. Expression Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rattner, A.; Sun, H.; Nathans, J. Molecular Genetics of Human Retinal Disease. Annu. Rev. Genet. 1999, 33, 89–131. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Leber Congenital Amaurosis. In Advances in Experimental Medicine and Biology; Springer International Publishing: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Allikmets, R. Leber congenital amaurosis: A genetic paradigm. Ophthalmic Genet. 2004, 25, 67–79. [Google Scholar] [CrossRef]
- Kondkar, A.A.; Abu-Amero, K.K. Leber congenital amaurosis: Current genetic basis, scope for genetic testing and personalized medicine. Exp. Eye Res. 2019, 189, 107834. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Lopez, I.; Allikmets, R.; Cremers, F.P.; Hollander, A.I.D. Genetics, phenotypes, mechanisms and treatments for Leber congenital amaurosis: A paradigm shift. Expert Rev. Ophthalmol. 2008, 3, 397–415. [Google Scholar] [CrossRef]
- Hollander, A.I.D.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Cremers, F.P.M.; Hurk, J.A.J.M.V.D.; Hollander, A.I.D. Molecular genetics of Leber congenital amaurosis. Hum. Mol. Genet. 2002, 11, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Ronquillo, C.; Bernstein, P.S.; Baehr, W. Senior–Løken syndrome: A syndromic form of retinal dystrophy associated with nephronophthisis. Vis. Res. 2012, 75, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Peter, V.G.; Quinodoz, M.; Pinto-Basto, J.; Sousa, S.B.; Di Gioia, S.A.; Soares, G.; Leal, G.F.; Silva, E.D.; Msc, R.P.G.; Miyake, N.; et al. The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in the PISD gene. Genet. Med. 2019, 21, 2734–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheck, L.; Davies, W.I.L.; Moradi, P.; Robson, A.G.; Kumaran, N.; Liasis, A.C.; Webster, A.R.; Moore, A.T.; Michaelides, M. Leber Congenital Amaurosis Associated with Mutations in CEP290, Clinical Phenotype, and Natural History in Preparation for Trials of Novel Therapies. Ophthalmology 2018, 125, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astuti, G.D.N.; Bertelsen, M.; Preising, M.N.; Ajmal, M.; Lorenz, B.; Faradz, S.M.H.; Qamar, R.; Collin, R.W.J.; Rosenberg, T.; Cremers, F.P.M. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur. J. Hum. Genet. 2016, 24, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoni, N.; Quinodoz, M.; Pinelli, M.; Cappuccio, G.; Torella, A.; Nigro, V.; Testa, F.; Simonelli, F.; Corton, M.; Lualdi, S.; et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum. Mol. Genet. 2020, 29, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef] [Green Version]
- Koenekoop, R.K.; Wang, H.; Majewski, J.; Wang, X.; Lopez, I.; Ren, H.; Chen, Y.; Li, Y.; Fishman, G.A.; Genead, M.; et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012, 44, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Chiang, P.-W.; Wang, J.; Chen, Y.; Fu, Q.; Zhong, J.; Chen, Y.; Yi, X.; Wu, R.; Gan, H.; Shi, Y.; et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet. 2012, 44, 972–974. [Google Scholar] [CrossRef]
- Coppieters, F.; Todeschini, A.L.; Fujimaki, T.; Baert, A.; De Bruyne, M.; Van Cauwenbergh, C.; Verdin, H.; Bauwens, M.; Ongenaert, M.; Kondo, M.; et al. Hidden Genetic Variation in LCA9-Associated Congenital Blindness Explained by 5′UTR Mutations and Copy-Number Variations of NMNAT1. Hum. Mutat. 2015, 36, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; Budde, B.S.; Nürnberg, P.; Kawalia, A.; Lenzner, S.; Bolz, H.J. Genome-wide linkage and sequence analysis challenge CCDC66 as a human retinal dystrophy candidate gene and support a distinct NMNAT1 -related fundus phenotype. Clin. Genet. 2018, 93, 149–154. [Google Scholar] [CrossRef]
- Magni, G.; Amici, A.; Emanuelli, M.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 1999, 73, 135–182. [Google Scholar] [PubMed]
- Berger, F.; Ramírez-Hernández, M.H.; Ziegler, M. The new life of a centenarian: Signalling functions of NAD(P). Trends Biochem. Sci. 2004, 29, 111–118. [Google Scholar] [CrossRef]
- Sasaki, Y.; Margolin, Z.; Borgo, B.; Havranek, J.J.; Milbrandt, J. Characterization of Leber Congenital Amaurosis-associated NMNAT1 Mutants. J. Biol. Chem. 2015, 290, 17228–17238. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özgül, R.K.; Bozkurt, B.; Kıratlı, H.; Oğüş, A.; Kiratli, H. Exclusion of LCA5 locus in a consanguineous Turkish family with macular coloboma-type LCA. Eye 2005, 20, 817–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad-Morales, V.; Burés-Jelstrup, A.; Navarro, R.; Ruiz-Nogales, S.; Méndez-Vendrell, P.; Corcóstegui, B.; Pomares, E. Characterization of the cone-rod dystrophy retinal phenotype caused by novel homozygous DRAM2 mutations. Exp. Eye Res. 2019, 187, 107752. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abad-Morales, V.; Wert, A.; Ruiz Gómez, M.Á.; Navarro, R.; Pomares, E. New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform. Int. J. Mol. Sci. 2021, 22, 2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052262
Abad-Morales V, Wert A, Ruiz Gómez MÁ, Navarro R, Pomares E. New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform. International Journal of Molecular Sciences. 2021; 22(5):2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052262
Chicago/Turabian StyleAbad-Morales, Víctor, Ana Wert, María Ángeles Ruiz Gómez, Rafael Navarro, and Esther Pomares. 2021. "New Insights on the Genetic Basis Underlying SHILCA Syndrome: Characterization of the NMNAT1 Pathological Alterations Due to Compound Heterozygous Mutations and Identification of a Novel Alternative Isoform" International Journal of Molecular Sciences 22, no. 5: 2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052262