
The CD44high Subpopulation of Multifraction Irradiation-Surviving NSCLC Cells Exhibits Partial EMT-Program Activation and DNA Damage Response Depending on Their p53 Status

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Radiosensitivity of A549IR and H1299IR Cells
2.2. DNA Repair Capacity of Parental Cells and Their MFR-Surviving Sublines Depending on Their p53 Status
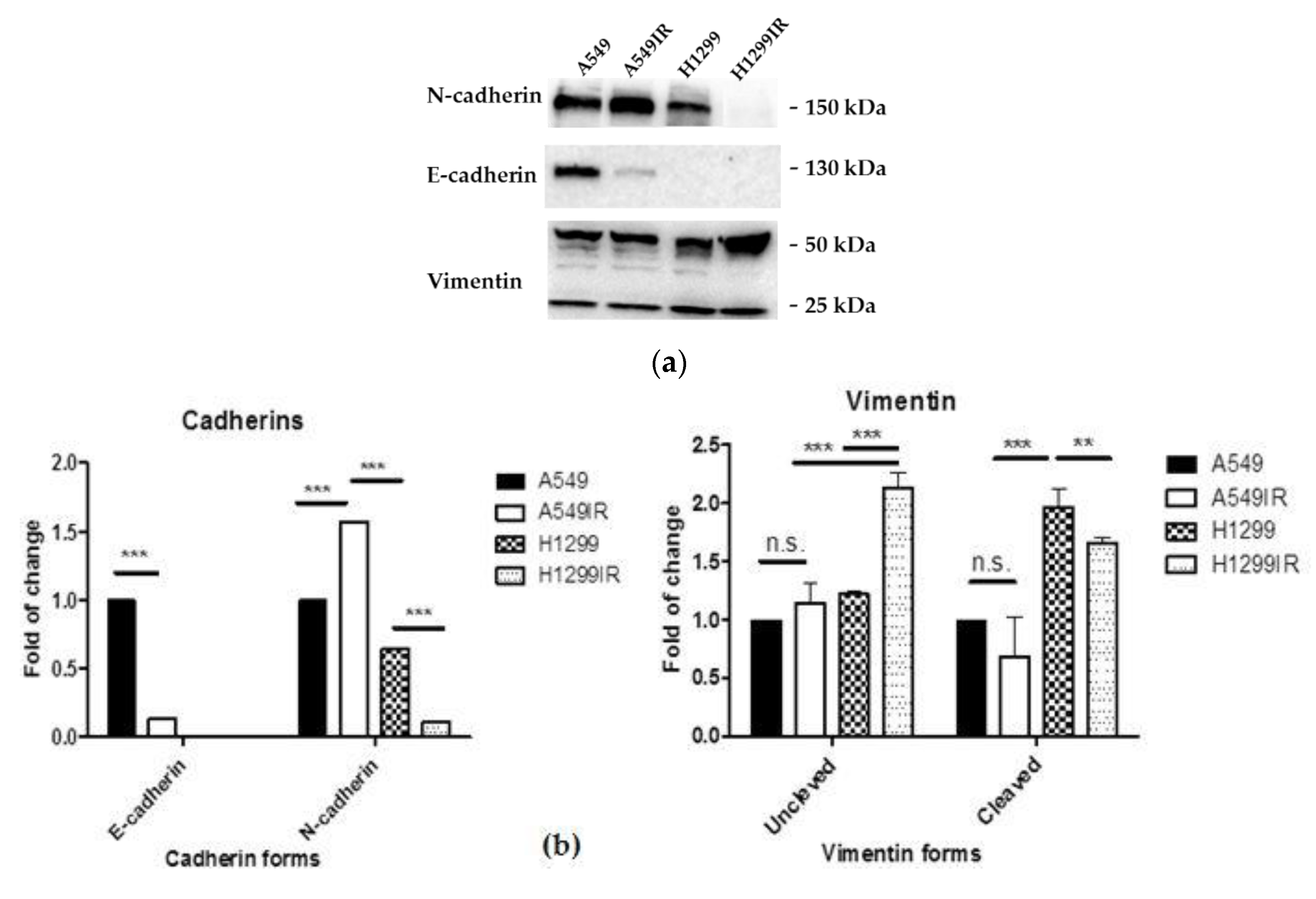
2.3. Partial EMT-Program Activation in Parental and MFR-Surviving Cells
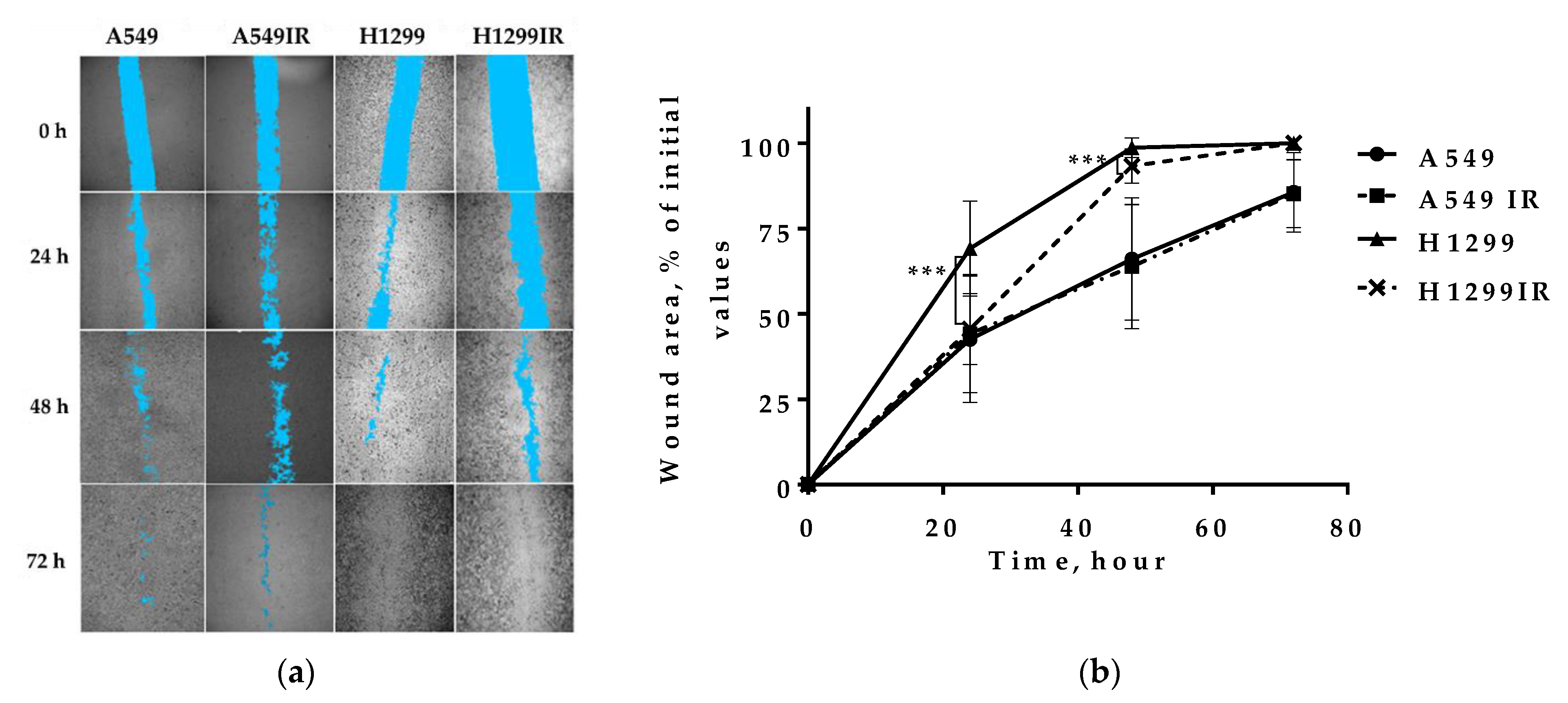
2.4. Migration Activity
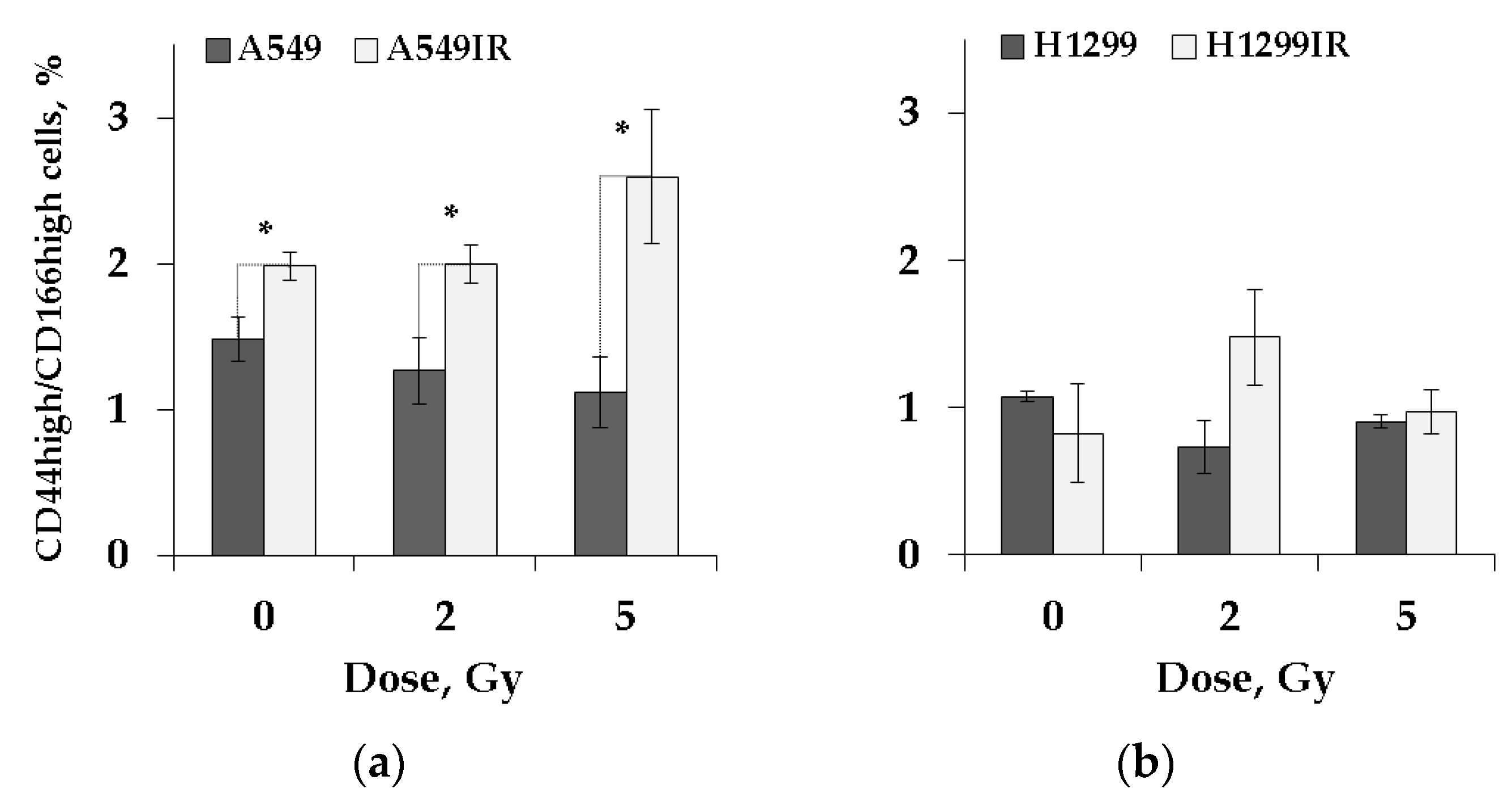
2.5. CD44high/CD166high (CSC-like) Population in Parental and MFR-Surviving Sublines after Additional Acute Irradiation
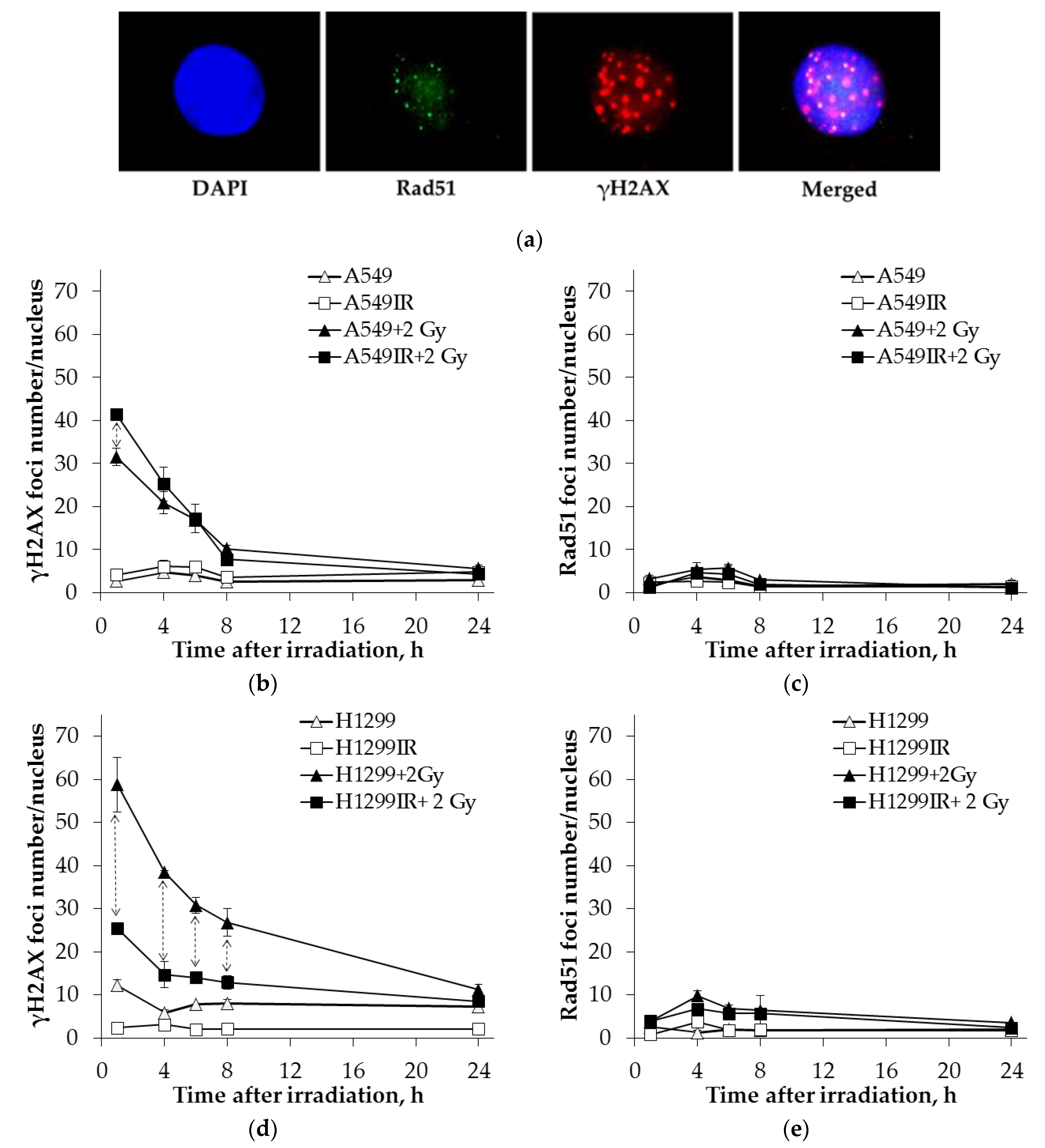
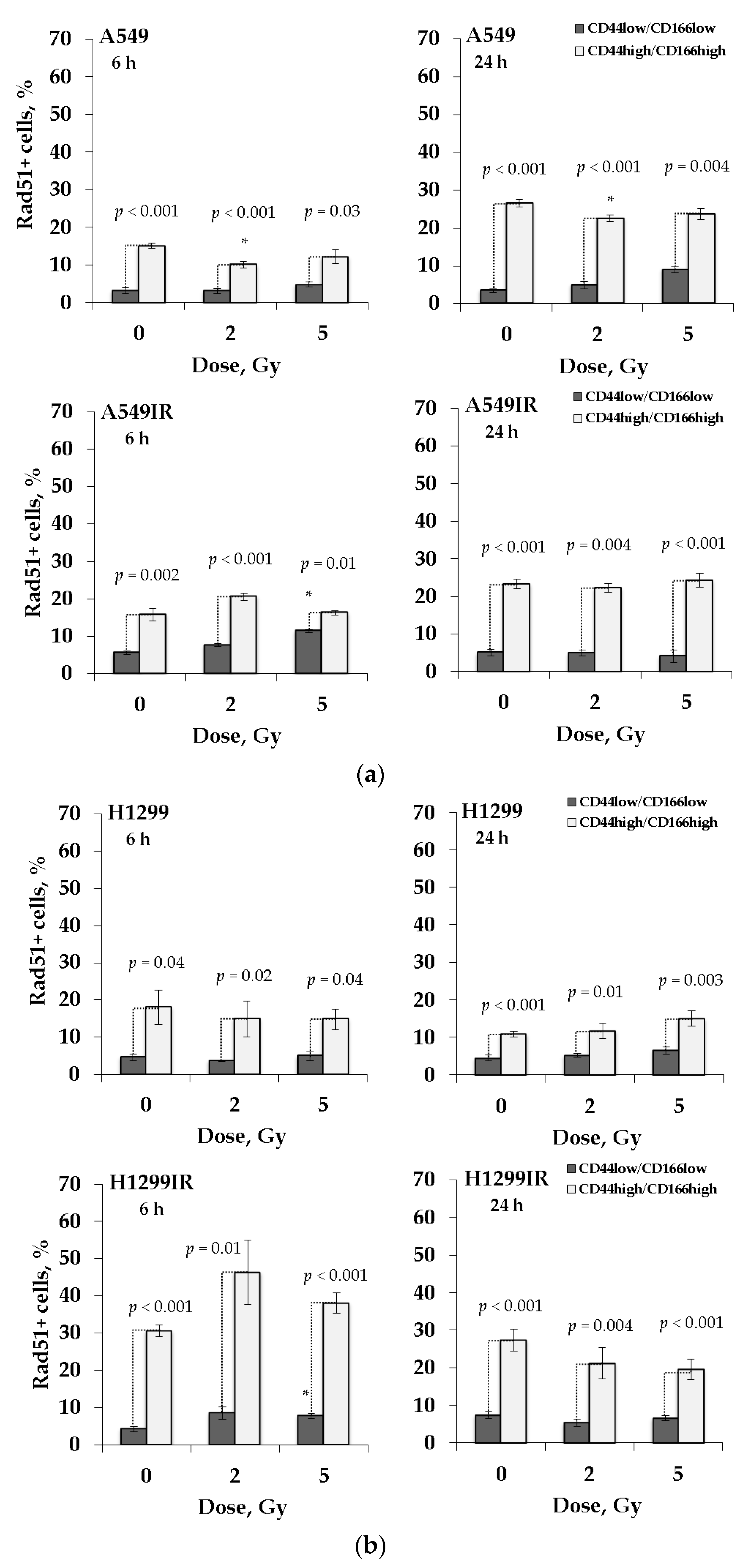
2.6. Rad51 Expression Indicates the Increased Activity of HR Pathway in CD44high/CD166high (CSC-Like) Population of Parental and MFR-Surviving Sublines
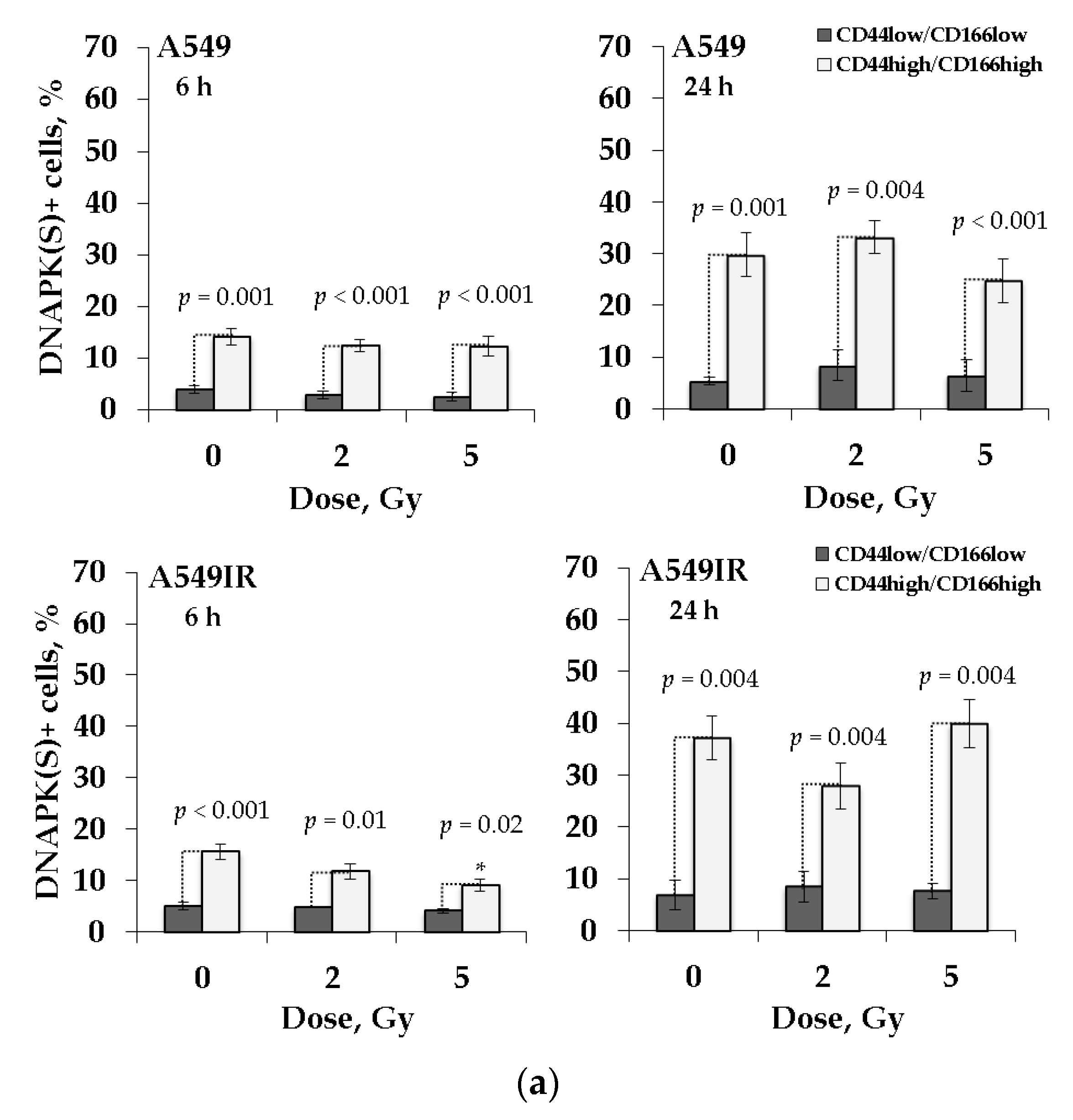
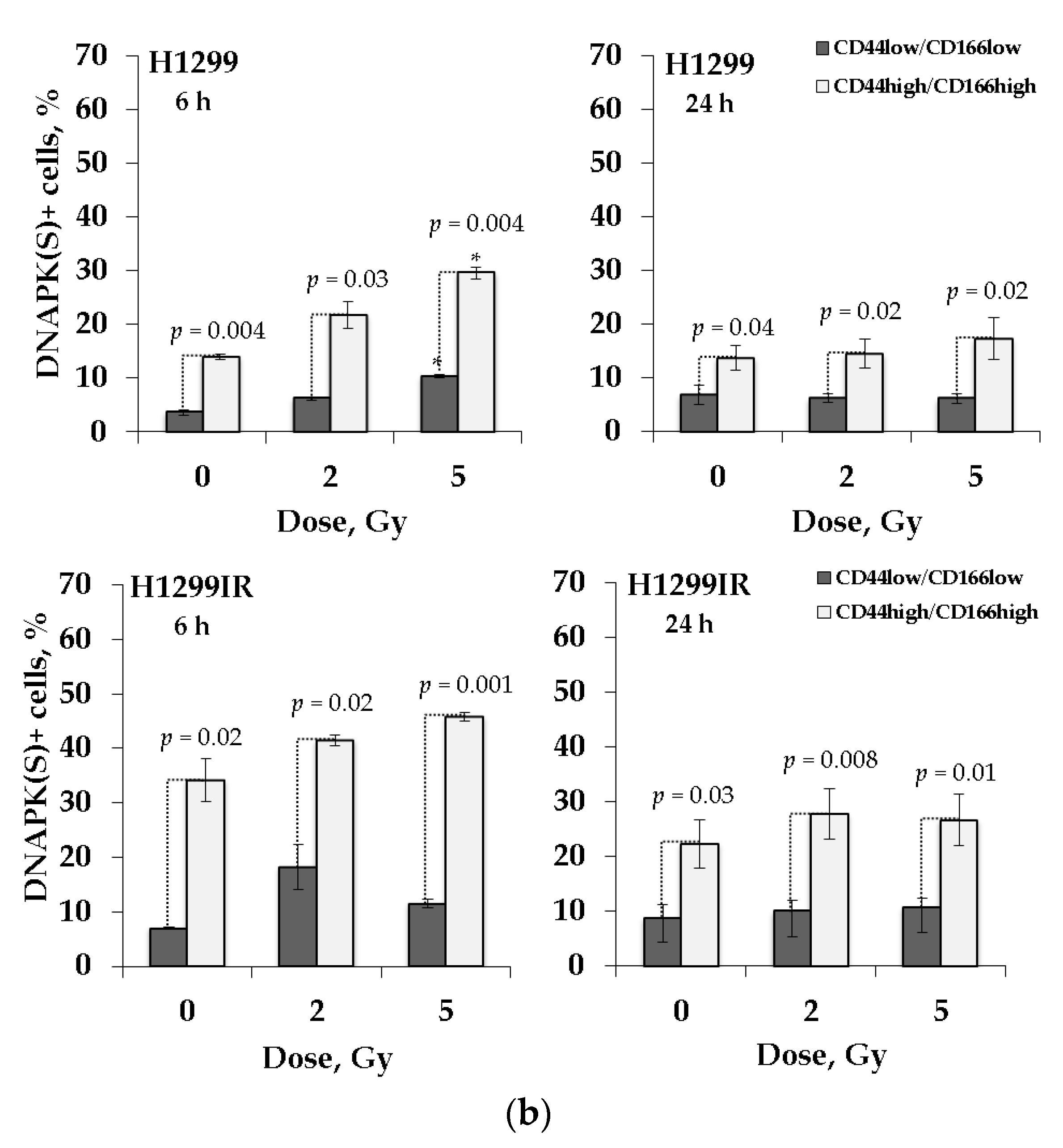
2.7. DNA-PK(S) Indicates an Attenuated DNA DSBs Repair by NHEJ in p53-Deficient Cells
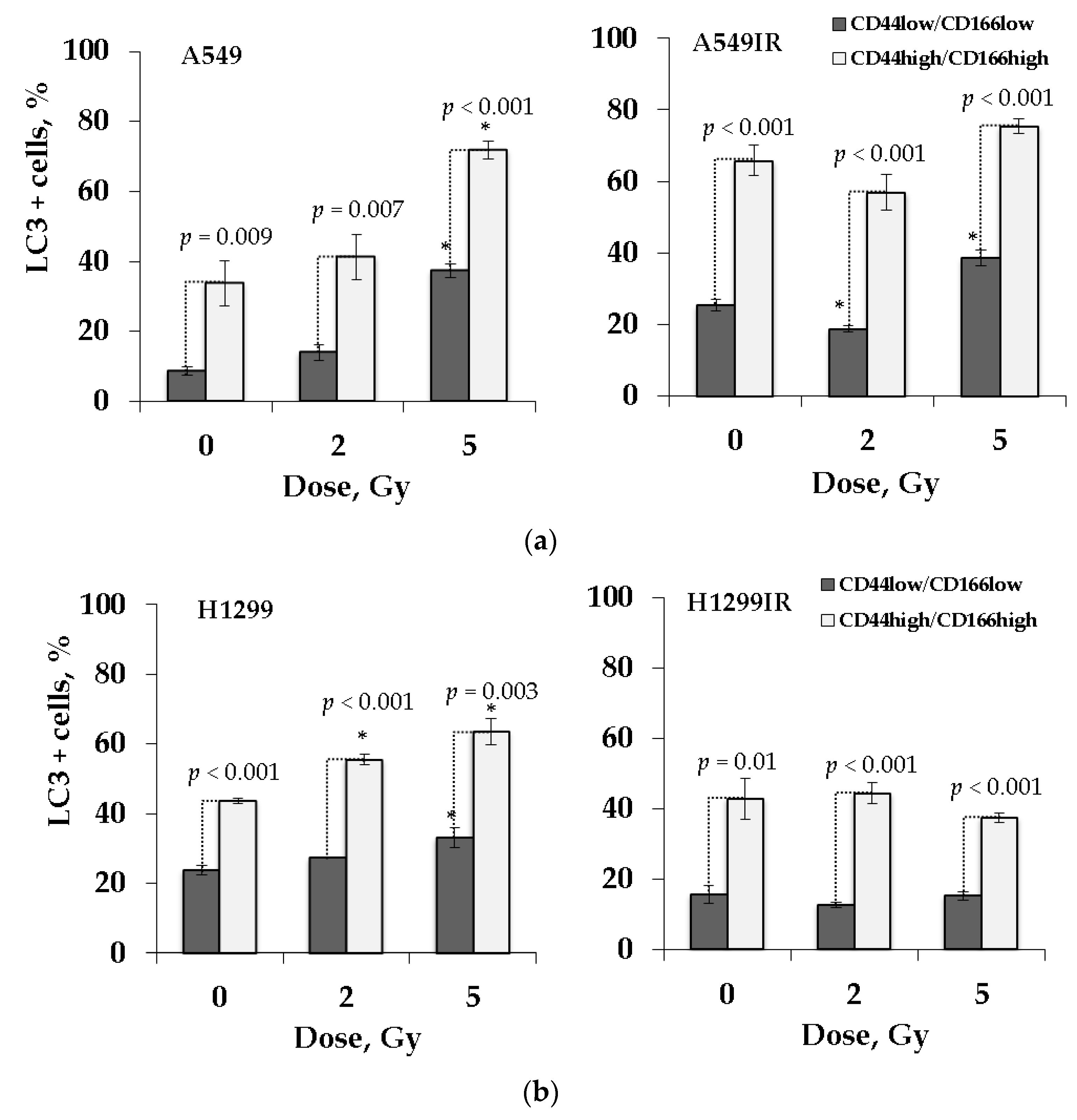
2.8. LC3 Activity Confers Cancer Stem-Like Cells Undergo Autophagy Process after Irradiation Exposure
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Irradiation
4.3. Colony Formation and Anchorage-Independent Soft Agar Assay
4.4. Immunofluorescence Staining
4.5. Flow Cytometry Analysis
4.6. Western Blotting
4.7. Migration Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Parsons, A.; Daley, A.; Begh, R.; Aveyard, P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. BMJ 2010, 340, b5569. [Google Scholar] [CrossRef] [Green Version]
- Onishi, H.; Shirato, H.; Nagata, Y.; Hiraoka, M.; Fujino, M.; Gomi, K.; Niibe, Y.; Karasawa, K.; Hayakawa, K.; Takai, Y.; et al. Hypofractionated stereotactic radiotherapy (HypoFXSRT) for stage I non-small cell lung cancer: Updated results of 257 patients in a Japanese multi-institutional study. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2007, 2, S94–S100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Urbano, M.A.; Grinan-Lison, C.; Marchal, J.A.; Nunez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef]
- Han, J.; Won, M.; Kim, J.H.; Jung, E.; Min, K.; Jangili, P.; Kim, J.S. Cancer stem cell-targeted bio-imaging and chemotherapeutic perspective. Chem. Soc. Rev. 2020, 49, 7856–7878. [Google Scholar] [CrossRef]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Fuller, M. Stem cells and cancer: Two faces of eve. Cell 2006, 124, 1111–1115. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassmann, A.; Carrillo Garcia, C.; Janzen, V.; Kraus, D.; Veit, N.; Winter, J.; Probstmeier, R. Staurosporine Induces the Generation of Polyploid Giant Cancer Cells in Non-Small-Cell Lung Carcinoma A549 Cells. Anal. Cell Pathol. (Amst.) 2018, 2018, 1754085. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.J.; Centuori, S.M.; Harman, M.W.; Putnam, C.W.; Wolgemuth, C.W.; Martinez, J.D. The induction of endoreduplication and polyploidy by elevated expression of 14-3-3gamma. Genes Cancer 2017, 8, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: Implications for treatment response. Clin. Cancer Res. 2005, 11, 2637–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef] [Green Version]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Babayan, N.; Vorobyeva, N.; Grigoryan, B.; Grekhova, A.; Pustovalova, M.; Rodneva, S.; Fedotov, Y.; Tsakanova, G.; Aroutiounian, R.; Osipov, A. Low Repair Capacity of DNA Double-Strand Breaks Induced by Laser-Driven Ultrashort Electron Beams in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9488. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Kholodenko, R.; Grekhova, A.; Suntsova, M.; Pustovalova, M.; Vorobyeva, N.; Kholodenko, I.; Malakhova, G.; Garazha, A.; Nedoluzhko, A.; et al. Acquired resistance to tyrosine kinase inhibitors may be linked with the decreased sensitivity to X-ray irradiation. Oncotarget 2018, 9, 5111–5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Jensen, R.B. DNA Strand Exchange to Monitor Human RAD51-Mediated Strand Invasion and Pairing. Methods Mol. Biol. 2021, 2153, 101–113. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Lobrich, M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 2015, 471, 1–11. [Google Scholar] [CrossRef]
- Ruiz, G.; Valencia-Gonzalez, H.A.; Leon-Galicia, I.; Garcia-Villa, E.; Garcia-Carranca, A.; Gariglio, P. Inhibition of RAD51 by siRNA and Resveratrol Sensitizes Cancer Stem Cells Derived from HeLa Cell Cultures to Apoptosis. Stem Cells Int. 2018, 2018, 2493869. [Google Scholar] [CrossRef]
- Short, S.C.; Giampieri, S.; Worku, M.; Alcaide-German, M.; Sioftanos, G.; Bourne, S.; Lio, K.I.; Shaked-Rabi, M.; Martindale, C. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor-derived cells. Neuro-Oncol. 2011, 13, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ma, N.; Yao, W.; Li, S.; Ren, Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019, 19, 356. [Google Scholar] [CrossRef] [PubMed]
- Tennstedt, P.; Fresow, R.; Simon, R.; Marx, A.; Terracciano, L.; Petersen, C.; Sauter, G.; Dikomey, E.; Borgmann, K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Cancer 2013, 132, 2118–2126. [Google Scholar] [CrossRef]
- Budke, B.; Lv, W.; Kozikowski, A.P.; Connell, P.P. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem 2016, 11, 2468–2473. [Google Scholar] [CrossRef] [Green Version]
- Aihara, H.; Ito, Y.; Kurumizaka, H.; Yokoyama, S.; Shibata, T. The N-terminal domain of the human Rad51 protein binds DNA: Structure and a DNA binding surface as revealed by NMR. J. Mol. Biol. 1999, 290, 495–504. [Google Scholar] [CrossRef]
- Bearss, D.J.; Lee, R.J.; Troyer, D.A.; Pestell, R.G.; Windle, J.J. Differential effects of p21(WAF1/CIP1) deficiency on MMTV-ras and MMTV-myc mammary tumor properties. Cancer Res. 2002, 62, 2077–2084. [Google Scholar]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pustovalova, M.; Alhaddad, L.; Smetanina, N.; Chigasova, A.; Blokhina, T.; Chuprov-Netochin, R.; Osipov, A.N.; Leonov, S. The p53-53BP1-Related Survival of A549 and H1299 Human Lung Cancer Cells after Multifractionated Radiotherapy Demonstrated Different Response to Additional Acute X-ray Exposure. Int. J. Mol. Sci. 2020, 21, 3342. [Google Scholar] [CrossRef]
- Lin, S.Q.; Jia, F.J.; Zhang, C.Y.; Liu, F.Y.; Ma, J.H.; Han, Z.; Xie, W.D.; Li, X. Actinomycin V Suppresses Human Non-Small-Cell Lung Carcinoma A549 Cells by Inducing G2/M Phase Arrest and Apoptosis via the p53-Dependent Pathway. Mar. Drugs 2019, 17, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Gu, Y.Y.; Li, Z.G.; Jia, Y.H.; Yuan, L.; Li, S.Y.; An, G.S.; Ni, J.H.; Jia, H.T. Exposure of human lung cancer cells to 8-chloro-adenosine induces G2/M arrest and mitotic catastrophe. Neoplasia 2004, 6, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Aroca, D.M.; Roche, O.; Sabater, S.; Pascual-Serra, R.; Ortega-Muelas, M.; Sanchez Perez, I.; Belandia, B.; Ruiz-Hidalgo, M.J.; Sanchez-Prieto, R. P53 pathway is a major determinant in the radiosensitizing effect of Palbociclib: Implication in cancer therapy. Cancer Lett. 2019, 451, 23–33. [Google Scholar] [CrossRef]
- Grekhova, A.K.; Pustovalova, M.V.; Eremin, P.S.; Ozerov, I.V.; Maksimova, O.A.; Gordeev, A.V.; Vorobyeva, N.Y.; Osipov, A.N. Evaluation of the Contribution of Homologous Recombination in DNA Double-Strand Break Repair in Human Fibroblasts after Exposure to Low and Intermediate Doses of X-ray Radiation. Biol. Bull. 2019, 46, 1496–1502. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [Green Version]
- de Jong, M.C.; Ten Hoeve, J.J.; Grenman, R.; Wessels, L.F.; Kerkhoven, R.; Te Riele, H.; van den Brekel, M.W.; Verheij, M.; Begg, A.C. Pretreatment microRNA Expression Impacting on Epithelial-to-Mesenchymal Transition Predicts Intrinsic Radiosensitivity in Head and Neck Cancer Cell Lines and Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5630–5638. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Zhang, M.; Zhou, C.; Wang, W.; Yang, H.; Ye, W. The role of epithelial-mesenchymal transition in regulating radioresistance. Crit. Rev. Oncol. /Hematol. 2020, 150, 102961. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.; Cheng, L.C.; Sihoe, A.D.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.P.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, P.; El-Deiry, W.S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, F.; Ghezzi, P.; Rumio, C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol. Cancer 2017, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Luo, H.; Zhou, X.; Zhu, B.; Wang, Y.; Bian, X. Identification of CD90 as a marker for lung cancer stem cells in A549 and H446 cell lines. Oncol. Rep. 2013, 30, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Sterlacci, W.; Savic, S.; Fiegl, M.; Obermann, E.; Tzankov, A. Putative stem cell markers in non-small-cell lung cancer: A clinicopathologic characterization. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2014, 9, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Hu, L.; Lau, S.H.; Tzang, C.H.; Wen, J.M.; Wang, W.; Xie, D.; Huang, M.; Wang, Y.; Wu, M.C.; Huang, J.F.; et al. Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene 2004, 23, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Schubert, J.; Brabletz, T. p53 Spreads out further: Suppression of EMT and stemness by activating miR-200c expression. Cell Res. 2011, 21, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Keck, T.; Brabletz, T. Under stress: p53 controls EMT and stemness in pancreatic epithelial cells. Cell Cycle 2011, 10, 1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef] [PubMed]
- Shahhoseini, E.; Feltis, B.N.; Nakayama, M.; Piva, T.J.; Pouniotis, D.; Alghamdi, S.S.; Geso, M. Combined Effects of Gold Nanoparticles and Ionizing Radiation on Human Prostate and Lung Cancer Cell Migration. Int. J. Mol. Sci. 2019, 20, 4488. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, K.; Tsuda, M.; Yazawa, N.; Nakamura, H.; Ishihara, S.; Haga, H.; Yasuda, M.; Yamazaki, R.; Shirato, H.; Kawaguchi, H.; et al. Increased motility and invasiveness in tumor cells that survive 10 Gy irradiation. Cell Struct. Funct. 2009, 34, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Moncharmont, C.; Levy, A.; Guy, J.B.; Falk, A.T.; Guilbert, M.; Trone, J.C.; Alphonse, G.; Gilormini, M.; Ardail, D.; Toillon, R.A.; et al. Radiation-enhanced cell migration/invasion process: A review. Crit. Rev. Oncol. Hematol. 2014, 92, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Akino, Y.; Teshima, T.; Kihara, A.; Kodera-Suzumoto, Y.; Inaoka, M.; Higashiyama, S.; Furusawa, Y.; Matsuura, N. Carbon-ion beam irradiation effectively suppresses migration and invasion of human non-small-cell lung cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Teshima, T.; Inaoka, M.; Minami, K.; Tsuchiya, T.; Isono, M.; Furusawa, Y.; Matsuura, N. Carbon ion irradiation suppresses metastatic potential of human non-small cell lung cancer A549 cells through the phosphatidylinositol-3-kinase/Akt signaling pathway. J. Radiat. Res. 2011, 52, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, M.; Wang, H.; Bocker, W.; Iliakis, G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell Physiol. 2005, 202, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Ulyanenko, S.; Pustovalova, M.; Koryakin, S.; Beketov, E.; Lychagin, A.; Ulyanenko, L.; Kaprin, A.; Grekhova, A.; Ozerova, A.M.; Ozerov, I.V.; et al. Formation of gammaH2AX and pATM Foci in Human Mesenchymal Stem Cells Exposed to Low Dose-Rate Gamma-Radiation. Int. J. Mol. Sci. 2019, 20, 2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Kashino, G.; Ono, K.; Watanabe, M. Phosphorylated H2AX foci in tumor cells have no correlation with their radiation sensitivities. J. Radiat. Res. 2009, 50, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Pustovalova, M.; Grekhova, A.; Astrelina capital Te, C.; Nikitina, V.; Dobrovolskaya, E.; Suchkova, Y.; Kobzeva, I.; Usupzhanova, D.; Vorobyeva, N.; Samoylov, A.; et al. Accumulation of spontaneous gammaH2AX foci in long-term cultured mesenchymal stromal cells. Aging 2016, 8, 3498–3506. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, P.L.; Banath, J.P. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Banath, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Y.; Li, Y.; An, G.S.; Ni, J.H.; Jia, H.T.; Li, S.Y. DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine. Int. J. Mol. Sci. 2018, 19, 1587. [Google Scholar] [CrossRef] [Green Version]
- Hannay, J.A.; Liu, J.; Zhu, Q.S.; Bolshakov, S.V.; Li, L.; Pisters, P.W.; Lazar, A.J.; Yu, D.; Pollock, R.E.; Lev, D. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: A role for p53/activator protein 2 transcriptional regulation. Mol. Cancer Ther. 2007, 6, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Sengupta, S.; Gurdziel, K.; Bell, G.W.; Jacks, T.; Flores, E.R. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009, 5, e1000680. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Yang, X. A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle 2013, 12, 3720–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, A.A.; Belkhiri, A.; Ecsedy, J.; Zaika, A.; El-Rifai, W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 2008, 68, 8998–9004. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia-Gonzalez, H.A.; Ruiz, G.; Ortiz-Sanchez, E.; Garcia-Carranca, A. Cancer Stem Cells from Tumor Cell Lines Activate the DNA Damage Response Pathway after Ionizing Radiation More Efficiently Than Noncancer Stem Cells. Stem Cells Int. 2019, 2019, 7038953. [Google Scholar] [CrossRef] [PubMed]
- Sneath, R.J.; Mangham, D.C. The normal structure and function of CD44 and its role in neoplasia. Mol. Pathol. Mp 1998, 51, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Chen, B.P.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Reddy, Y.V.; Wang, W.; Woods, T.; Douglas, P.; Ramsden, D.A.; Lees-Miller, S.P.; Meek, K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 2003, 23, 5836–5848. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Chi, L.; So, S.; Lee, K.J.; Mori, E.; Fattah, K.; Yang, J.; Chen, D.J. BRCA1 modulates the autophosphorylation status of DNA-PKcs in S phase of the cell cycle. Nucleic Acids Res. 2014, 42, 11487–11501. [Google Scholar] [CrossRef]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: Inhibition not equal deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. Jci Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegue, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef]
- Lomonaco, S.L.; Finniss, S.; Xiang, C.; Decarvalho, A.; Umansky, F.; Kalkanis, S.N.; Mikkelsen, T.; Brodie, C. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 2009, 125, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Shvets, E.; Fass, E.; Scherz-Shouval, R.; Elazar, Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J. Cell Sci. 2008, 121, 2685–2695. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, D.; Sun, Z.; Ye, T.; Li, J.; Zeng, B.; Zhao, Q.; Rosie Xing, H. Autophagy augments the self-renewal of lung cancer stem cells by the degradation of ubiquitinated p53. Cell Death Dis. 2021, 12, 98. [Google Scholar] [CrossRef]
- Xu, J.; Patel, N.H.; Saleh, T.; Cudjoe, E.K., Jr.; Alotaibi, M.; Wu, Y.; Lima, S.; Hawkridge, A.M.; Gewirtz, D.A. Differential Radiation Sensitivity in p53 Wild-Type and p53-Deficient Tumor Cells Associated with Senescence but not Apoptosis or (Nonprotective) Autophagy. Radiat. Res. 2018, 190, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; Antunes, F.; Rangel, M.C.; Chammas, R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression—Implications for Cancer Therapy. Front. Oncol. 2020, 10, 606436. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, e51998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkova, A.; Ozerov, I.V.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyeva, N.; Eremin, I.; Pulin, A.; Zorin, V.; Kopnin, P.; et al. gammaH2AX, 53BP1 and Rad51 protein foci changes in mesenchymal stem cells during prolonged X-ray irradiation. Oncotarget 2017, 8, 64317–64329. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.N.; Grekhova, A.; Pustovalova, M.; Ozerov, I.V.; Eremin, P.; Vorobyeva, N.; Lazareva, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; et al. Activation of homologous recombination DNA repair in human skin fibroblasts continuously exposed to X-ray radiation. Oncotarget 2015, 6, 26876–26885. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pustovalova, M.; Alhaddad, L.; Blokhina, T.; Smetanina, N.; Chigasova, A.; Chuprov-Netochin, R.; Eremin, P.; Gilmutdinova, I.; Osipov, A.N.; Leonov, S. The CD44high Subpopulation of Multifraction Irradiation-Surviving NSCLC Cells Exhibits Partial EMT-Program Activation and DNA Damage Response Depending on Their p53 Status. Int. J. Mol. Sci. 2021, 22, 2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052369
Pustovalova M, Alhaddad L, Blokhina T, Smetanina N, Chigasova A, Chuprov-Netochin R, Eremin P, Gilmutdinova I, Osipov AN, Leonov S. The CD44high Subpopulation of Multifraction Irradiation-Surviving NSCLC Cells Exhibits Partial EMT-Program Activation and DNA Damage Response Depending on Their p53 Status. International Journal of Molecular Sciences. 2021; 22(5):2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052369
Chicago/Turabian StylePustovalova, Margarita, Lina Alhaddad, Taisia Blokhina, Nadezhda Smetanina, Anna Chigasova, Roman Chuprov-Netochin, Petr Eremin, Ilmira Gilmutdinova, Andreyan N. Osipov, and Sergey Leonov. 2021. "The CD44high Subpopulation of Multifraction Irradiation-Surviving NSCLC Cells Exhibits Partial EMT-Program Activation and DNA Damage Response Depending on Their p53 Status" International Journal of Molecular Sciences 22, no. 5: 2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052369