
Phenylalanine-Derived β-Lactam TRPM8 Modulators. Configuration Effect on the Antagonist Activity

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
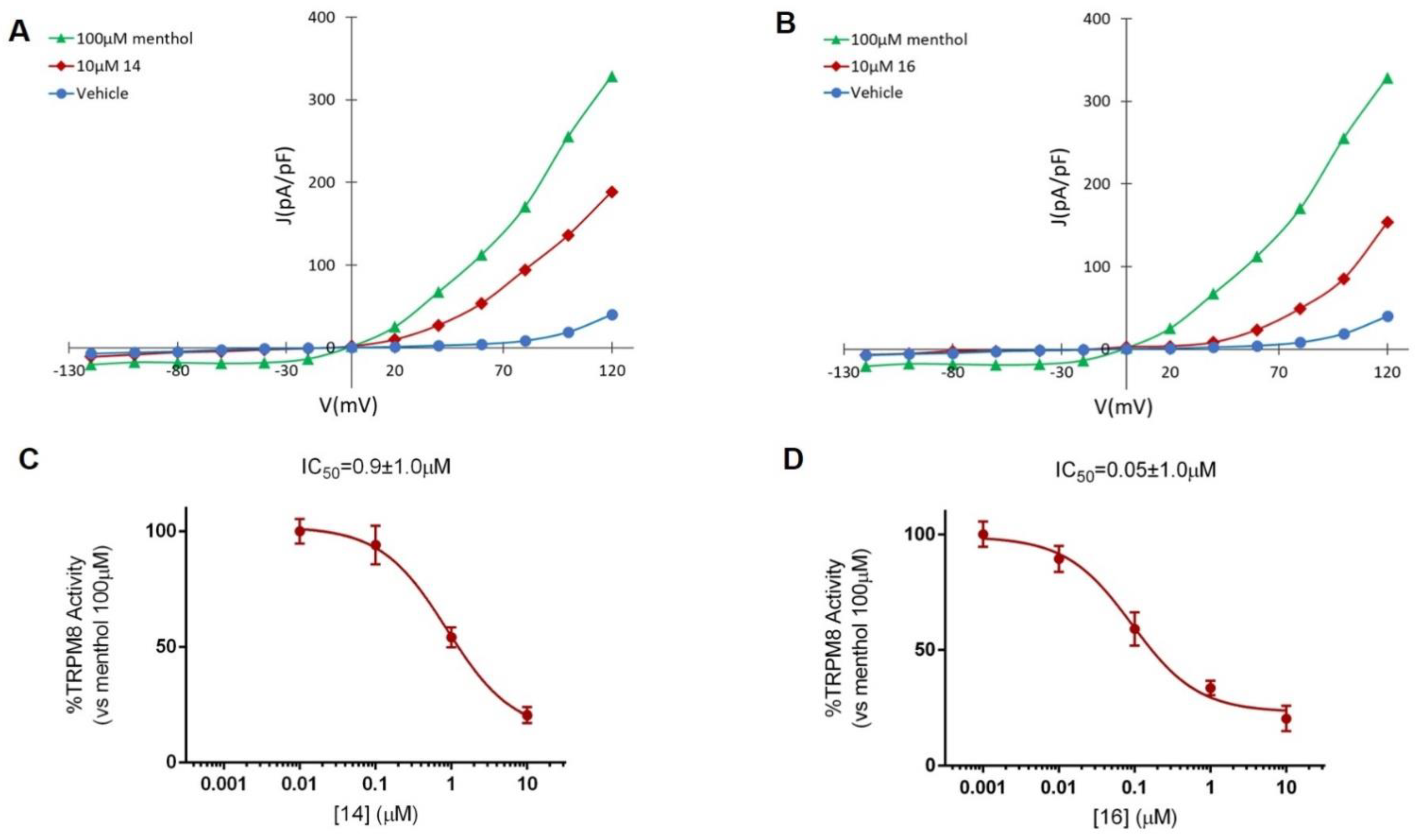
2. Results and Discussion
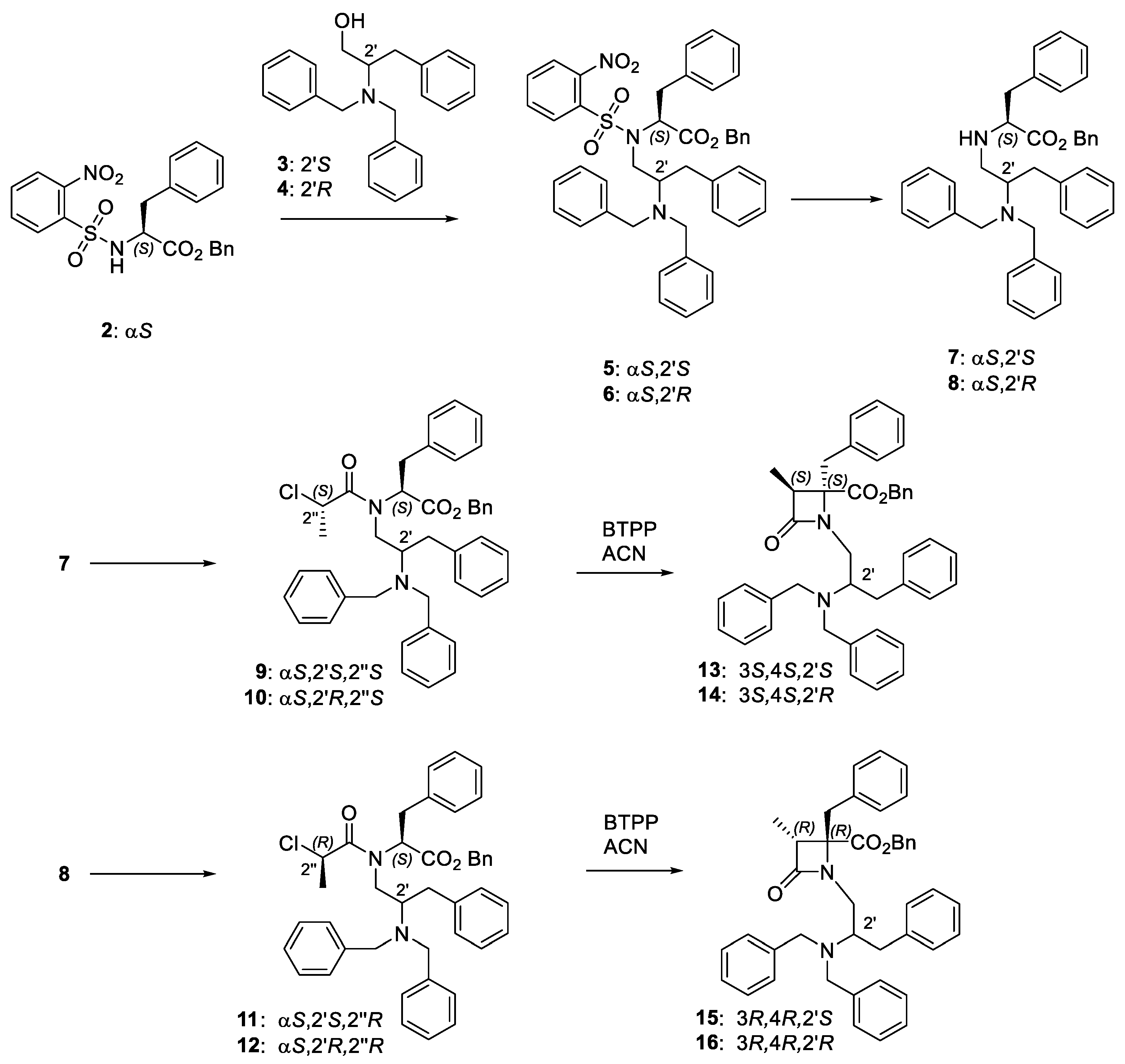
2.1. Chemistry
2.2. Biological Activity
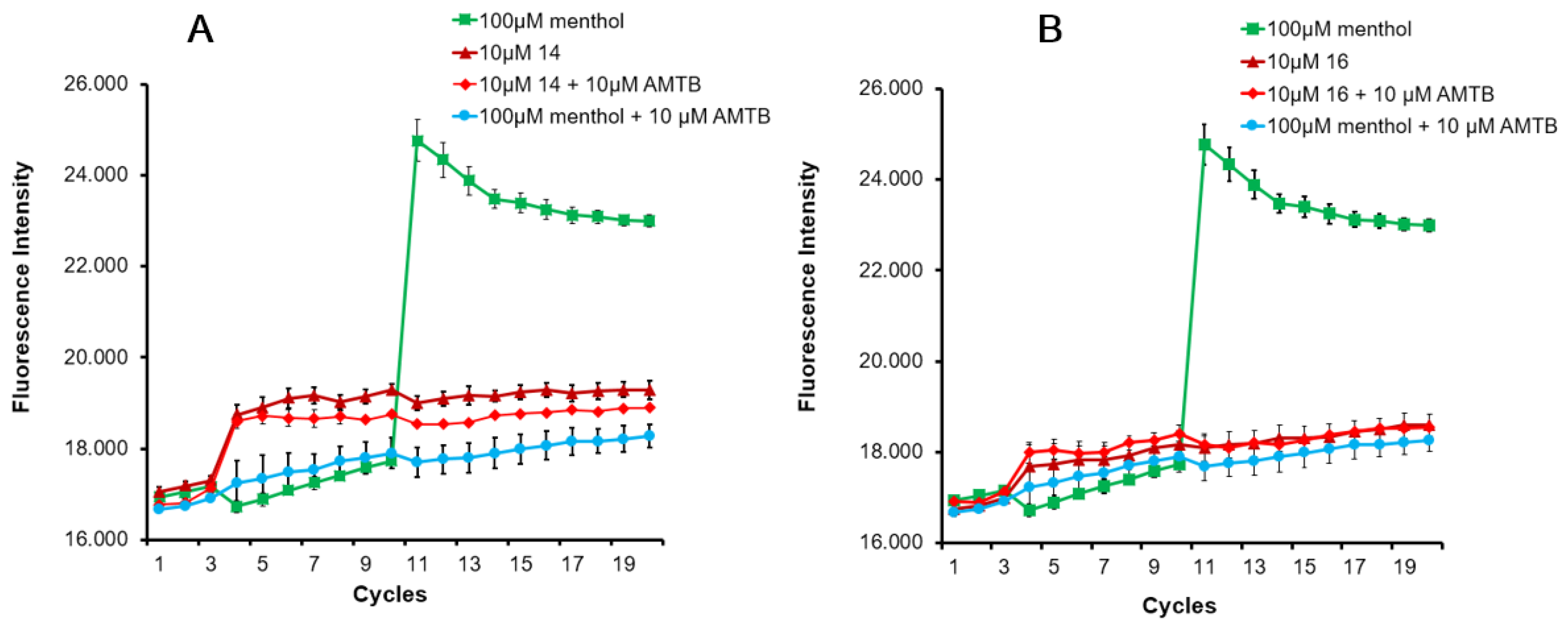
2.2.1. Ca2+ Intracellular Influx Assay
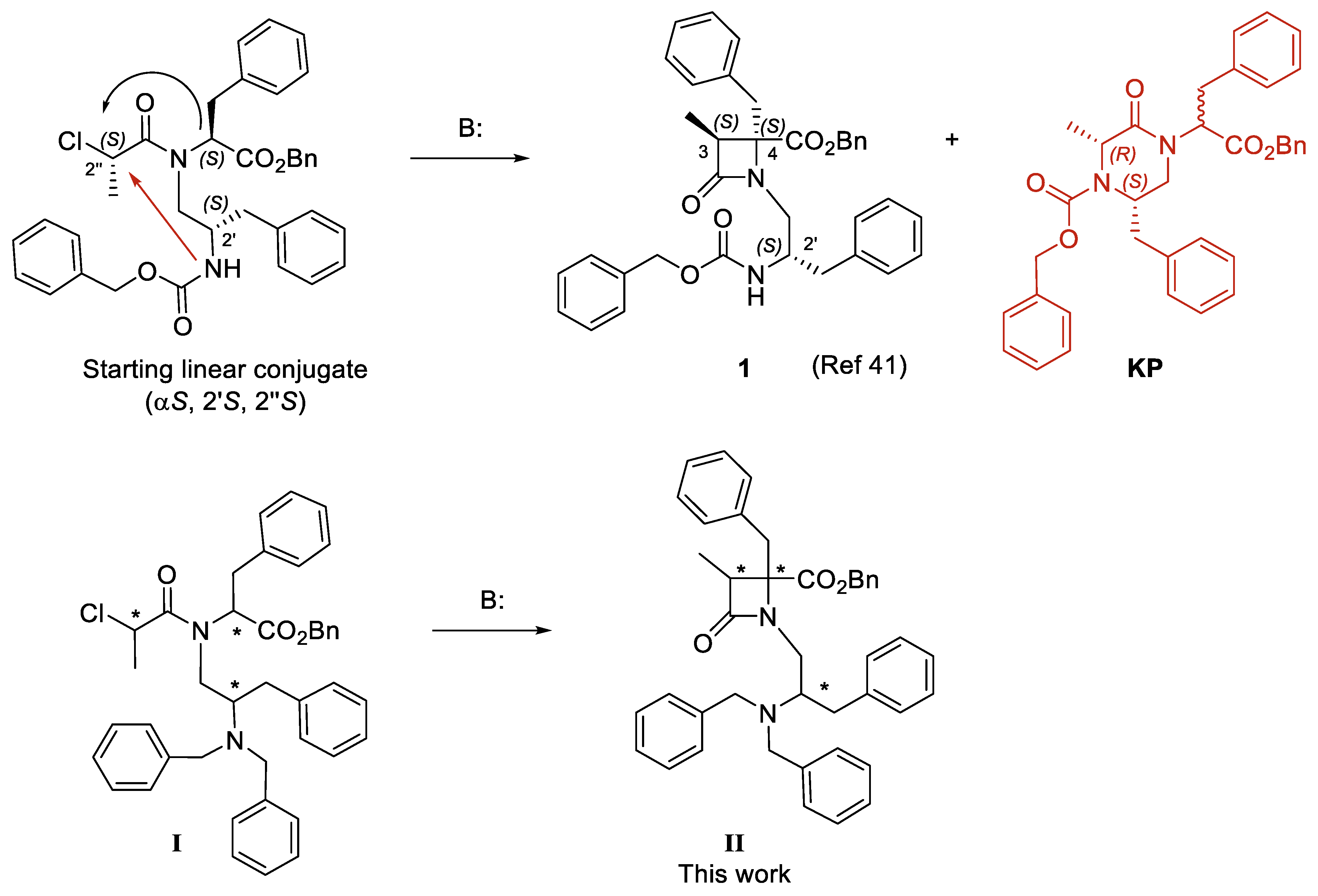
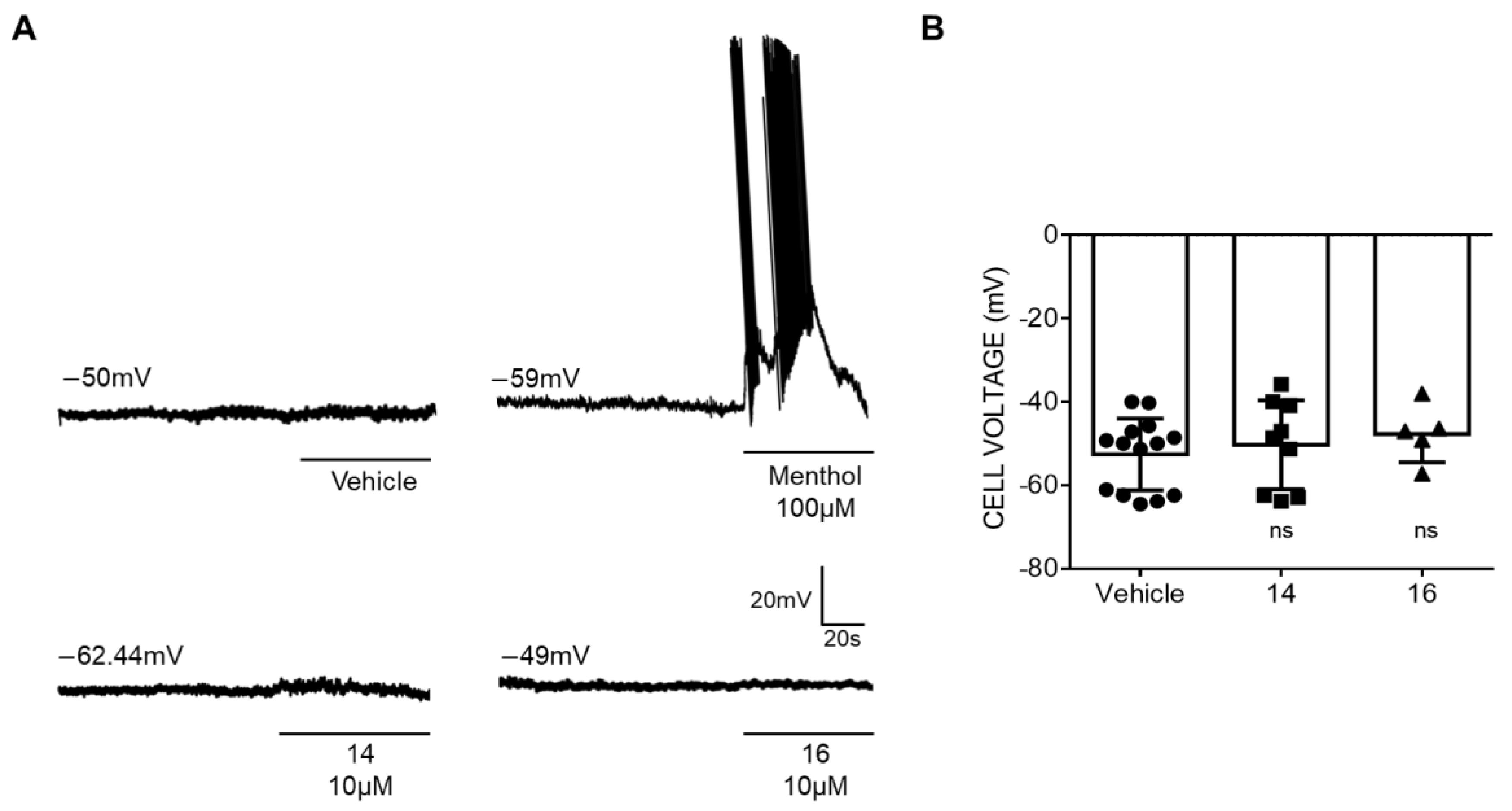
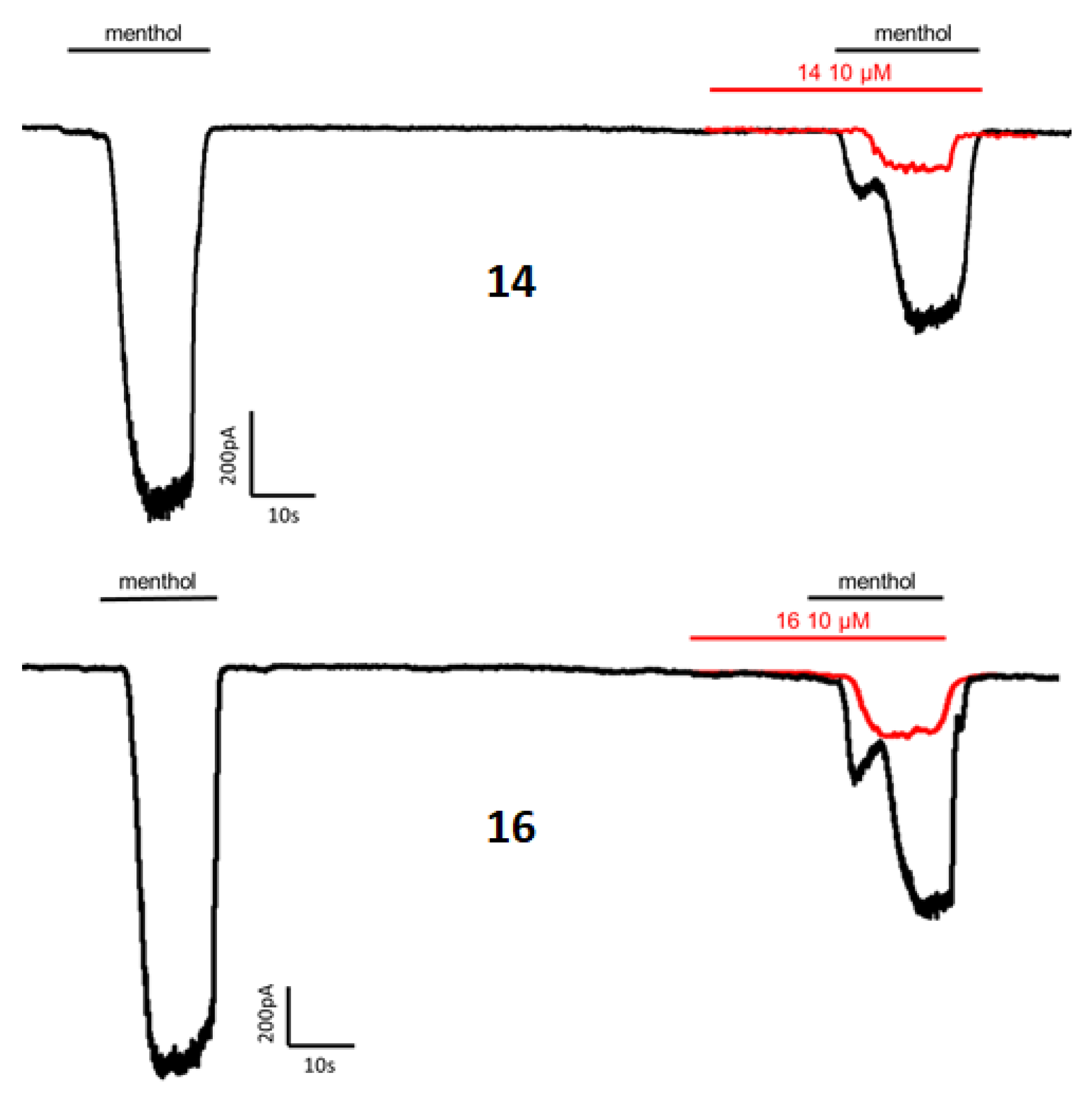
2.2.2. Patch–Clamp Experiments
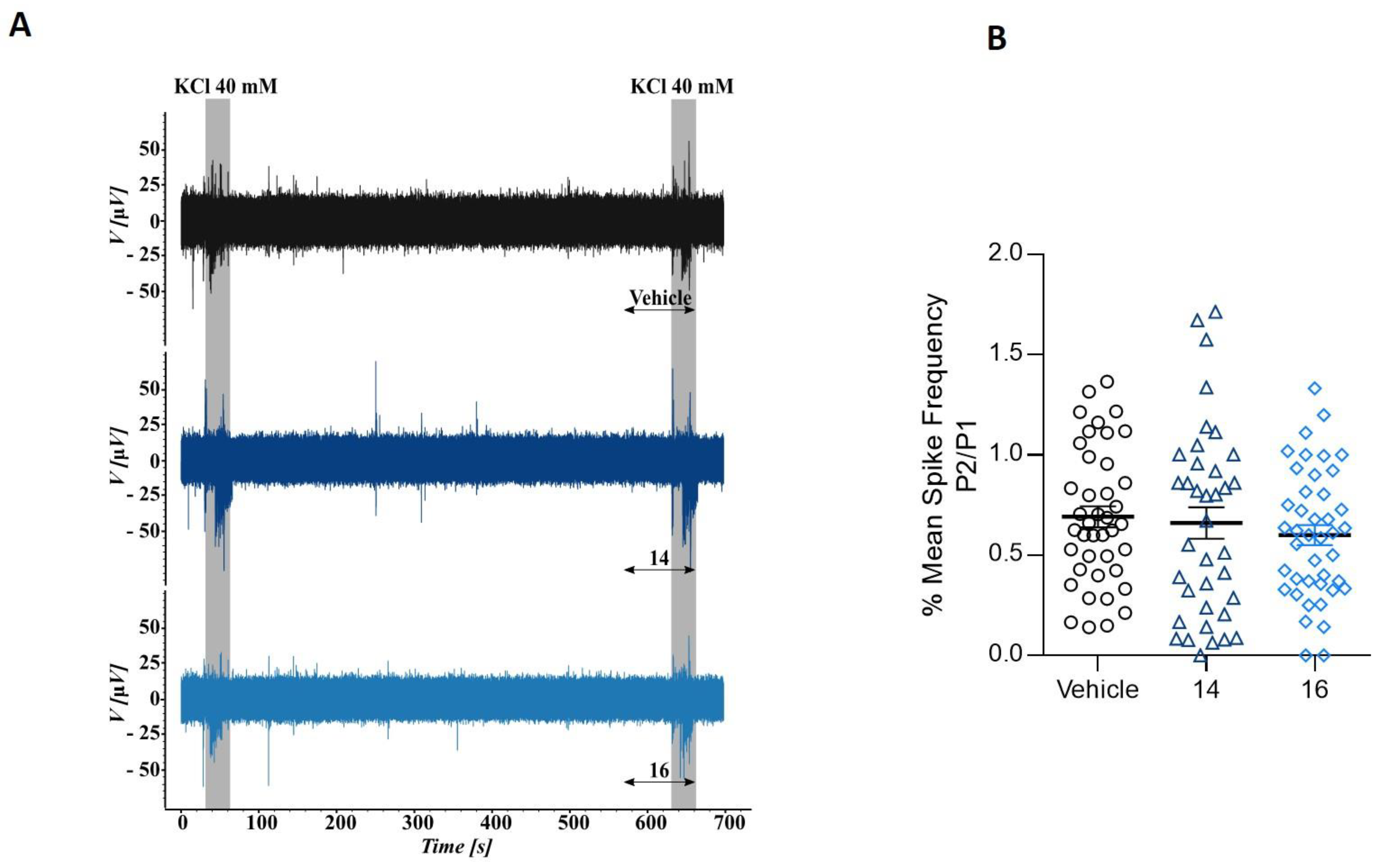
2.2.3. Microelectrode Array Experiments
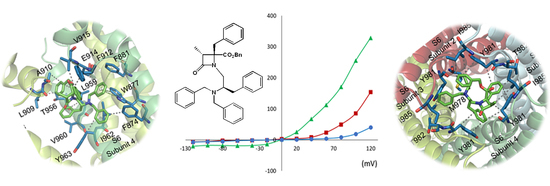
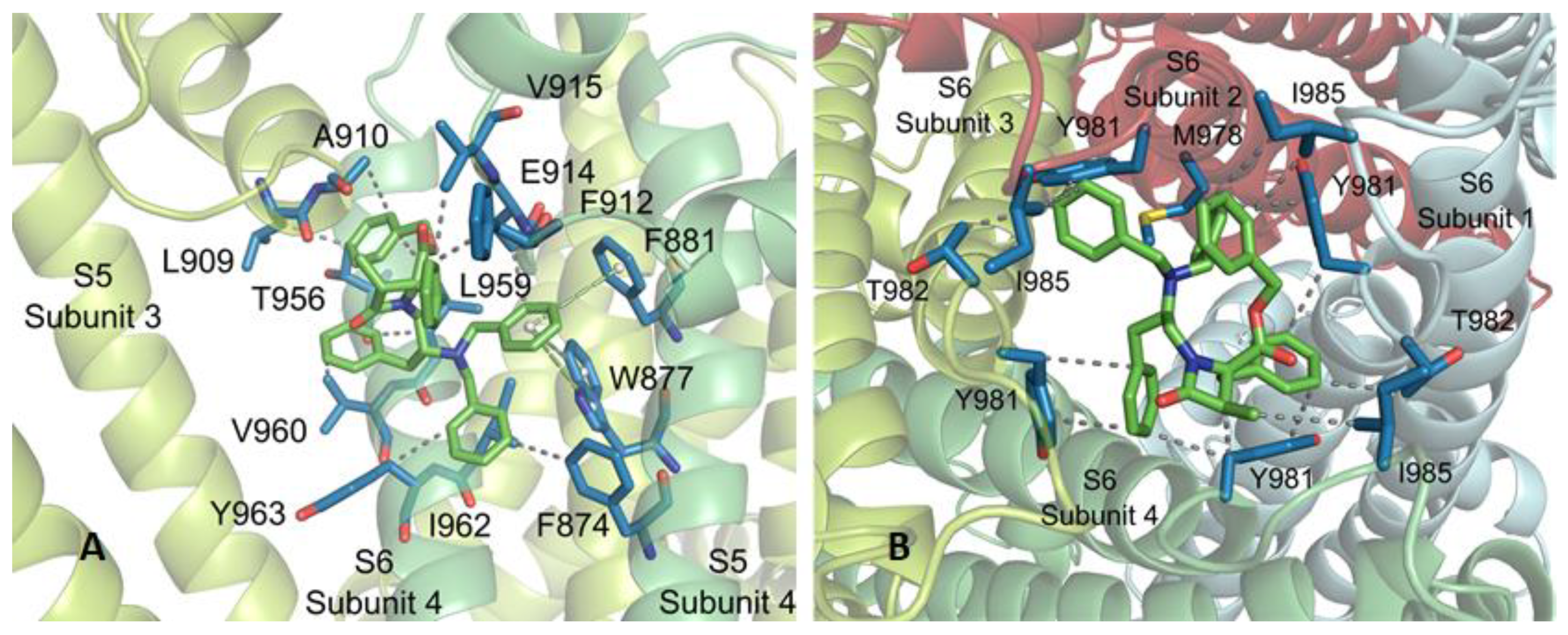
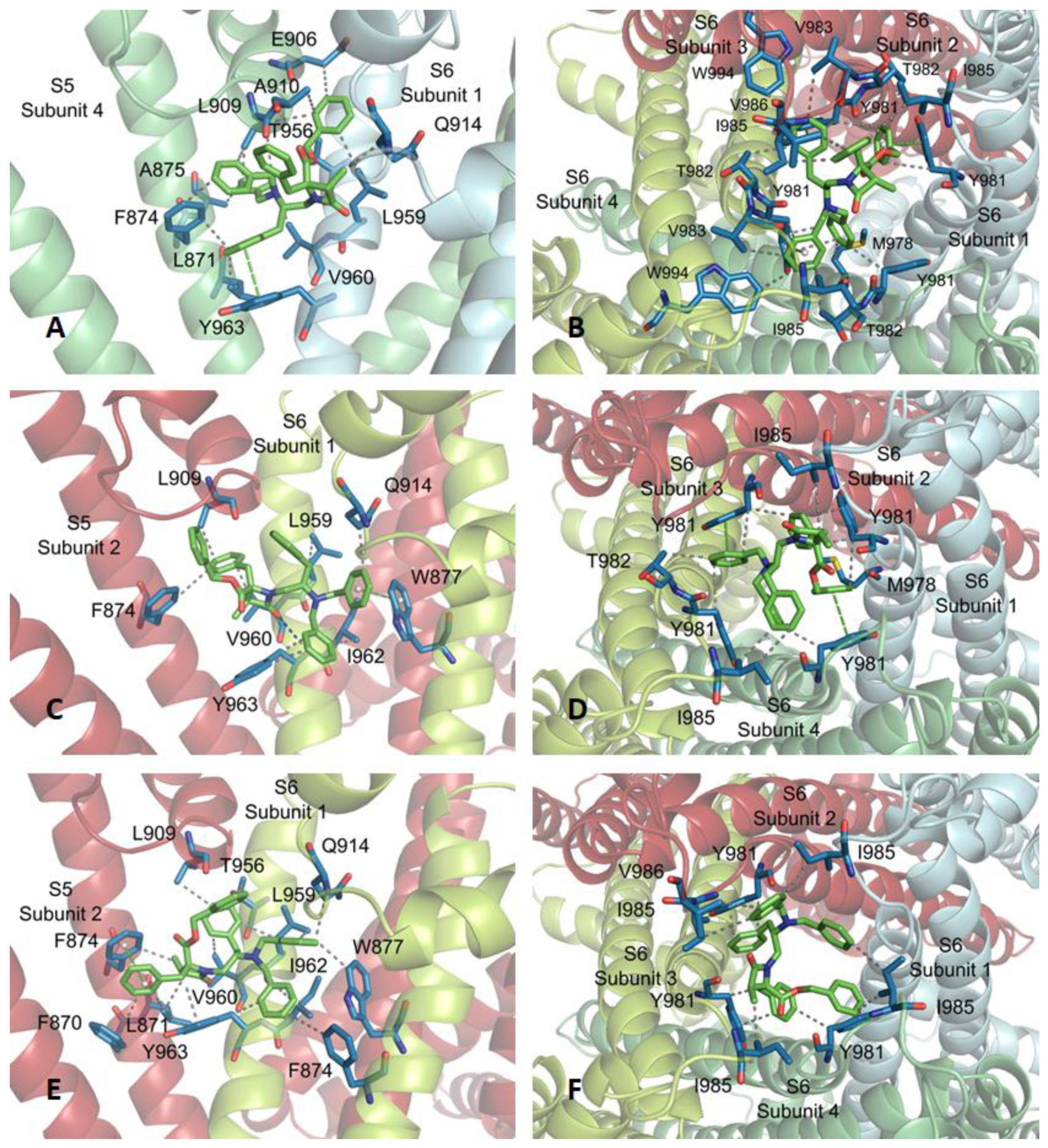
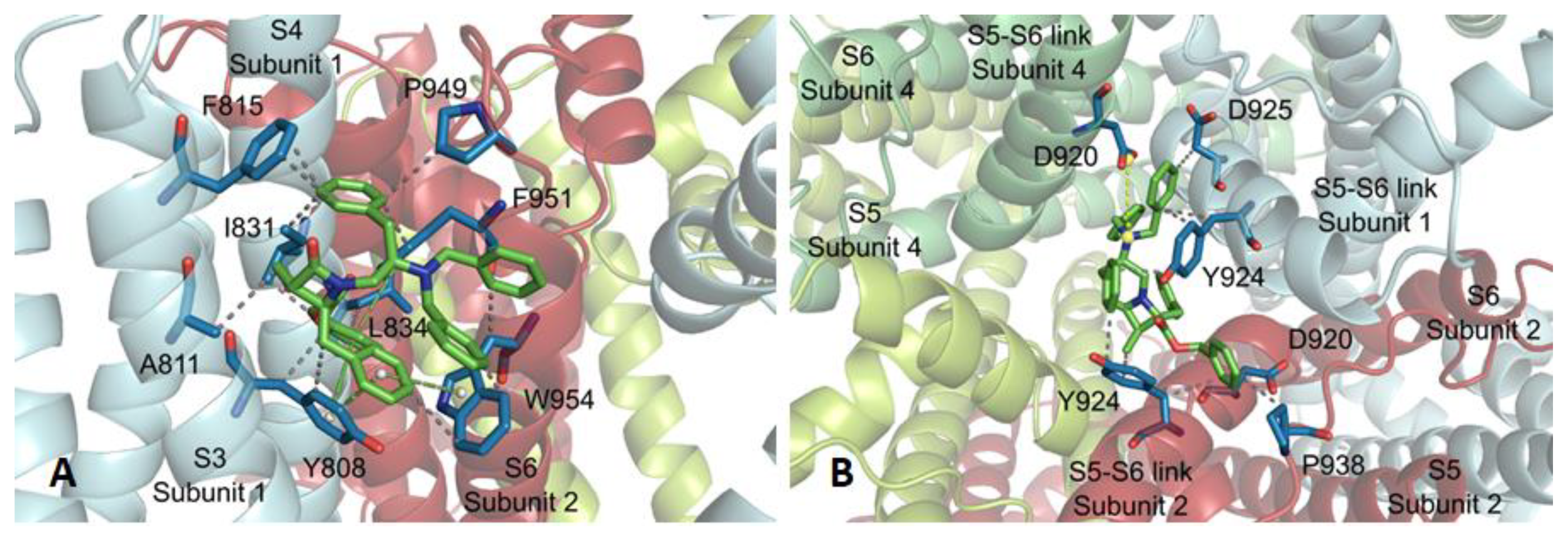
2.3. Molecular Modeling Studies
3. Materials and Methods
3.1. β-Lactam Formation
3.1.1. 4S-Benzyl-1-[(2′S-dibenzylamino-3′-phenyl-)prop-1′-yl]-4-benzyloxicarbonyl-3S-methyl-2-oxoazetidine (13)
3.1.2. 4S-Benzyl-1-[(2′R-dibenzylamino-3′-phenyl)prop-1′-yl]-4-benzyloxicarbonyl-3S-methyl-2-oxoazetidine (14)
3.1.3. 4R-Benzyl-1-[(2′S-dibenzylamino-3′-phenyl)prop-1′-yl]-4-benzyloxicarbonyl- 3R-methyl-2-oxoazetidine (15)
3.1.4. 4R-Benzyl-1-[(2′R-dibenzylamino-3′-phenyl-)prop-1′-yl]-4-benzyloxicarbonyl- 3R-methyl-2-oxoazetidine (16)
3.2. Functional Assays by Calcium Microfluorimetry
3.3. Functional Assays by Patch-Clamp Electrophysiology
3.4. Functional Assays by Microelectrode Arrays
3.5. Docking Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMTB | N-(3-Aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide |
| Bn | Benzyl |
| BTPP | Phosphazene base P1-t-Bu-tris(tetramethylene) |
| HEK | Human embryo kidney cells |
| HPLC | High-performance liquid chromatography |
| KP | 2-Ketopiperazine |
| NMR | Nuclear magnetic resonance |
| Ns | Nosyl |
| Phe | Phenylalanine |
| PIP2 | Phosphatidylinositol bisphosphate |
| SD | Standard deviation |
| TRP | Transient receptor potential channel |
| TRPM8 | Transient receptor potential melastatin type 8 channel |
| Z | Benzyloxycarbonyl |
Appendix A
Appendix A.1. Chemistry, General Procedures
Appendix A.2. Preparation of Synthetic Intermediates on the Road to Β-Lactam Derivatives
Appendix A.2.1. Synthesis of N-Ns-N-alkyl-Phe-OBn Derivatives
N-[(2′S-Dibenzylamino-3-phenyl-)prop-1′-yl]-Ns-L-Phe-OBn (5)
N-[(2′R-Dibenzylamino-3-phenyl-)prop-1′-yl]-Ns-L-Phe-OBn (6)
Appendix A.2.2. Removal of the Ns Group
N-[(2′S-Dibenzylamino-3′-phenyl-)prop-1′-yl]-L-Phe-OBn (7)
N-[(2′R-Dibenzylamino-3′-phenyl-)prop-1′-yl]-L-Phe-OBn (8)
Appendix A.2.3. Synthesis of N-Alkyl-N-Chloropropionyl-Xaa Derivatives
N-(2″S-Chloropropanoyl)-N-[2′S-dibenzylamino)-3′-phenyl)prop-1′-yl]-L-Phe-OBn (9)
N-(2″S-Chloropropanoyl)-N-[2′R-dibenzylamino)-3′-phenyl)prop-1′-yl]-L-Phe-OBn (10)
N-(2″R-Chloropropanoyl)-N-[2′S-dibenzylamino)-3′-phenyl)prop-1′-yl]-L-Phe-OBn (11)
N-(2″R-Chloropropanoyl)-N-[2′R-dibenzylamino)-3′-phenyl)prop-1′-yl] -L-Phe-OBn (12)




References
- Liu, Y.; Qin, N. TRPM8 in health and disease: Cold sensing and beyond. Adv. Exp. Med. Biol. 2011, 704, 185–208. [Google Scholar]
- Zakharian, E.; Cao, C.; Rohacs, T. Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J. Neurosci. 2010, 30, 12526–12534. [Google Scholar] [CrossRef] [Green Version]
- Daniels, R.L.; Takashima, Y.; McKemy, D.D. Activity of the Neuronal Cold Sensor TRPM8 Is Regulated by Phospholipase C via the Phospholipid Phosphoinositol 4,5-Bisphosphate. J. Biol. Chem. 2009, 284, 1570–1582. [Google Scholar] [CrossRef] [Green Version]
- Asuthkar, S.; Demirkhanyan, L.; Sun, X.; Velpula, K.K.; Zakharian, E.; Elustondo, P.A.; Krishnan, V.; Baskaran, P.; Thyagarajan, B.; Pavlov, E.V. The TRPM8 protein is a testosterone receptor: II. Functional evidence for an ionotropic effect of testosterone on TRPM8. J. Biol. Chem. 2015, 290, 2670–2688. [Google Scholar] [CrossRef] [Green Version]
- Lippoldt, E.K.; Elmes, R.R.; McCoy, D.D.; Knowlton, W.M.; McKemy, D.D. Artemin, a glial cell line-derived neurotrophic factor family member, induces TRPM8-dependent cold pain. J. Neurosci. 2013, 33, 12543–12552. [Google Scholar] [CrossRef]
- Tang, Z.; Kim, A.; Masuch, T.; Park, K.; Weng, H.; Wetzel, C.; Dong, X. Pirt functions as an endogenous regulator of TRPM8. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winchester, W.J.; Gore, K.; Glatt, S.; Petit, W.; Gardiner, J.C.; Conlon, K.; Postlethwaite, M.; Saintot, P.P.; Roberts, S.; Gosset, J.R.; et al. Inhibition of TRPM8 channels reduces pain in the cold pressor test in humans. J. Pharmacol. Exp. Ther. 2014, 351, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. NoIdentification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Kobayashi, K.; Fukuoka, T.; Obata, K.; Yamanaka, H.; Dai, Y.; Tokunaga, A.; Noguchi, K. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J. Comp. Neurol. 2005, 493, 596–606. [Google Scholar] [CrossRef]
- Weyer, A.; Lehto, S. Development of TRPM8 Antagonists to Treat Chronic Pain and Migraine. Pharmaceuticals 2017, 10, 37. [Google Scholar] [CrossRef]
- Raineroa, I.; Rovetab, F.; Vaccac, A.; Noviellob, C.; Rubinoc, E. Migraine pathways and the identification of novel therapeutic targets Rubinoc. Expert Opin. Ther. Target 2020, 3, 245–253. [Google Scholar] [CrossRef]
- Benemei, S.; Dussor, G. TRP Channels and Migraine: Recent Developments and New Therapeutic Opportunities. Pharmaceuticals 2019, 12, 54. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.; Coudore-Civiale, M.-A.; Balayssac, D.; Eschalier, A.; Coudore, F.; Authier, N.; Ling, B.; Coudore-Civiale, M.-A.; Balayssac, D.; Eschalier, A.; et al. Behavioral and Immunohistological Assessment of Painful Neuropathy Induced by a Single Oxaliplatin Injection in the Rat. Toxicology 2007, 234, 176–184. [Google Scholar] [CrossRef]
- Ling, B.; Authier, N.; Balayssac, D.; Eschalier, A.; Coudore, F. Behavioral and Pharmacological Description of Oxaliplatin-Induced Painful Neuropathy in Rat. Pain 2007, 128, 225–234. [Google Scholar] [CrossRef]
- Mizoguchi, S.; Andoh, T.; Yakura, T.; Kuraishi, Y. Involvement of C-Myc-Mediated Transient Receptor Potential Melastatin 8 Expression in Oxaliplatin-Induced Cold Allodynia in Mice. Pharmacol. Rep. 2016, 68, 645–648. [Google Scholar] [CrossRef]
- Sabnis, A.S. Expression and Characterization of the TRPM8 Receptor in Lung Epithelial Cells. Cell Mol. Biol. 2008, 39, 466–474. [Google Scholar]
- Yu, W.; Hill, W.G.; Apodaca, G.; Zeidel, M.L. Expression and Distribution of Transient Receptor Potential (TRP) Channels in Bladder Epithelium. Am. J. Physiol. 2011, 300, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Noyer, L.; Grolez, G.P.; Prevarskaya, N.; Gkika, D.; Lemonnier, L.; Noyer, L.; Grolez, G.P.; Prevarskaya, N.; Gkika, D.; Lemonnier, L. TRPM8 and Prostate: A Cold Case? Pflugers Arch. 2018, 470, 1419–1429. [Google Scholar] [CrossRef]
- Ordas, P.; Hernandez-Ortego, P.; Vara, H.; Fernandez-Pena, C.; Morenilla-Palao, C.; Gomis, A.; Viana, F.; Reimundez, A.; Senaris, R.; Guadano-Ferraz, A.; et al. Expression of the cold thermoreceptor TRPM8 in rodent brain thermoregulatory circuits. J. Comp. Neurol. 2019, 529, 234–256. [Google Scholar] [CrossRef]
- Alcalde, I.; Íñigo-Portugués, A.; González-González, O.; Almaraz, L.; Artime, E.; Morenilla-Palao, C.; Gallar, J.; Viana, F.; Merayo-Lloves, J.; Belmonte, C. Morphological and functional changes in TRPM8-expressing corneal cold thermoreceptor neurons during aging and their impact on tearing in mice. J. Comp. Neurol. 2018, 526, 1859–1874. [Google Scholar] [CrossRef]
- Liu, X.; Ong, H.L.; Ambudkar, I. TRP Channel Involvement in Salivary Glands-Some Good, Some Bad. Cells 2018, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Berdugo, D.; Rofes, L.; Casamitjana, J.F.; Enrique, A.; Chamizo, J.; Vina, C.; Pollan, C.M.; Clave, P. TRPM8, ASIC1, and ASIC3 localization and expression in the human oropharynx. Neurogastroenterol. Motil. 2018, 30, e13398. [Google Scholar] [CrossRef]
- Mohandass, A.; Krishnan, V.; Gribkova, E.D.; Asuthkar, S.; Baskaran, P.; Nersesyan, Y.; Hussain, Z.; Wise, L.M.; George, R.E.; Stokes, N.; et al. TRPM8 as the rapid testosterone signaling receptor: Implications in the regulation of dimorphic sexual and social behaviors. FASEB J. 2020, 34, 10887–10906. [Google Scholar] [CrossRef] [PubMed]
- Lidao, B.; Yi, W.; Ruilian, M.; Xianhua, R.; Rui, C.; Agula, B. Apoptosis-inducing effects of lentinan on the proliferation of human bladder cancer T24 cells. Pakistan Pharm. Sci. 2015, 8, 1595–1600. [Google Scholar]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 Inhibits Endothelial Cell Migration via a Nonchannel Function by Trapping the Small GTPase Rap. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef] [Green Version]
- Arcas, J.M.; Gonzalez, A.; Gers-Barlag, K.; Gonzalez-Gonzalez, O.; Bech, F.; Belmonte, C.; Gomis, A.; Viana, F.; Gonzalez-Gonzalez, O.; Bech, F.; et al. The Immunosuppressant Macrolide Tacrolimus Activates Cold-Sensing TRPM8 Channels. J. Neurosci. 2019, 39, 949–969. [Google Scholar] [CrossRef]
- Perez de Vega, M.J.; Gomez-Monterrey, I.; Ferrer-Montiel, A.; Gonzalez-Muniz, R. Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem. 2016, 59, 10006–10029. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Muniz, R.; Bonache, M.A.; Martin-Escura, C.; Gomez-Monterrey, I. Recent Progress in TRPM8 Modulation: An Update. Int. J. Mol. Sci. 2019, 20, 1628. [Google Scholar] [CrossRef] [Green Version]
- Legay, C.M.; Gorobets, E.; Iftinca, M.; Ramachandran, R.; Altier, C.; Derksen, D.J. Natural-Product-Derived Transient Receptor Potential Melastatin 8 (TRPM8) Channel Modulators. Org. Lett. 2016, 18, 2746–2749. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Arroyo, F.J.; Orlando, P.; Schiano Moriello, A.; Vitale, R.M.; Amodeo, P.; Sanchez, A.; Roncero, C.; Bianchini, G.; Martin, M.A.; et al. Tetrahydroisoquinoline-Derived Urea and 2,5-Diketopiperazine Derivatives as Selective Antagonists of the Transient Receptor Potential Melastatin 8 (TRPM8) Channel Receptor and Antiprostate Cancer Agents. J. Med. Chem. 2016, 59, 5661–5683. [Google Scholar] [CrossRef] [PubMed]
- Beccari, A.R.; Gemei, M.; Monte, M.L.; Menegatti, N.; Fanton, M.; Pedretti, A.; Bovolenta, S.; Nucci, C.; Molteni, A.; Rossignoli, A. Novel Selective, Potent Naphthyl TRPM8 Antagonists Identified through a Combined Ligand-and Structure-Based Virtual Screening Approach. Sci. Rep. 2017, 7, 10999. [Google Scholar] [CrossRef] [Green Version]
- Bertamino, A.; Ostacolo, C.; Ambrosino, P.; Musella, S.; Di Sarno, V.; Ciaglia, T.; Soldovieri, M.V.; Iraci, N.; Fernandez Carvajal, A.; De La Torre-Martinez, R.; et al. Tryptamine-Based Derivatives as Transient Receptor Potential Melastatin Type 8 (TRPM8) Channel Modulators. J. Med. Chem. 2016, 59, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Bertamino, A.; Iraci, N.; Ostacolo, C.; Ambrosino, P.; Musella, S.; Di Sarno, V.; Ciaglia, T.; Pepe, G.; Sala, M.; Soldovieri, M.V.; et al. Identification of a Potent Tryptophan-Based TRPM8 Antagonist with in Vivo Analgesic Activity. J. Med. Chem. 2018, 61, 6140–6152. [Google Scholar] [CrossRef]
- Journigan, V.B.; Feng, Z.; Rahman, S.; Wang, Y.; Amin, A.R.M.R.; Heffner, C.E.; Bachtel, N.; Wang, S.; Gonzalez-Rodriguez, S.; Fernández-Carvajal, A.; et al. Structure-Based Design of Novel Biphenyl Amide Antagonists of Human Transient Receptor Potential Cation Channel Subfamily M Member 8 Channels with Potential Implications in the Treatment of Sensory Neuropathies. ACS Chem. Neurosci. 2020, 11, 268–290. [Google Scholar] [CrossRef]
- Andrews, M.D.; Af Forselles, K.; Beaumont, K.; Galan, S.R.; Glossop, P.A.; Grenie, M.; Jessiman, A.; Kenyon, A.S.; Lunn, G.; Maw, G.; et al. Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain. ACS Med. Chem. Lett. 2015, 6, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Horne, D.B.; Biswas, K.; Brown, J.; Bartberger, M.D.; Clarine, J.; Davis, C.D.; Gore, V.K.; Harried, S.; Horner, M.; Kaller, M.R.; et al. Discovery of TRPM8 Antagonist (S)-6-(((3-Fluoro-4-(Trifluoromethoxy)Phenyl)(3-Fluoropyridin-2-Yl)Methyl)Carbamoyl)Nicotinic Acid (AMG 333), a Clinical Candidate for the Treatment of Migraine. J. Med. Chem. 2018, 61, 8186–8201. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Lee, S.-Y. Current View of Ligand and Lipid Recognition by the Menthol Receptor TRPM8. Trends Biochem. Sci. 2020, 45, 806–819. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the coldactivated TRPM8 channel. Science 2019, 363. [Google Scholar] [CrossRef]
- De la Torre-Martinez, R.; Bonache, M.A.; Llabres-Campaner, P.J.; Balsera, B.; Fernandez-Carvajal, A.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; Perez de Vega, M.J.; Gonzalez-Muniz, R. Synthesis, high-throughput screening and pharmacological characterization of β-lactam derivatives as TRPM8 antagonists. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bonache, M.A.; Martin-Escura, C.; de la Torre Martinez, R.; Medina, A.; Gonzalez-Rodriguez, S.; Francesch, A.; Cuevas, C.; Roa, A.M.; Fernandez-Ballester, G.; Ferrer-Montiel, A.; et al. Highly functionalized β-lactams and 2-ketopiperazines as TRPM8 antagonists with antiallodynic activity. Sci. Rep. 2020, 10, 14154. [Google Scholar] [CrossRef]
- Bertamino, A.; Ostacolo, C.; Medina, A.; Di Sarno, V.; Lauro, G.; Ciaglia, T.; Vestuto, V.; Pepe, G.; Basilicata, M.G.; Musella, S.; et al. Exploration of TRPM8 Binding Sites by β-Carboline-Based Antagonists and Their In Vitro Characterization and In Vivo Analgesic Activities. J. Med. Chem. 2020, 63, 9672–9694. [Google Scholar] [CrossRef] [PubMed]
- Perez-Faginas, P.; O’Reilly, F.; O’Byrne, A.; Garcia-Aparicio, C.; Martin-Martinez, M.; Perez de Vega, M.J.; Garcia-Lopez, M.T.; Gonzalez-Muniz, R. Exceptional Stereoselectivity in the Synthesis of 1,3,4-Trisubstituted 4-Carboxy β-Lactam Derivatives from Amino Acids. Org. Lett. 2007, 9, 1593–1596. [Google Scholar] [CrossRef]
- Perez-Faginas, P.; Alkorta, I.; Garcia-Lopez, M.T.; Gonzalez-Muniz, R. From theoretical calculations to the enantioselective synthesis of a 1,3,4-trisubstituted Gly-derived 2-azetidinone. Tetrahedron Lett. 2008, 49, 215–218. [Google Scholar] [CrossRef]
- Lampe, T.; Alonso-Alija, C.; Beck, H.; Rosentreter, U.; Sandner, P.; Stahl, E.; Stelte-Ludwig, B. Preparation of substituted 2-benzyloxy-benzoic acid amide derivatives as Cold Menthol Receptor 1 (CMR-1) modulators for treating and preventing urol. diseases or disorders. PCT International Application WO 2007/017093, 15 February 2007. [Google Scholar]
- Lashinger, E.S.R.; Steiginga, M.S.; Hieble, J.P.; Leon, L.A.; Gardner, S.D.; Nagilla, R.; Davenport, E.A.; Hoffman, B.E.; Laping, N.J.; Su, X. AMTB, a TRPM8 channel blocker: Evidence in rats for activity in overactive bladder and painful bladder syndrome. Am. J. Physiol. Renal Physiol. 2008, 295, 803–810. [Google Scholar] [CrossRef]
- Noncovich, A.; Priest, C.; Ung, J.; Patron, A.P.; Servant, G.; Brust, P.; Servant, N.; Faber, N.; Liu, H.; Gonsalves, N.S.; et al. Discovery and development of a novel class of phenoxyacetyl amides as highly potent TRPM8 agonists for use as cooling agents. Bioorg. Med. Chem. Lett. 2017, 27, 3931–3938. [Google Scholar] [CrossRef]
- Chaudhari, S.S.; Kadam, A.B.; Khairatkar-Joshi, N.; Mukhopadhyay, I.; Karnik, P.V.; Raghuram, A.; Rao, S.S.; Vaiyapuri, T.S.; Wale, D.P.; Bhosale, V.M.; et al. Synthesis and pharmacological evaluation of novel N-aryl-3,4-dihydro-1′H-spiro[chromene-2,4′-piperidine]-1′-carboxamides as TRPM8 antagonists. Bioorg. Med. Chem. 2013, 21, 6542–6553. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Hirasawa, H.; Fujimori, Y.; Nakanishi, O.; Kamada, N.; Ikeda, T.; Yamamoto, A.; Kanbe, H. Identification of N-acyl-N-indanyl-α-phenylglycinamides as selective TRPM8 antagonists designed to mitigate the risk of adverse effects. Bioorg. Med. Chem. 2021, 30, 115903. [Google Scholar] [CrossRef]
- Kistner, K.; Siklosi, N.; Babes, A.; Khalil, M.; Selescu, T.; Zimmermann, K.; Wirtz, S.; Becker, C.; Neurath, M.F.; Reeh, P.W.; et al. Systemic desensitization through TRPA1 channels by capsazepine and mustard oil—A novel strategy against inflammation and pain. Sci. Rep. 2016, 6, 28621. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with yasara NOVA-a self-parameterizing force field. Proteins Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Diver, M.M.; Cheng, Y.; Julius, D. Structural insights into TRPM8 inhibition and desensitization. Science 2019, 365, 1434–1440. [Google Scholar] [CrossRef]
- Nikolaeva-Koleva, M.; Butron, L.; Gonzalez-Rodriguez, S.; Devesa, I.; Valente, P.; Serafini, M.; Genazzani, A.A.; Pirali, T.; Ballester, G.F.; Fernandez-Carvajal, A.; et al. A capsaicinoid-based soft drug, AG1529, for attenuating TRPV1-mediated histaminergic and inflammatory sensory neuron excitability. Sci. Rep. 2021, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Canutescu, A.A.; Dunbrack, R.L.J. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003, 12, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compoundd | Configuration | % Blockade 50 μM | % Blockade 5 μM | IC50 (μM) | 95% Confidence Intervals |
|---|---|---|---|---|---|
| 13 | 3S,4S,2′S | 86.1 ± 5.0 | 57.7 ± 7.2 | 3.1 ± 1.1 | 2.574 to 3.997 |
| 14 | 3S,4S,2′R | 87.7 ± 3.1 | 78.9 ± 1.7 | 0.3 ± 1.0 | 0.2162 to 0.2804 |
| 15 | 3R,4R,2′S | 90.3 ± 2.8 | 71.1 ± 4.3 | 0.7 ± 1.1 | 0.6376 to 0.9071 |
| 16 | 3R,4R,2′R | 98.5 ± 3.2 | 87.3 ± 8.1 | 0.02 ± 1.1 | 0.010 to 0.019 |
| 1 a | 3S,4S,2′S | 2.4 ± 1.2 | |||
| AMTB a | _ | 7.3 ± 1.5 |
| Compoundd | Configuration | % TRPV1 Blockade at 50 μM | % TRPA1 Blockade at 50 μM |
|---|---|---|---|
| 13 | 3S,4S,2′S | 7.9 ± 3.2 | 3.7 ± 4.1 |
| 14 | 3S,4S,2′R | 2.8 ± 4.1 | 2.4 ± 1.4 |
| 15 | 3R,4R,2′S | 5.3 ± 3.4 | 2.6 ± 1.9 |
| 16 | 3R,4R,2′R | 4.2 ± 3.2 | 0.5± 4.1 |
| Compound | Configuration | % Blockade at 10 μM | IC50 (μM) |
|---|---|---|---|
| 14 | 3S,4S,2′R | 77.2 ± 2.7 | 0.9 ± 1.0 |
| 16 | 3R,4R,2′R | 79.3 ± 2.7 | 0.05 ± 1.0 |
| 1 | 3S,4S,2′S | 1.4 ± 1.1 |
| Subsite | Location | % of Docking Solutions (Estimated Binding Energies, kcal/mol) | |||
|---|---|---|---|---|---|
| 13 | 14 | 15 | 16 | ||
| 1 | Inner pore, S5S6, S6 | 40% (10.64) | 42% (10.53) | 50% (10.58) | 52% (10.26) |
| 2 | Internal mouth pore | 34% (11.44) | 18% (10.15) | 22% (10.14) | 20% (11.40) |
| 3 | Pore, high S3-S4, S6 | 12% (9.95) | 24% (10.24) | 10% (9.24) | 12% (10.32) |
| 4 | Pore, external tower | 4% (10.20) | 16% (10.43) | 18% (11.46) | 16% (10.41) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonache, M.Á.; Llabrés, P.J.; Martín-Escura, C.; De la Torre-Martínez, R.; Medina-Peris, A.; Butrón, L.; Gómez-Monterrey, I.; Roa, A.M.; Fernández-Ballester, G.; Ferrer-Montiel, A.; et al. Phenylalanine-Derived β-Lactam TRPM8 Modulators. Configuration Effect on the Antagonist Activity. Int. J. Mol. Sci. 2021, 22, 2370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052370
Bonache MÁ, Llabrés PJ, Martín-Escura C, De la Torre-Martínez R, Medina-Peris A, Butrón L, Gómez-Monterrey I, Roa AM, Fernández-Ballester G, Ferrer-Montiel A, et al. Phenylalanine-Derived β-Lactam TRPM8 Modulators. Configuration Effect on the Antagonist Activity. International Journal of Molecular Sciences. 2021; 22(5):2370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052370
Chicago/Turabian StyleBonache, María Ángeles, Pedro Juan Llabrés, Cristina Martín-Escura, Roberto De la Torre-Martínez, Alicia Medina-Peris, Laura Butrón, Isabel Gómez-Monterrey, Ana María Roa, Gregorio Fernández-Ballester, Antonio Ferrer-Montiel, and et al. 2021. "Phenylalanine-Derived β-Lactam TRPM8 Modulators. Configuration Effect on the Antagonist Activity" International Journal of Molecular Sciences 22, no. 5: 2370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052370