
Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa

 , ,
, ,
Abstract
:1. Introduction
2. Results
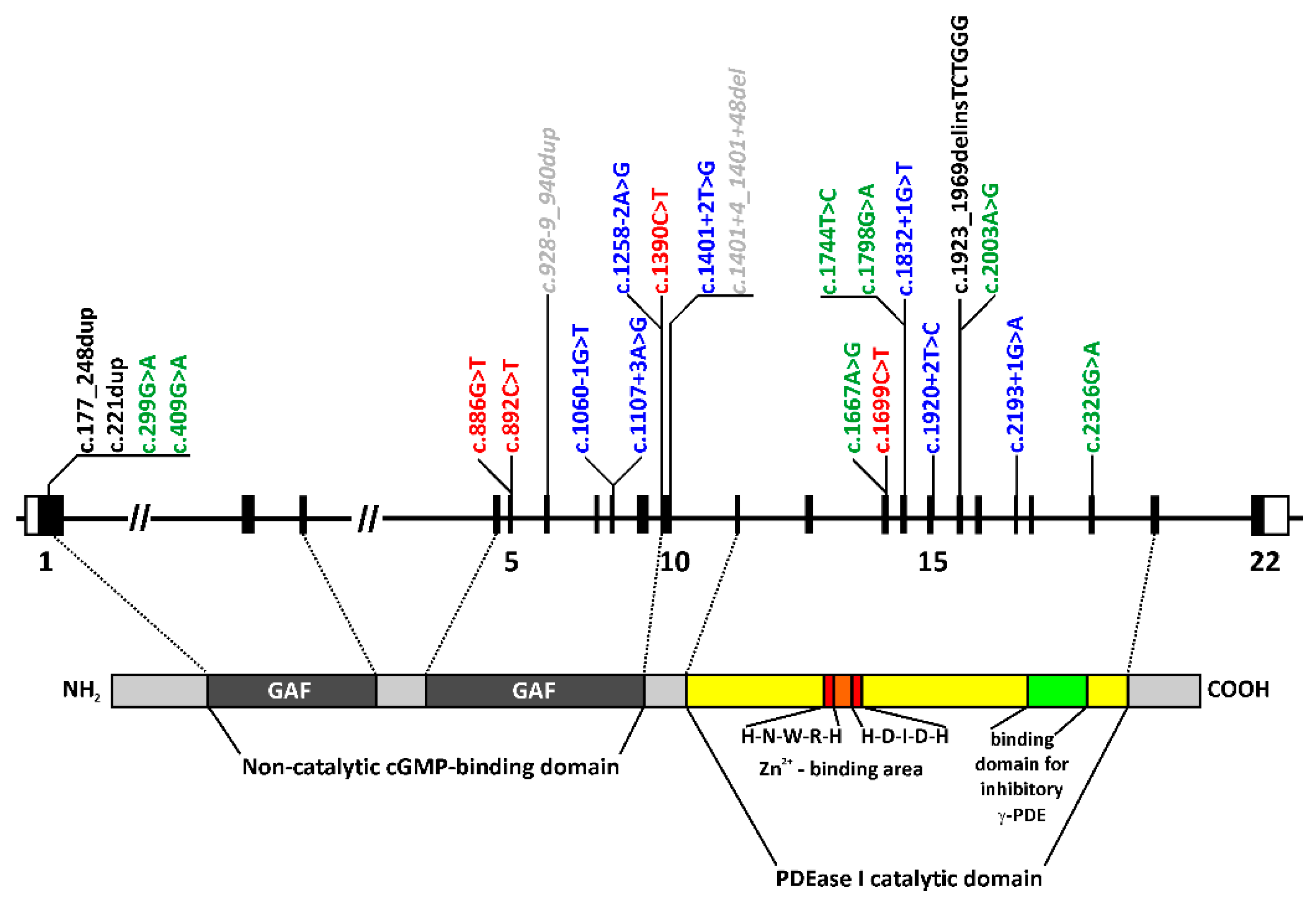
2.1. Genetic Findings
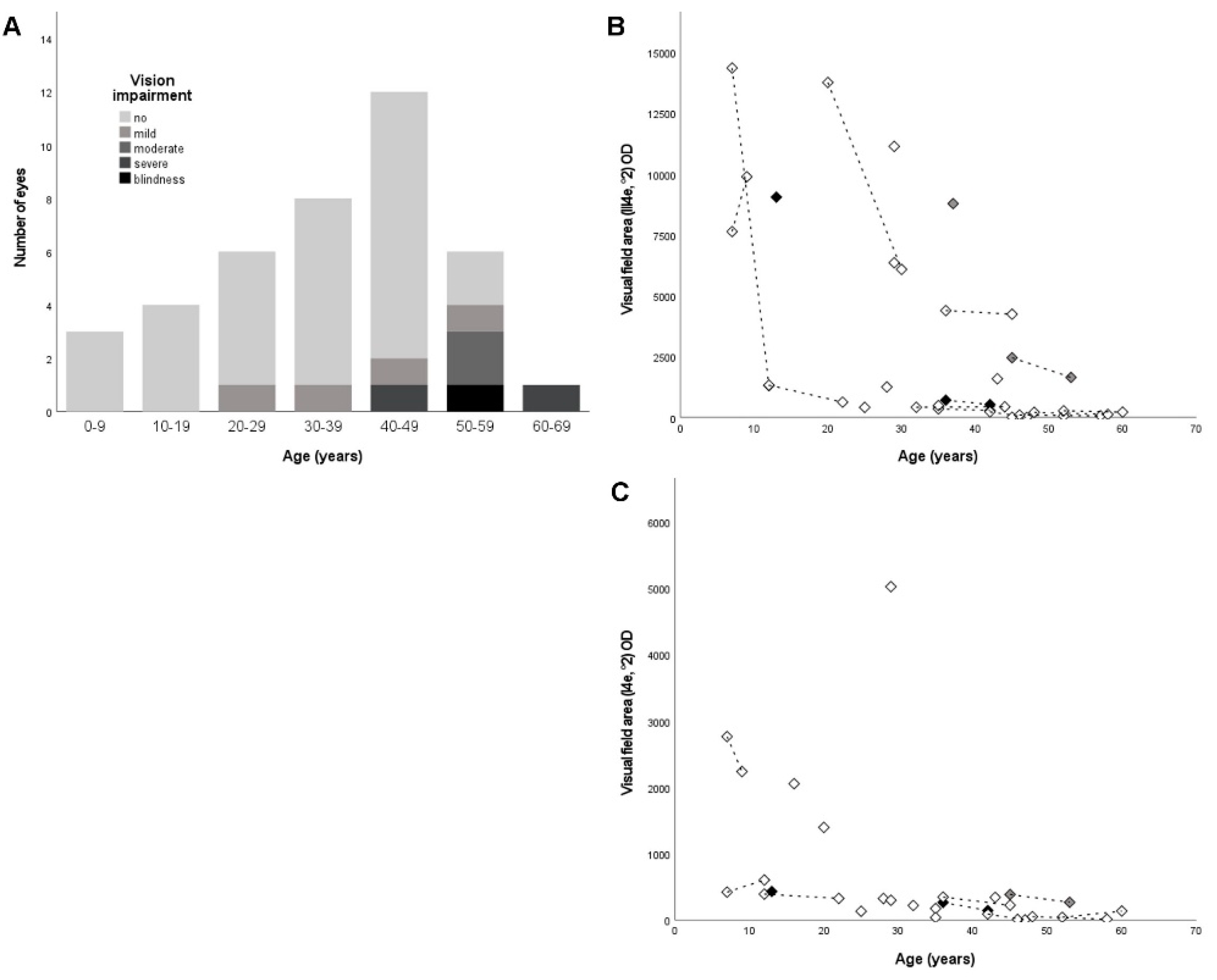
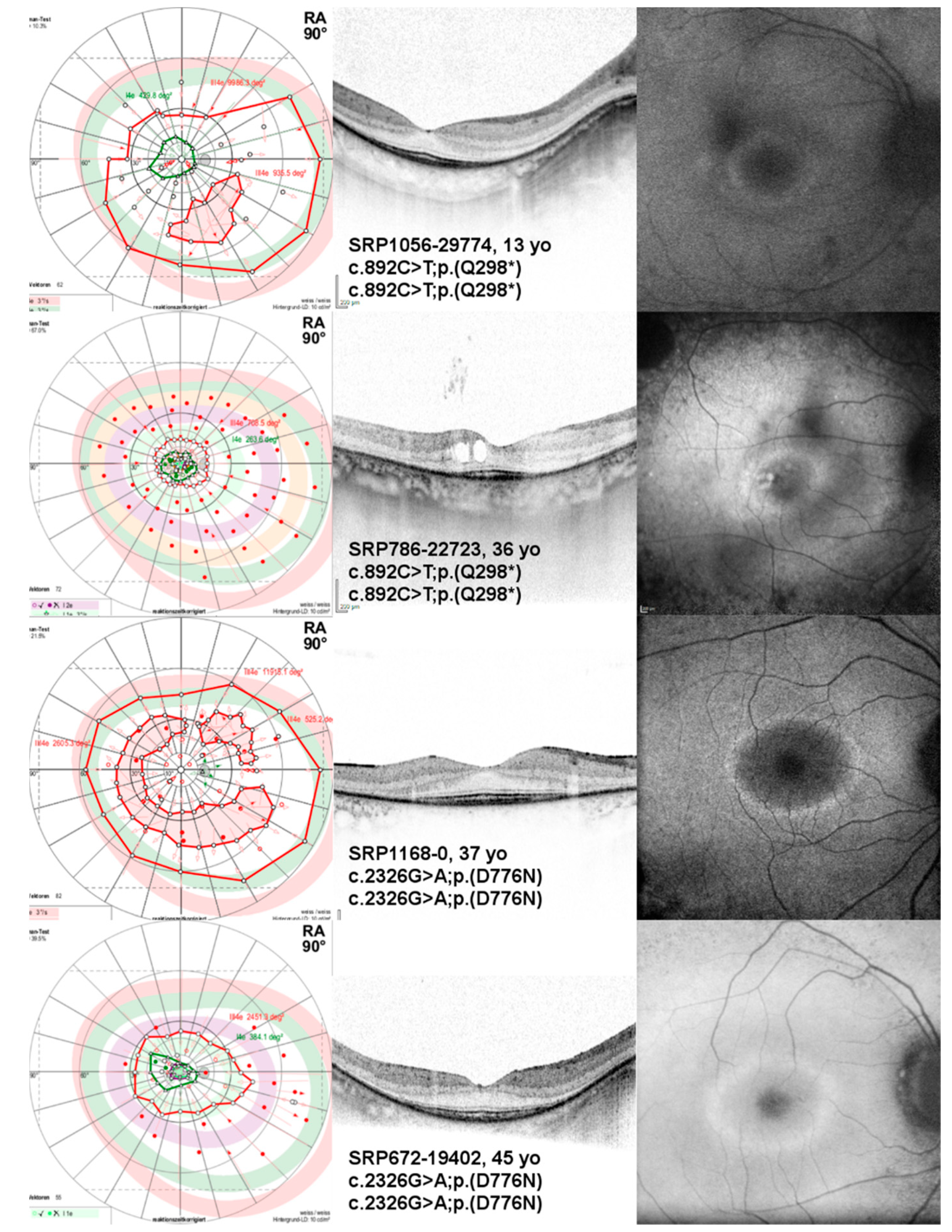
2.2. Ophthalmological Findings
3. Discussion
3.1. Genetic Findings
3.2. Clinical Findings
4. Materials and Methods
4.1. Ophthalmological Testing
4.2. Genetic Testing and Variant Classification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Cote, R.H. cGMP signaling in vertebrate retinal photoreceptor cells. Front. Biosci. 2005, 10, 1191. [Google Scholar] [CrossRef] [Green Version]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal degeneration in the rd mouse is caused by a defect in the β subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Pittler, S.J.; Baehr, W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase β-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. USA 1991, 88, 8322–8326. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Hawes, N.L.; Pardue, M.T.; German, A.M.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Rengarajan, K.; Boyd, A.P.; Sidney, S.S.; et al. Two mouse retinal degenerations caused by missense mutations in the β-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007, 47, 624–633. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, M.E.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the β–subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993, 4, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.B.; Danciger, J.S.; Aguirre, G. The β subunit of cyclic GMP phosphodiesterase mRNA is deficient in canine rod-cone dysplasia 1. Neuron 1992, 9, 349–356. [Google Scholar] [CrossRef]
- Suber, M.L.; Pittler, S.J.; Qin, N.; Wright, G.C.; Holcombe, V.; Lee, R.H.; Craft, C.M.; Lolley, R.N.; Baehr, W.; Hurwitz, R.L. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase β-subunit gene. Proc. Natl. Acad. Sci. USA 1993, 90, 3968–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayés, M.; Giordano, M.; Balcells, S.; Grinberg, D.; Vilageliu, L.; Martínez, I.; Ayuso, C.; Benítez, J.; Ramos-Arroyo, M.A.; Chivelet, P.; et al. Homozygous tandem duplication within the gene encoding the β-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum. Mutat. 1995, 5, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Hmani-Aifa, M.; Benzina, Z.; Zulfiqar, F.; Dhouib, H.; Shahzadi, A.; Ghorbel, A.; Rebaï, A.; Söderkvist, P.; Riazuddin, S.; Kimberling, W.J.; et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur. J. Hum. Genet. 2009, 17, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Orth, U.; Baehr, W.; Schwinger, E.; Rosenberg, T. Heterozygous missense mutation in the rod cGMP phosphodiesterase β–subunit gene in autosomal dominant stationary night blindness. Nat. Genet. 1994, 7, 64–68. [Google Scholar] [CrossRef]
- Tsang, S.H.; Woodruff, M.L.; Jun, L.; Mahajan, V.; Yamashita, C.K.; Pedersen, R.; Lin, C.S.; Goff, S.P.; Rosenberg, T.; Larsen, M.; et al. Transgenic mice carrying the H258N mutation in the gene encoding the β-subunit of phosphodiesterase-6 (PDE6B) provide a model for human congenital stationary night blindness. Hum. Mutat. 2007, 28, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khramtsov, N.V.; Feshchenko, E.A.; Suslova, V.A.; Shmukler, B.E.; Terpugov, B.E.; Rakitina, T.V.; Atabekova, N.V.; Lipkin, V.M. The human rod photoreceptor cGMP phosphodiesterase β-subunit. Structural studies of its cDNA and gene. FEBS Lett. 1993, 327, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.; Tanabe, T.; Sun, D.; Zeng, Y.; Kjeldbye, H.; Gouras, P.; Maguire, A.M. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat. Med. 1996, 2, 649–654. [Google Scholar] [CrossRef]
- Pang, J.J.; Boye, S.L.; Kumar, A.; Dinculescu, A.; Deng, W.; Li, J.; Li, Q.; Rani, A.; Foster, T.C.; Chang, B.; et al. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEβ mutation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4278–4283. [Google Scholar] [CrossRef] [Green Version]
- Petit, L.; Lhériteau, E.; Weber, M.; Le Meur, G.; Deschamps, J.-Y.; Provost, N.; Mendes-Madeira, A.; Libeau, L.; Guihal, C.; Colle, M.-A.; et al. Restoration of vision in the pde6β-deficient dog, a large animal model of rod-cone dystrophy. Mol. Ther. 2012, 20, 2019–2030. [Google Scholar] [CrossRef] [Green Version]
- Pichard, V.; Provost, N.; Mendes-Madeira, A.; Libeau, L.; Hulin, P.; Tshilenge, K.-T.; Biget, M.; Ameline, B.; Deschamps, J.-Y.; Weber, M.; et al. AAV-mediated Gene Therapy Halts Retinal Degeneration in PDE6β-deficient Dogs. Mol. Ther. 2016, 24, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Mahato, B.; Kaya, K.D.; Fan, Y.; Sumien, N.; Shetty, R.A.; Zhang, W.; Davis, D.; Mock, T.; Batabyal, S.; Ni, A.; et al. Pharmacologic fibroblast reprogramming into photoreceptors restores vision. Nature 2020, 581, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Manes, G.; Cheguru, P.; Majumder, A.; Bocquet, B.; Sénéchal, A.; Artemyev, N.O.; Hamel, C.P.; Brabet, P. A truncated form of rod photoreceptor PDE6 β-subunit causes autosomal dominant congenital stationary night blindness by interfering with the inhibitory activity of the γ-subunit. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Beheshtian, M.; Saee Rad, S.; Babanejad, M.; Mohseni, M.; Hashemi, H.; Eshghabadi, A.; Hajizadeh, F.; Akbari, M.; Kahrizi, K.; Riazi Esfahani, M.; et al. Impact of whole exome sequencing among Iranian patients with autosomal recessive retinitis pigmentosa. Arch. Iran Med. 2015, 18, 776–785. [Google Scholar] [PubMed]
- Van Huet, R.A.C.; Pierrache, L.H.M.; Meester-Smoor, M.A.; Klaver, C.C.W.; van den Born, L.I.; Hoyng, C.B.; de Wijs, I.J.; Collin, R.W.J.; Hoefsloot, L.H.; Klevering, B.J. The efficacy of microarray screening for autosomal recessive retinitis pigmentosa in routine clinical practice. Mol. Vis. 2015, 21, 461–476. [Google Scholar]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Issa, P.C.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Tsui, I.; Chou, C.L.; Zernant, J.; Haamer, E.; Iranmanesh, R.; Tosi, J.; Allikmets, R. A Novel Mutation and Phenotypes in Phosphodiesterase 6 Deficiency. Am. J. Ophthalmol. 2008, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farwell, K.D.; Shahmirzadi, L.; El-Khechen, D.; Powis, Z.; Chao, E.C.; Tippin Davis, B.; Baxter, R.M.; Zeng, W.; Mroske, C.; Parra, M.C.; et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 2015, 17, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Mclaughlin, M.E.; Ehrhart, T.L.; Berson, E.L.; Dryja, T.P. Mutation spectrum of the gene encoding the β subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 3249–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khateb, S.; Nassisi, M.; Bujakowska, K.M.; Méjécase, C.; Condroyer, C.; Antonio, A.; Foussard, M.; Démontant, V.; Mohand-Saïd, S.; Sahel, J.-A.; et al. Longitudinal Clinical Follow-up and Genetic Spectrum of Patients With Rod-Cone Dystrophy Associated With Mutations in PDE6A and PDE6B. JAMA Ophthalmol. 2019, 137, 669–679. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.J.; Gilissen, C.; Van Huet, R.A.C.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; De Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Kuehlewein, L.; Zobor, D.; Andreasson, S.O.; Ayuso, C.; Banfi, S.; Bocquet, B.; Bernd, A.S.; Biskup, S.; Boon, C.J.F.; Downes, S.M.; et al. Clinical Phenotype and Course of PDE6A -Associated Retinitis Pigmentosa Disease, Characterized in Preparation for a Gene Supplementation Trial. JAMA Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Hood, D.C.; Ramachandran, R.; Holopigian, K.; Lazow, M.; Birch, D.G.; Greenstein, V.C. Method for deriving visual field boundaries from OCT scans of patients with retinitis pigmentosa. Biomed. Opt. Express 2011, 2, 1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Lima, L.H.; Tsang, S.H. Structural disease progression in PDE6-associated autosomal recessive retinitis pigmentosa. Ophthalmic Genet. 2018, 39, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Danciger, M.; Heilbron, V.; Gao, Y.Q.; Zhao, D.Y.; Jacobson, S.G.; Farber, D.B. A homozygous PDE6B mutation in a family with autosomal recessive retinitis pigmentosa. Mol. Vis. 1996, 2, 10. [Google Scholar]
- Jacobson, S.G.; Sumaroka, A.; Aleman, T.S.; Cideciyan, A.V.; Danciger, M.; Farber, D.B. Evidence for retinal remodelling in retinitis pigmentosa caused by PDE6B mutation. Br. J. Ophthalmol. 2007, 91, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Riazuddin, S.A.; Shahzadi, A.; Nasir, I.A.; Khan, S.N.; Husnain, T.; Akram, J.; Sieving, P.A.; Hejtmancik, J.F.; Riazuddin, S. Mutations in the β-subunit of rod phosphodiesterase identified in consanguineous Pakistani families with autosomal recessive retinitis pigmentosa. Mol. Vis. 2011, 17, 1373–1380. [Google Scholar] [PubMed]
- Kim, C.; Kim, K.J.; Bok, J.; Lee, E.J.; Kim, D.J.; Oh, J.H.; Park, S.P.; Shin, J.Y.; Lee, J.Y.; Yu, H.G. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol. Vis. 2012, 18, 2398–2410. [Google Scholar]
- Tatour, Y.; Tamaiev, J.; Shamaly, S.; Colombo, R.; Bril, E.; Rabinowitz, T.; Yaakobi, A.; Mezer, E.; Leibu, R.; Tiosano, B.; et al. A novel intronic mutation of PDE6B is a major cause of autosomal recessive retinitis pigmentosa among caucasus jews. Mol. Vis. 2019, 25, 155–164. [Google Scholar] [PubMed]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Stingl, K.; Stingl, K.; Nowomiejska, K.; Kuehlewein, L.; Kohl, S.; Kempf, M.; Strasser, T.; Jung, R.; Wilhelm, B.; Peters, T.; et al. Clinical protocols for the evaluation of rod function. Ophthalmologica 2020. [Google Scholar] [CrossRef]
- Schulze-Bonsel, K.; Feltgen, N.; Burau, H.; Hansen, L.; Bach, M. Visual acuities “hand motion” and “counting fingers” can be quantified with the freiburg visual acuity test. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1236–1240. [Google Scholar] [CrossRef]
- Ramachandran, R.; Cai, C.X.; Lee, D.; Epstein, B.C.; Locke, K.G.; Birch, D.G.; Hood, D.C. Reliability of a manual procedure for marking the EZ endpoint location in patients with retinitis pigmentosa. Transl. Vis. Sci. Technol. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Feldhaus, B.; Khan, M.I.; Cremers, F.P.M.; Kohl, S.; Wissinger, B.; Zobor, D. Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ID | Gender | Age | Disease Onset | Variant 1 | Variant 2 | BCVA (logMAR) OD/OS | VF (III4e, °2) OD/OS | VF (I4e, °2) OD/OS |
|---|---|---|---|---|---|---|---|---|
| LCA120-28043 | M | 7 | 1st decade | c.892C>T;p.(Q298*) | c.886G>T;p.(E296*) | 0.1/0.1 | 7636/7651 | 2764/2345 |
| ARRP260-25421§ | M | 7 | 1st decade | c.177_248dup;p.(L60_L83dup) | c.1401+2T>G;p.(?) | 0.2/0.1 | 14359/9883 | 420/672 |
| ARRP260-25423§ | M | 12 | 2nd decade | c.177_248dup;p.(L60_L83dup) | c.1401+2T>G;p.(?) | 0.2/0.1 | 1325/1545 | 390/417 |
| SRP1056-29774§ | M | 13 | 1st decade | c.892C>T;p.(Q298*) | c.892C>T;p.(Q298*) | 0.0/-0.1 | 9051/8358 | 430/413 |
| ARRP395-30213 | F | 16 | 2nd decade | c.1832+1G>T;p.(?) | c.1832+1G>T;p.(?) | 0.3/0.2 | NP/NP | 2054/1502 |
| SRP759-19456 | F | 20 | 3rd decade | c.1258-2A>G;p.(?) | c.2193+1G>A;p.(?) | 0.2/0.4 | 1376/13149 | 1395/1221 |
| ARRP411-30491 | M | 25 | 1st decade | c.1060-1G>T;p.(?) | c.1060-1G>T;p.(?) | 0.0/0.0 | 412/493 | 131/162 |
| ARRP75-5835§ | M | 28 | 1st decade | c.1699C>T;p.(Q567*) | c.1699C>T;p.(Q567*) | 0.1/0.6 | 1249/1801 | 323/204 |
| SRP1035-29494 | M | 29 | 1st decade | c.1107+3A>G;p.(?) | c.1920+2T>C;p.(?) | 0.3/0.3 | 6354/10143 | 297/225 |
| SRP612-17465 | F | 29 | 1st decade | c.1667A>G;p.(Y556C) | c.1667A>G;p.(Y556C) | 0.3/0.3 | 11137/9484 | 5025/5129 |
| SRP774-22406 | F | 32 | Birth | c.1390C>T;p.(Q464*) | c.1390C>T;p.(Q464*) | 0.1/0.2 | 416/440 | 218/277 |
| ARRP26-21885§ | M | 35 | 1st decade | c.892C>T;p.(Q298*) | c.2003A>G;(p.D668G) | 0.3/0.3 | 349/427 | 172/33 |
| SRP823-26156 | M | 35 | 2nd decade | c.2193+1G>A;p.(?) | c.2193+1G>A;p.(?) | 0.2/0.2 | 484/420 | 36/18 |
| SRP786-22723 | F | 36 | 1st decade | c.892C>T;p.(Q298*) | c.892C>T;p.(Q298*) | 0.1/0.0 | 709/588 | 264/53 |
| SRP778-22500 | F | 36 | 1st decade | c.1923_1969delinsTCTGGG;p.(N643Gfs*29) | c.2326G>A;p.(D776N) | 0.0/0.0 | 4388/7223 | 345/398 |
| SRP1168-0 | F | 37 | ND | c.2326G>A;p.(D776N) | c.2326G>A;p.(D776N) | -0.1/-0.1 | 8785/7893 | NP/NP |
| ARRP209-23862 | F | 42 | 1st decade | c.[299G>A;1401+4_1401+48del];p.[(R100H);(?)] | c.[299G>A;1401+4_1401+48del];p.[(R100H);(?)] | 0.3/0.6 | 228/239 | 88/66 |
| ARRP26-18556§ | F | 43 | 2nd decade | c.892C>T;p.(Q298*) | c.2003A>G;(p.D668G) | 0.2/0.7 | 1586/1579 | 341/398 |
| SRP672-19402 | M | 45 | 1st decade | c.2326G>A;p.(D776N) | c.2326G>A;p.(D776N) | 0.1/0.1 | 2451/2259 | 384/272 |
| ARRP209-22048 | M | 45 | 1st decade | c.[299G>A;1401+4_1401+48del];p.[(R100H);(?)] | c.[299G>A;1401+4_1401+48del];p.[(R100H);(?)] | 0.3/0.1 | 9/3 | NP/NP |
| ARRP171-15079# | F | 46 | 2nd decade | c.1798G>A;p.(D600N) | c.1798G>A;p.(D600N) | 1.2/1.2 | 96/226 | 8/ND |
| SRP960-28509 | F | 48 | 1st decade | c.[409G>A;928-9_940dup];p.[(G137R);(?)] | c.[409G>A;928-9_940dup];p.[(G137R);(?)] | 0.1/0.1 | 208/287 | 49/37 |
| SRP341-24713 | M | 52 | 1st decade | c.1744T>C;p.(Y582H) | c.1744T>C;p.(Y582H) | 0.6/0.4 | 54/34 | NP/NP |
| SRP754-21728 | M | 52 | 1st decade | c.221dup;p.(V75Rfs*91) | c.892C>T;p.(Q298*) | 1.0/0.6 | 269/185 | 40/23 |
| PDE6B cDNA (NM_000283.3) | PDE6B Protein (NP_000274.2) | Number of Alleles | Reference (dbSNP & Literature) | Location of Missense Variant in Protein Domain | Consensus Predictions of Various Software § | gnomAD MAF | ACMG Prediction [18] | ACMG Classification [18] |
|---|---|---|---|---|---|---|---|---|
| c.177_248dup | p.(L60_L83dup) | 2 | This study | in-frame duplication | n.d. | Pathogenic (IIIa) | PM2, PM4, PP1, PM3 | |
| c.221dup | p.(V75Rfs*91) | 1 | This study | frame-shift, PTC, NMD | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.299G>A | p.(R100H) | 4 | rs555600300 [12] | GAF1 | damaging | 0.0057% | VUS | PM2 |
| c.409G>A | p.(G137R) | 2 | rs781658083 [19] | GAF1 | damaging | 0.00092% | VUS | PM2 |
| c.886G>T | p.(E296*) | 1 | rs1064797304 This study | PTC, NMD | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.892C>T | p.(Q298*) | 8 | rs121918579 [5] | PTC, NMD | 0.005% | Pathogenic (Ib) | PM2, PVS1, PM3 | |
| c.928-9_940dup | p.(?) | 2 | rs539992414 [20] | benign | 0.50% | Likely pathogenic (I) | PM2, PVS1 | |
| c.1060-1G>T | p.(?) | 2 | rs863223339 [21] | missplicing | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.1107+3A>G | p.(?) | 1 | rs370898371 [12] | missplicing | 0.0042% | VUS | PM2 | |
| c.1258-2A>G | p.(?) | 1 | This study | missplicing | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.1390C>T | p.(Q464*) | 2 | This study | PTC, NMD | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.1401+2T>G | p.(?) | 2 | This study | missplicing | n.d. | Pathogenic (Ia) | PM2, PVS1, PP1, PM3 | |
| c.1401+4_1401+48del | p.(?) | 4 | rs768567008 [22] | possibly missplicing | 3.56% | VUS | PM2 | |
| c.1667A>G | p.(Y556C) | 2 | rs755577875 This study | PDEase I catalytic domain | damaging | 0.001% | VUS | PM2 |
| c.1699C>T | p.(Q567*) | 2 | rs772057239 [23] | PTC, NMD | 0.0011% | Pathogenic (Ib) | PM2, PVS1, PM3 | |
| c.1744T>C | p.(Y582H) | 2 | This study | PDEase I catalytic domain | damaging | n.d. | VUS | PM2 |
| c.1798G>A | p.(D600N) | 2 | rs764605140 [24] | PDEase I catalytic domain second Zn2+ binding motif | damaging | 0.0056% | VUS | PM2 |
| c.1832+1G>T | p.(?) | 2 | This study | missplicing | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.1920+2T>C | p.(?) | 1 | rs763996159 [12] | missplicing | 0.0008% | Likely pathogenic (I) | PM2, PVS1 | |
| c.1923_1969delinsTCTGGG | p.(N643Gfs*29) | 1 | [25] | frame-shift, PTC, NMD | n.d. | Likely pathogenic (I) | PM2, PVS1 | |
| c.2003A>G | (p.D668G) | 2 | This study | PDEase I catalytic domain | damaging | n.d. | VUS | PM2, PM3 |
| c.2193+1G>A | p.(?) | 3 | rs727504075 [26] | missplicing | 0.0072% | Likely pathogenic (I) | PM2, PVS1 | |
| c.2326G>A | p.(D776N) | 5 | rs141563823 [12] | PDEase I catalytic domain | damaging | 0.0057% | VUS | PM2 |
| Frequency | N | |||||||
|---|---|---|---|---|---|---|---|---|
| ERM | 67% | 32/48 | ||||||
| CME | 35% | 17/48 | ||||||
| Macular atrophy | 19% | 9/48 | ||||||
| Posterior staphyloma associated with myopia | 10% | 5/48 | ||||||
| Papillomacular retinal thickening | 8% | 4/48 | ||||||
| IHRM | 6% | 3/48 | ||||||
| Lamellar macular hole | 2% | 1/48 | ||||||
| N | Median | Mean | SD | Min | Max | Symmetry | ||
| EZ-width OD (µm) | 18/24 | 2042 | 2042 | 874 | 696 | 3810 | R2 = 0.942 | |
| EZ-width OS (µm) | 16/24 | 1964 | 1924 | 844 | 627 | 3474 | ||
| FT OD (µm) | 22/24 | 231 | 264 | 115 | 115 | 526 | R2 = 0.603 | |
| FT OS (µm) | 21/24 | 225 | 233 | 109 | 36 | 567 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuehlewein, L.; Zobor, D.; Stingl, K.; Kempf, M.; Nasser, F.; Bernd, A.; Biskup, S.; Cremers, F.P.M.; Khan, M.I.; Mazzola, P.; et al. Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa. Int. J. Mol. Sci. 2021, 22, 2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052374
Kuehlewein L, Zobor D, Stingl K, Kempf M, Nasser F, Bernd A, Biskup S, Cremers FPM, Khan MI, Mazzola P, et al. Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa. International Journal of Molecular Sciences. 2021; 22(5):2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052374
Chicago/Turabian StyleKuehlewein, Laura, Ditta Zobor, Katarina Stingl, Melanie Kempf, Fadi Nasser, Antje Bernd, Saskia Biskup, Frans P.M. Cremers, Muhammad Imran Khan, Pascale Mazzola, and et al. 2021. "Clinical Phenotype of PDE6B-Associated Retinitis Pigmentosa" International Journal of Molecular Sciences 22, no. 5: 2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052374