Combined Transcriptomic and Metabolomic Analysis Reveals the Role of Phenylpropanoid Biosynthesis Pathway in the Salt Tolerance Process of Sophora alopecuroides




 and
and
Abstract
:
1. Introduction
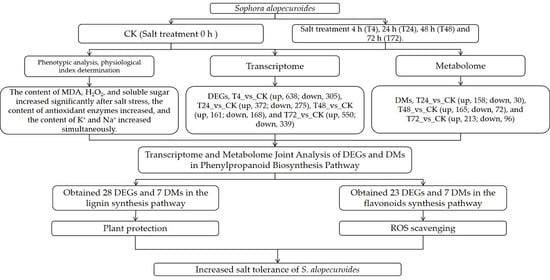
2. Results
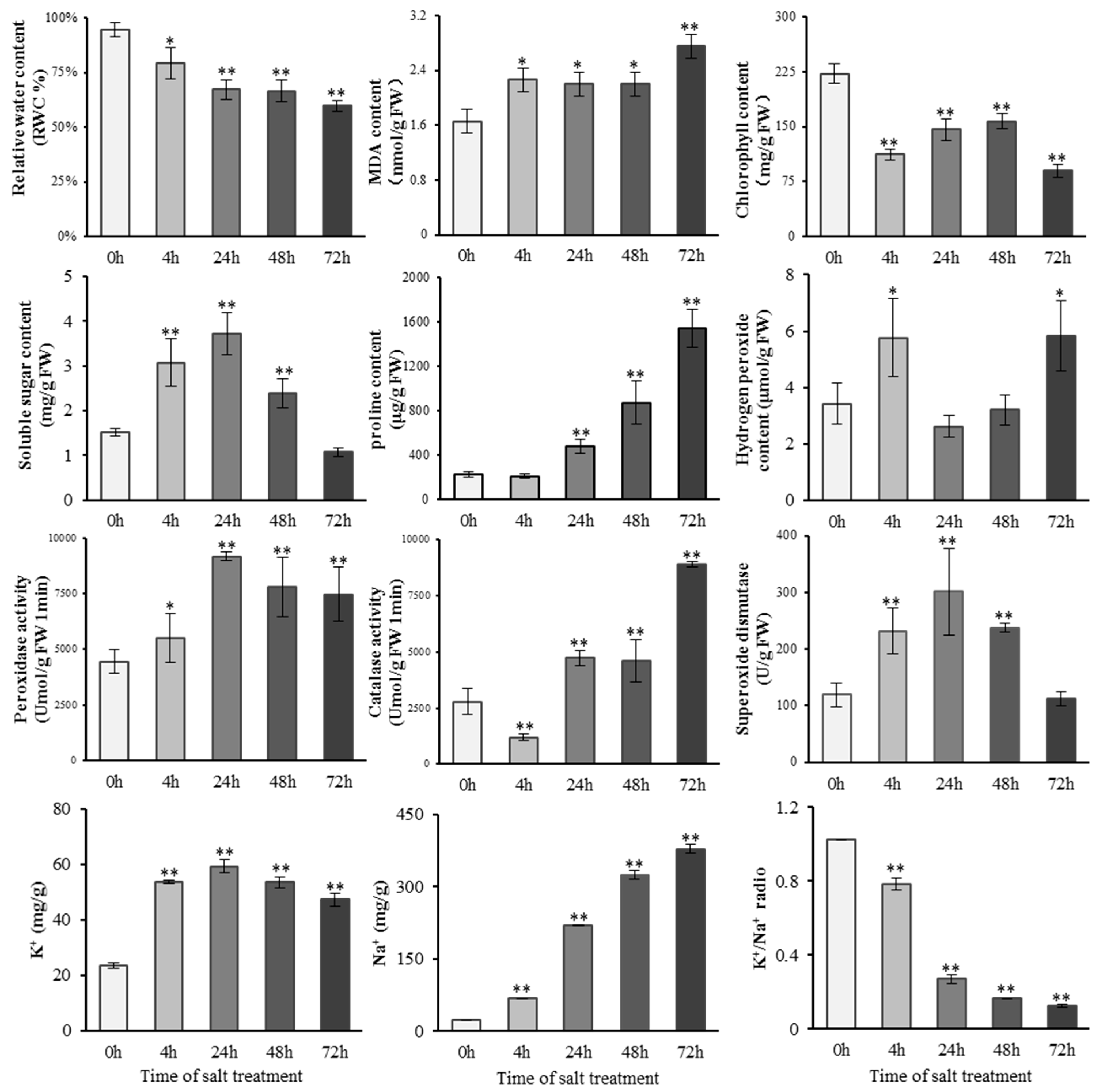
2.1. Physiological Changes of S. alopecuroides under Salt Stress
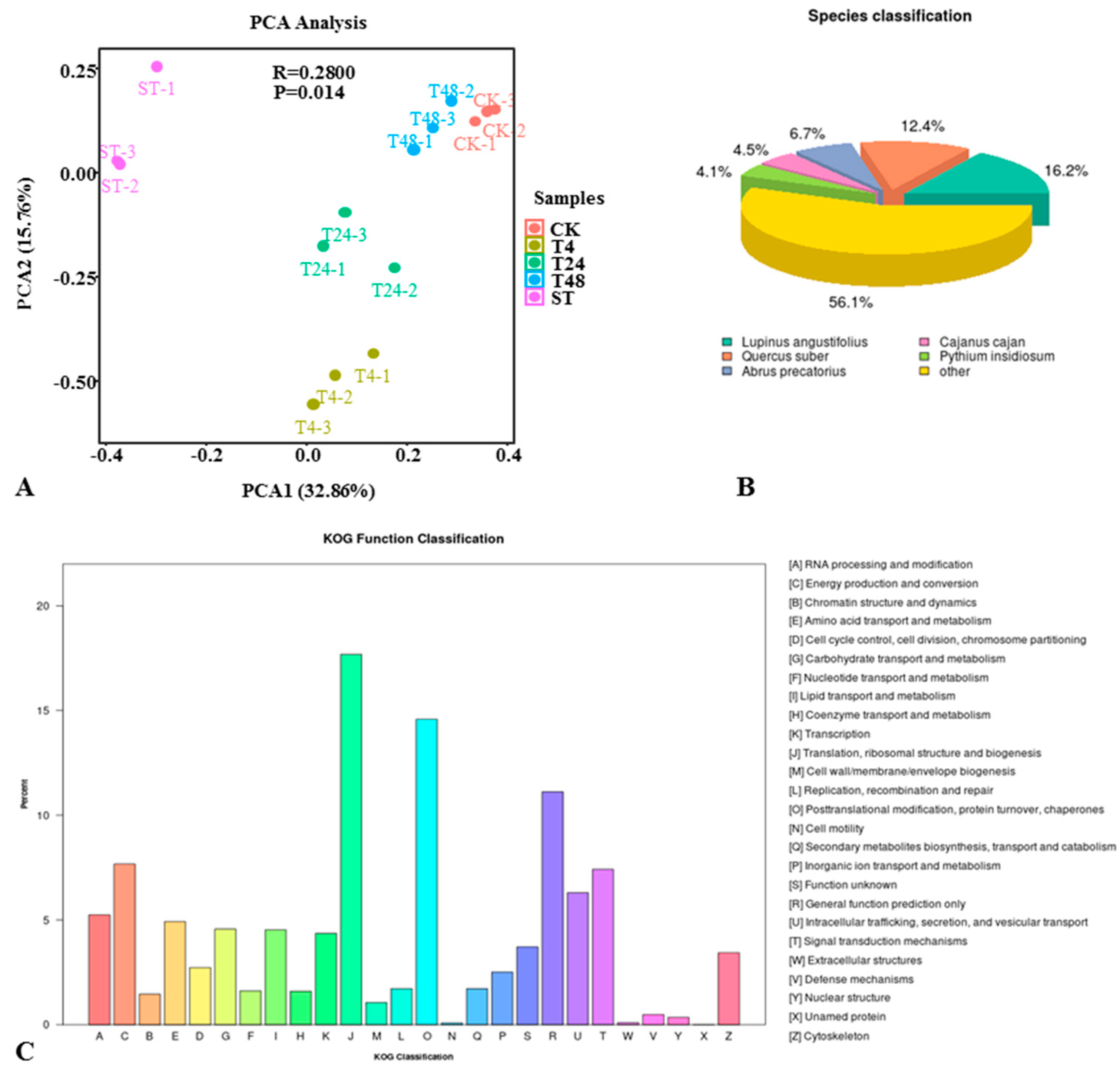
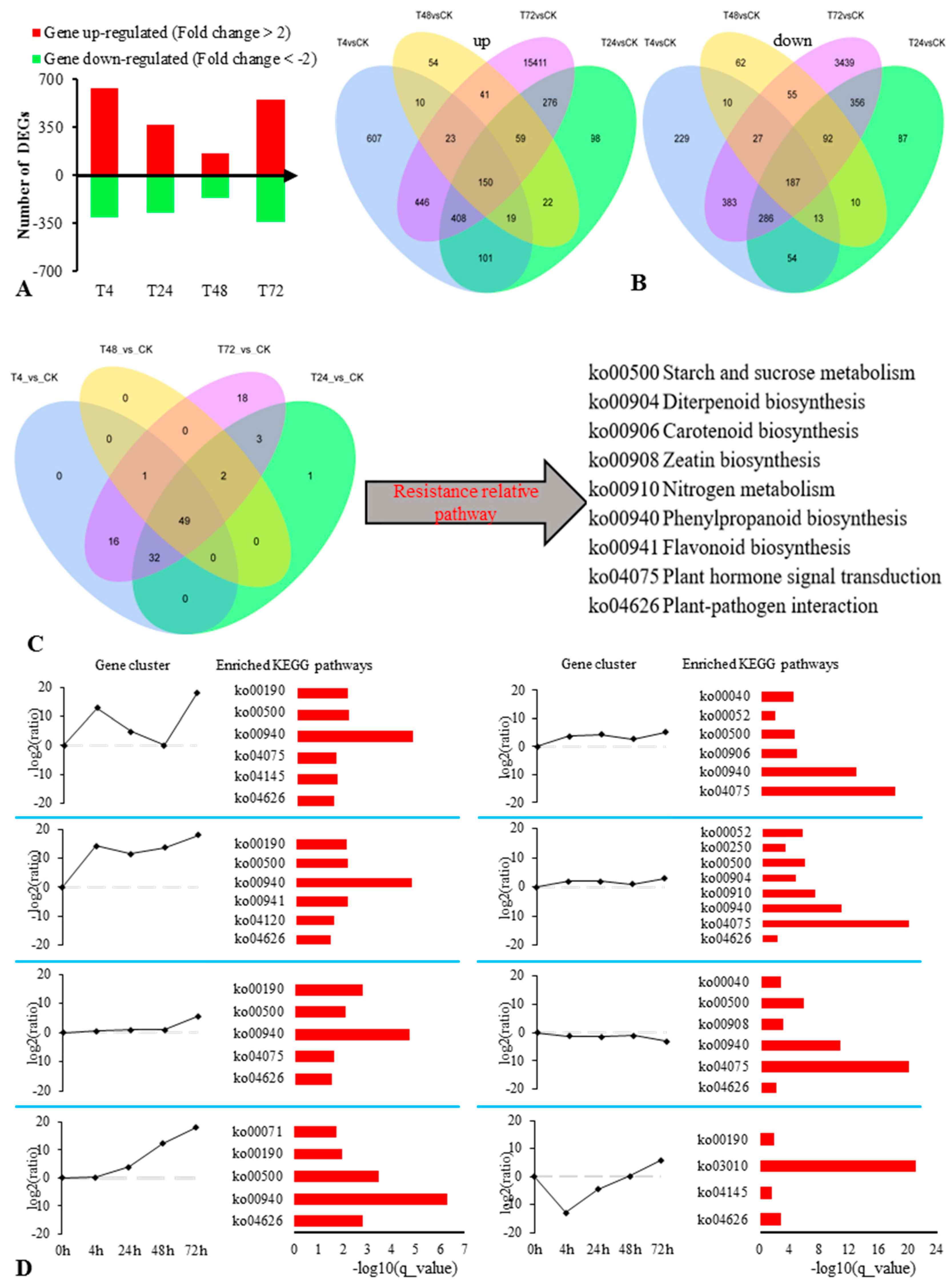
2.2. Transcriptomic Analysis of the Response of S. alopecuroides to Salt Stress
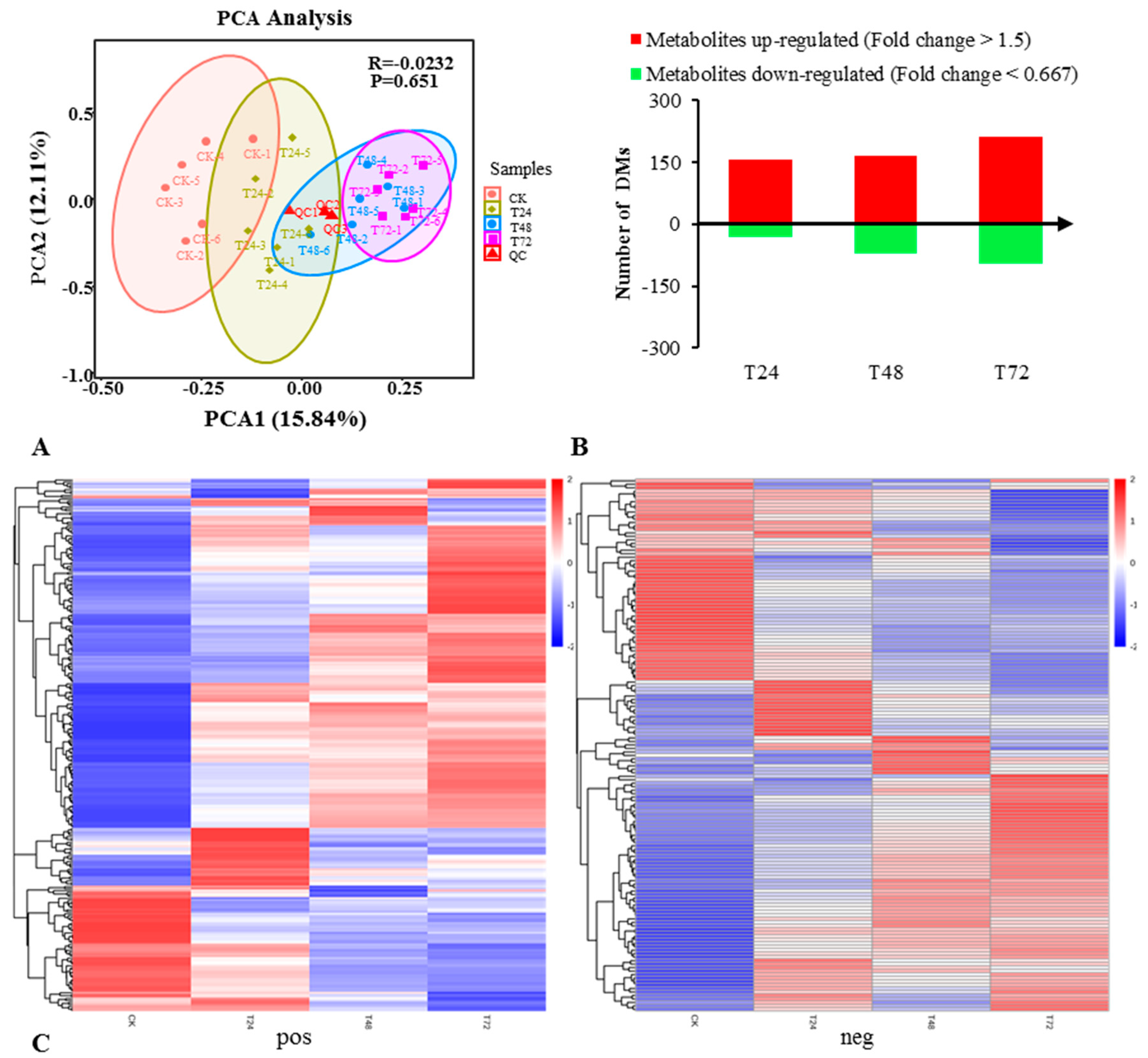
2.3. Metabonomic Analysis on the Response of S. alopecuroides to Salt Stress
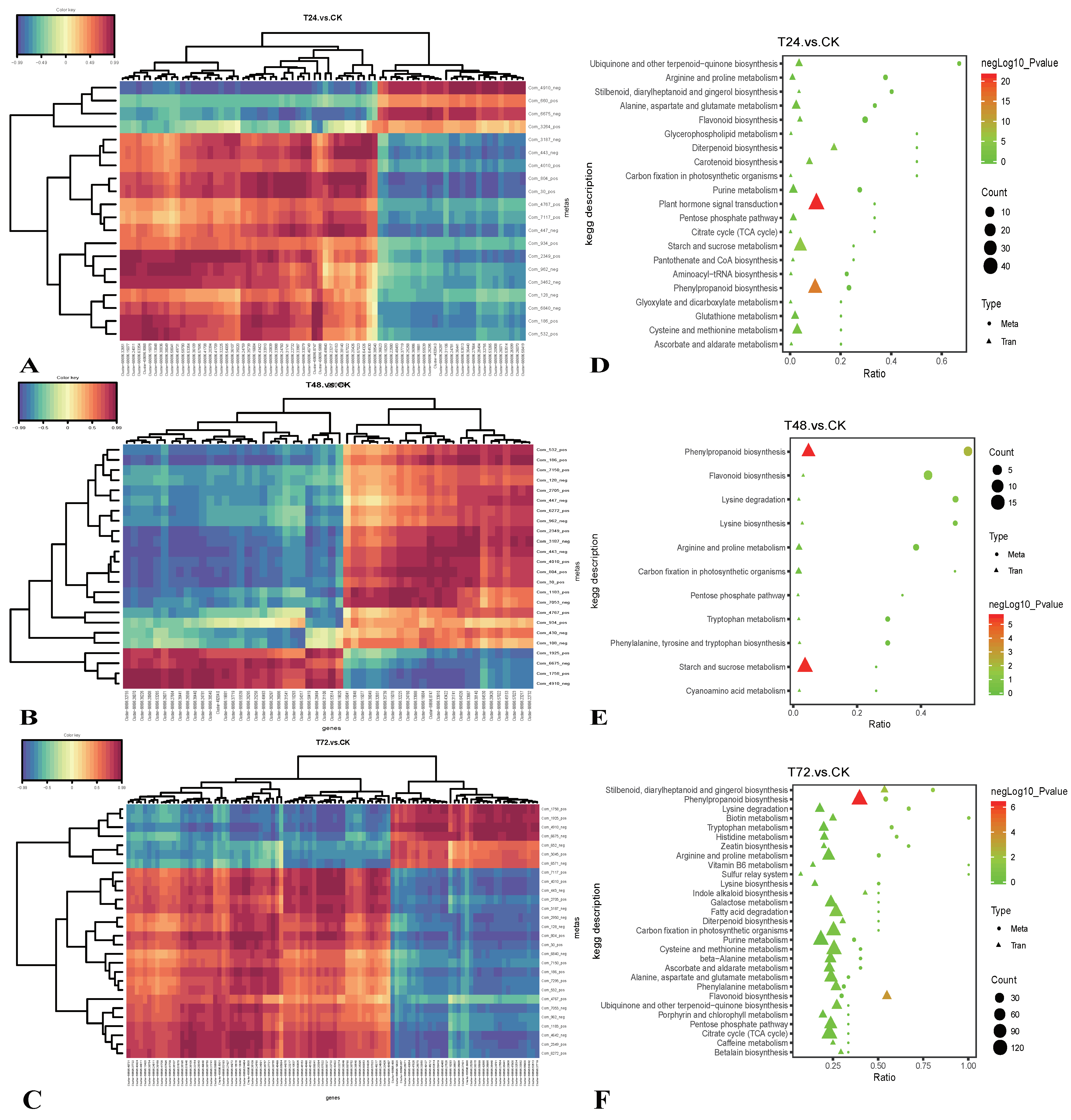
2.4. Integrated Transcriptomic and Metabolomic Analysis of the Response of S. alopecuroides to Salt Stress
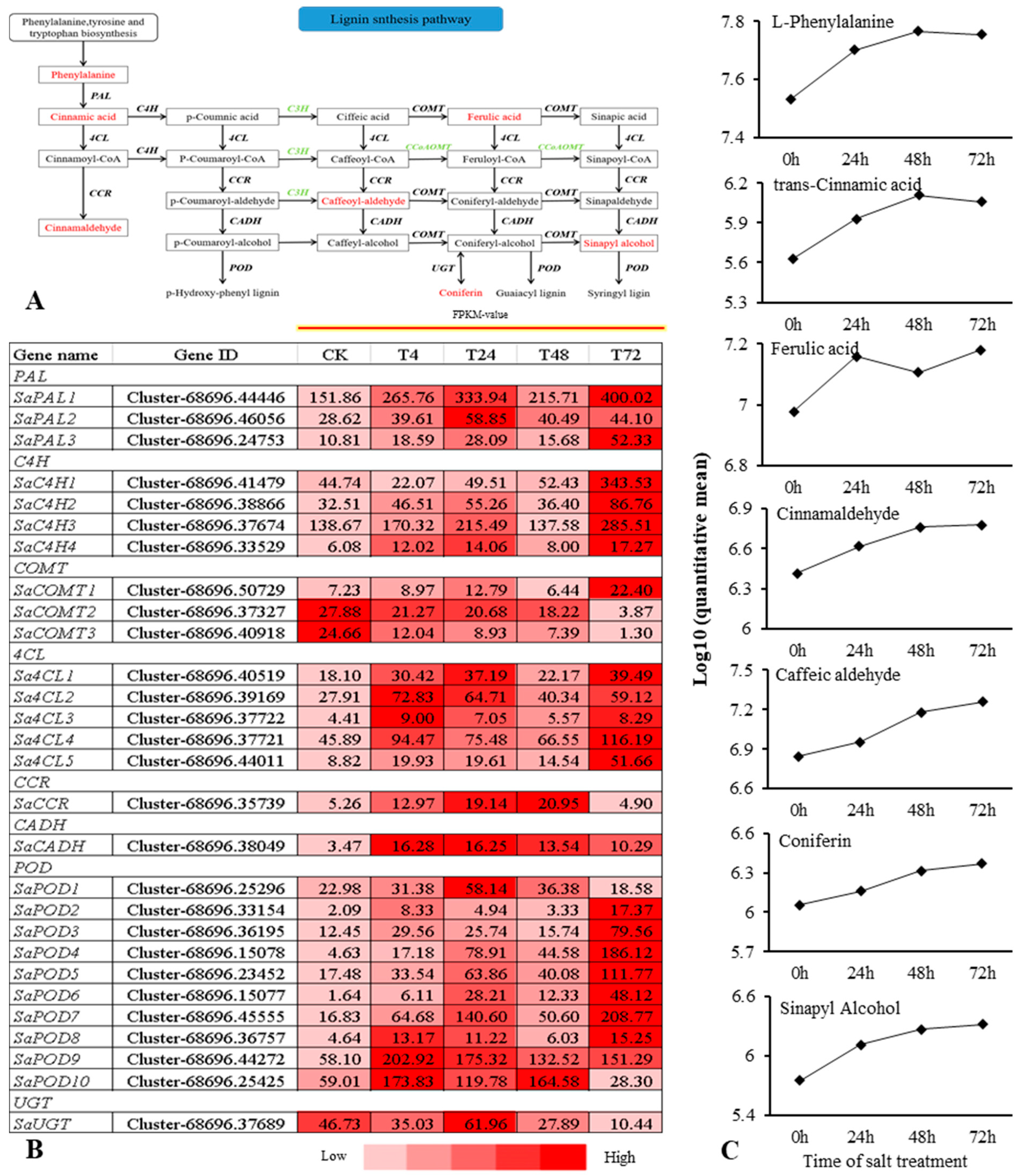
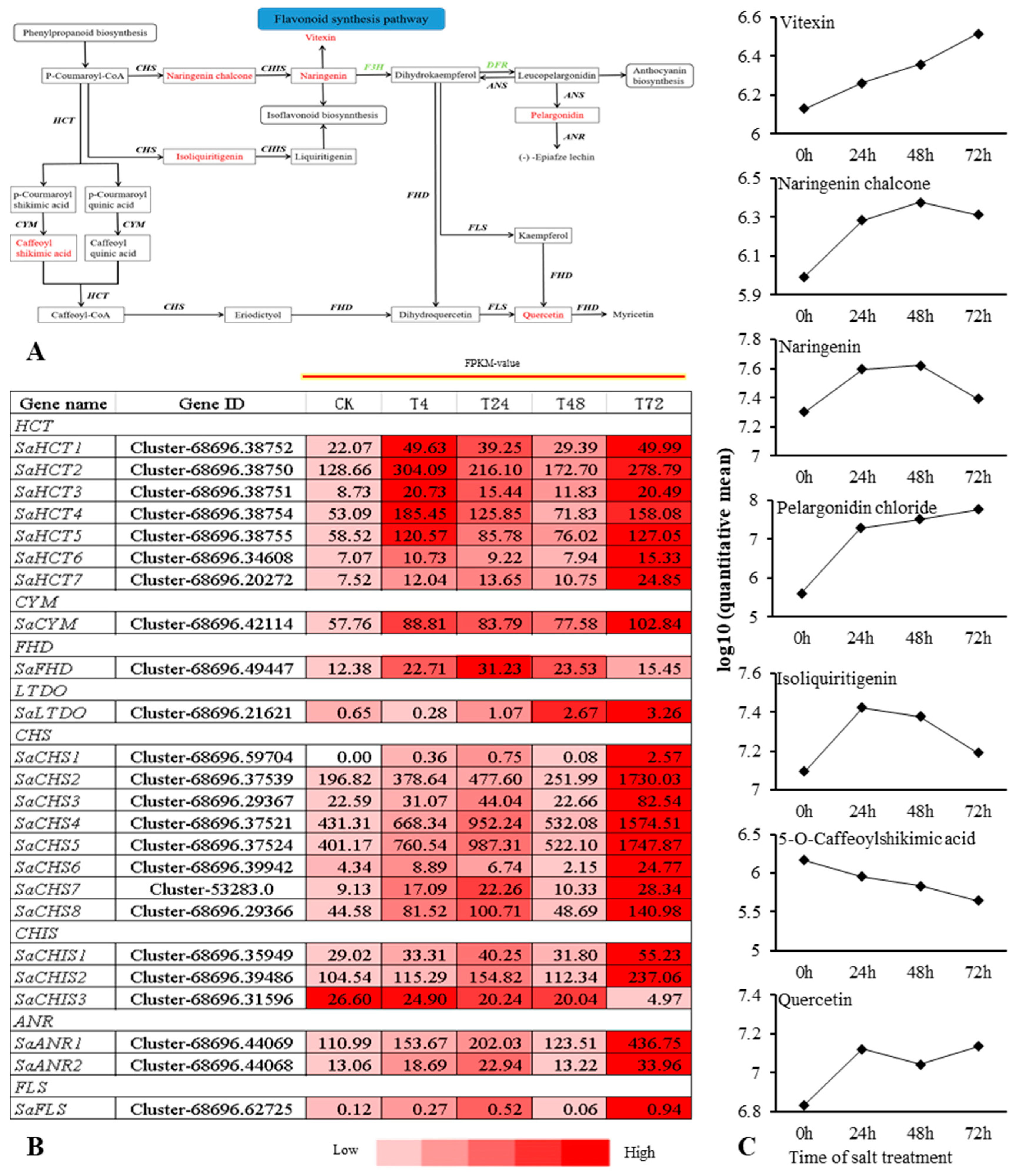
2.5. Combined Transcriptomic and Metabolomic Analysis on the Response of S. alopecuroides in the LSP under Salt Stress
2.6. Combined Transcriptomic and Metabolomic Analysis on the Response of S. alopecuroides in the FSP under Salt Stress
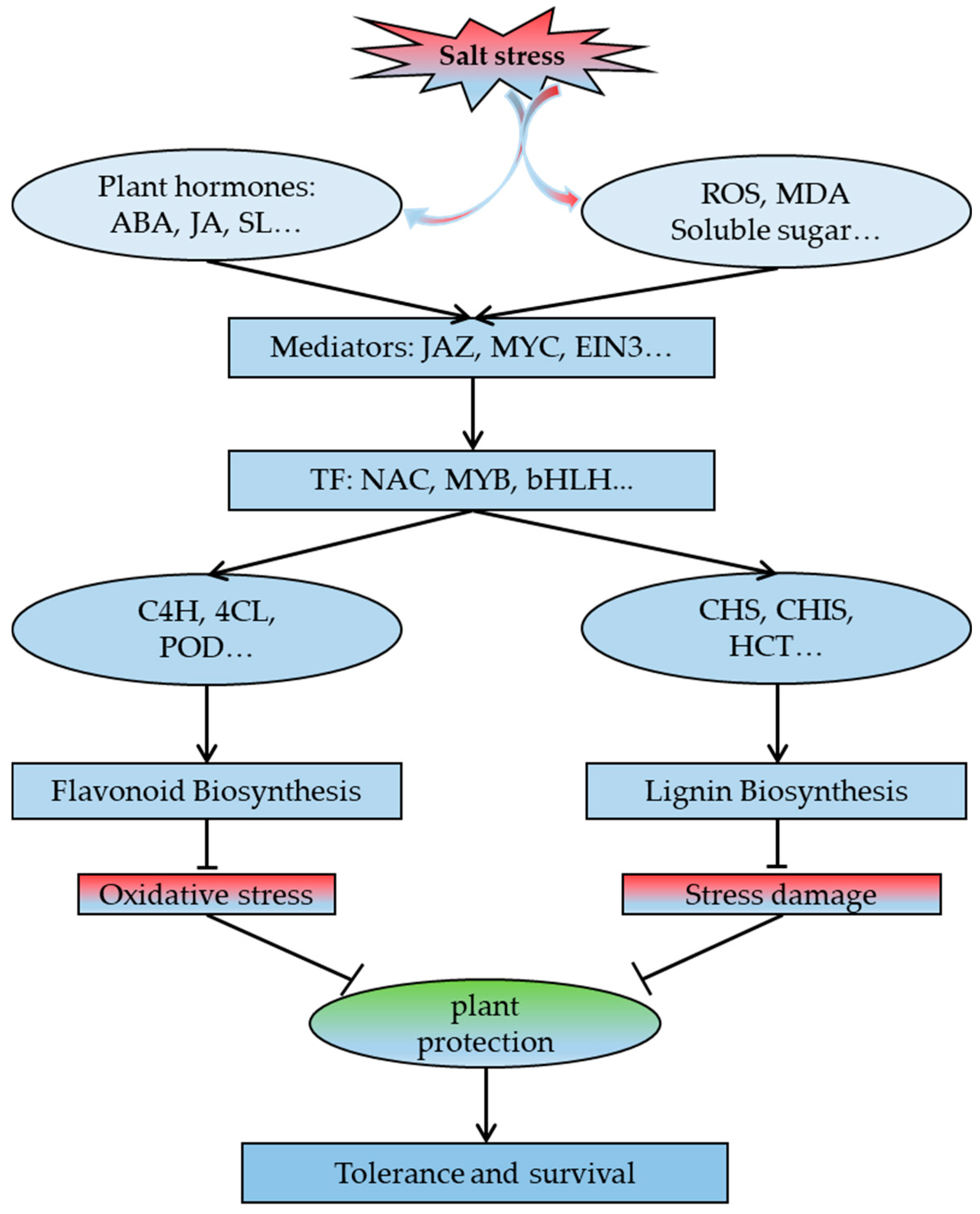
3. Discussion
4. Material and Methods
4.1. Plant Growth and Salt Stress
4.2. Measurement and Analysis of Physiological Indices
4.3. Transcriptome Sequencing and Data Analysis
4.4. Metabolite Profiling Analysis
4.5. Metabolome and Transcriptome Co-Expression Network Analysis
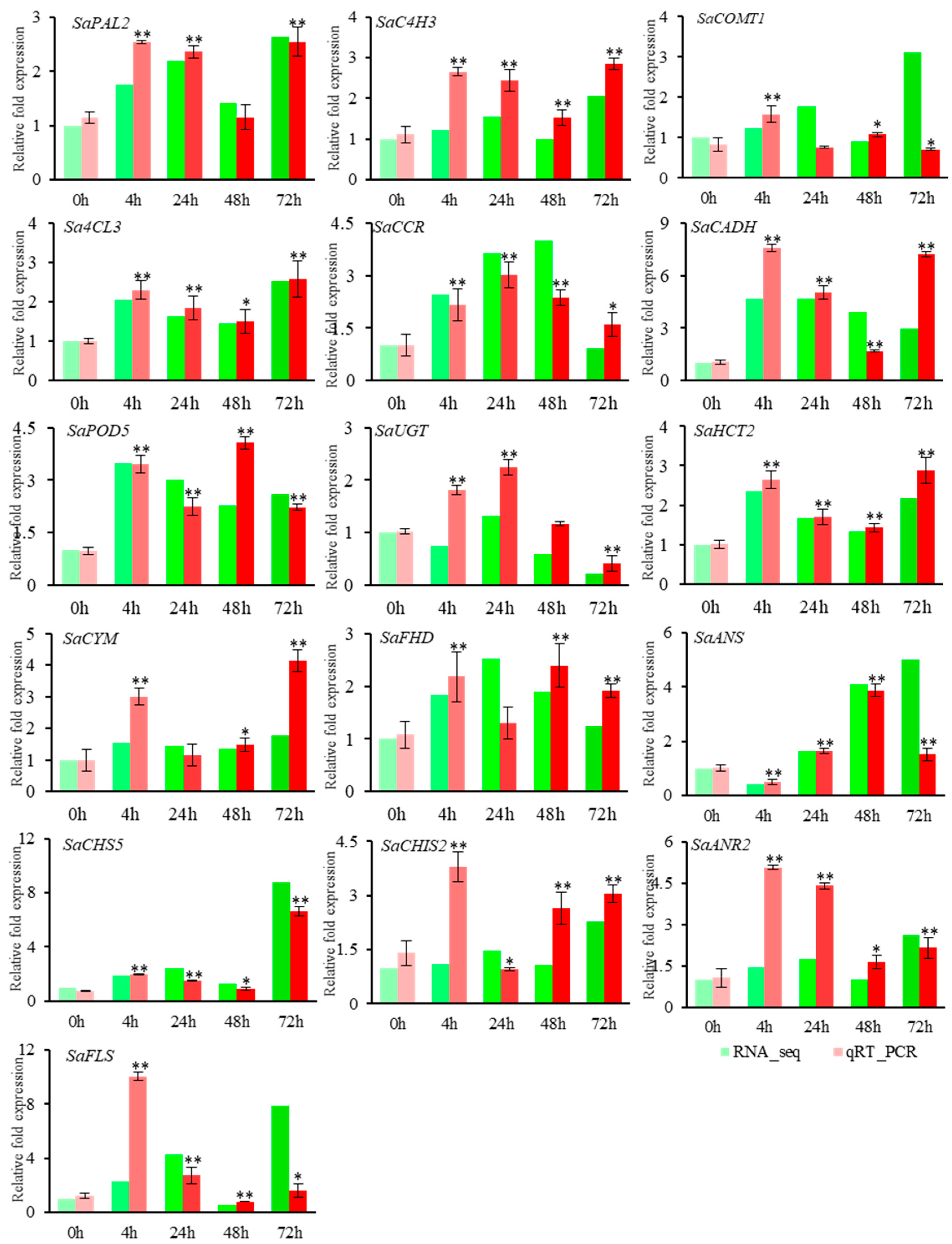
4.6. The qRT-PCR Validation for DEGs
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RWC | Relative water content |
| MDA | Malondialdehyde |
| ROS | Reactive oxygen species |
| DEGs | Differentially expressed genes |
| DMs | Differential metabolites |
| H2O2 | Hydrogen peroxide |
| POD | Peroxidase |
| CAT | Catalase |
| SOD | Superoxide dismutase |
| GR | Glutathione reductase |
| GST | Glutathione S-transferase |
| ABA | Abscisic acid |
| CKs | Cytokinins |
| SLs | Strigolactones |
| GA | Gibberellin |
| JA | Jasmonic acid |
| SA | Salicylic acid |
| PHB | Phenylpropanoid Biosynthesis |
| FLB | Flavonoid Biosynthesis |
| CHS | Chalcone synthase |
| CHIS | Chalcone isomerase |
| CPM | Cytochrome P450 monooxygenase |
| VIP | Variable Importance in the Projection |
| FC | Fold change |
| HCA | Hierarchical clustering analysis |
| PAL | Phenylalanine ammonia-lyase |
| PCA | Principal component analysis |
| C4H | Trans-cinnamate 4-monooxygenase |
| COMT | Caffeic acid 3-O-methyltransferase |
| 4CL | 4-coumarate–CoA ligase |
| CCR | Cinnamoyl-CoA reductase |
| CADH | Cinnamyl-alcohol dehydrogenase |
| UGT | Coniferyl-alcohol glucosyltransferase |
| HCT | O-hydroxycinnamoyltransferase |
| CYM | Coumaroylquinate (coumaroylshikimate) 3′-monooxygenase |
| FHD | Flavonoid 3′,5′-hydroxylase |
| FLS | Flavonol synthase |
| ANR | Anthocyanidin reductase |
| ANS | Leucoanthocyanidin dioxygenase |
| FW | Fresh weight |
| SW | Saturated weight |
| DW | Dry weight |
References
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2017, 217, 523–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, S.A.; Zaman, M.; Heng, L. Soil Salinity: Historical Perspectives and a World Overview of the Problem. Guideline for Salinity Assessment, Mitigation and Adaptation Using Nuclear and Related Techniques; Springer International Publishing: Cham, Switzerland, 2018; pp. 43–53. [Google Scholar]
- Zhu, J.-K. Genetic Analysis of Plant Salt Tolerance Using Arabidopsis: Figure 1. Plant Physiol. 2000, 124, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, B.; Huang, B. Mechanism of Salinity Tolerance in Plants: Physiological, Biochemical, and Molecular Characterization. Int. J. Genom. 2014, 2014, 1–18. [Google Scholar] [CrossRef]
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.; Zhang, Y.; Dai, S. Mechanisms of Plant Salt Response: Insights from Proteomics. J. Proteome Res. 2011, 11, 49–67. [Google Scholar] [CrossRef]
- Sun, H.; Chen, L.; Li, J.; Hu, M.; Ullah, A.; Heng, S.; Yang, X.; Zhang, X. The JASMONATE ZIM-Domain Gene Family Mediates JA Signaling and Stress Response in Cotton. Plant Cell Physiol. 2017, 58, 2139–2154. [Google Scholar] [CrossRef]
- Ullah, A.; Sun, H.; Yang, X.; Zhang, X. A novel cotton WRKY gene, GhWRKY6-like, improves salt tolerance by activating the ABA signaling pathway and scavenging of reactive oxygen species. Physiol. Plant. 2018, 162, 439–454. [Google Scholar] [CrossRef]
- Apse, M.P.; Sottosanto, J.B.; Blumwald, E. Vacuolar cation/H+exchange, ion homeostasis, and leaf development are altered in a T-DNA insertional mutant ofAtNHX1, the Arabidopsis vacuolar Na+/H+antiporter. Plant J. 2003, 36, 229–239. [Google Scholar] [CrossRef]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenes. Tissue Repair 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandehagh, A.; Salekdeh, G.H.; Toorchi, M.; Mohammadi, A.; Komatsu, S. Comparative proteomic analysis of canola leaves under salinity stress. Proteomics 2011, 11, 1965–1975. [Google Scholar] [CrossRef]
- Huang, C.; He, W.; Guo, J.; Chang, X.; Su, P.; Zhang, L. Increased sensitivity to salt stress in an ascorbate-deficient Arabidopsis mutant. J. Exp. Bot. 2005, 56, 3041–3049. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Su, W.; Zhang, H.; Zhan, Y.; Zeng, F. Fraxinus mandshurica 4-coumarate-CoA ligase 2 enhances drought and osmotic stress tolerance of tobacco by increasing coniferyl alcohol content. Plant Physiol. Biochem. 2020, 155, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, Y.; Khadr, A.; Wang, Y.H.; Xu, Z.S.; Xiong, A.S. DcDREB1A, a DREB-binding transcription factor from Daucus carota, enhances drought tolerance in transgenic Arabidopsis thaliana and modulates lignin levels by regulating lignin-biosynthesis-related genes. Environ. Exp. Bot. 2020, 169, 103896. [Google Scholar] [CrossRef]
- Aoki, T.; Akashi, T.; Ayabe, S.-I. Flavonoids of Leguminous Plants: Structure, Biological Activity, and Biosynthesis. J. Plant Res. 2000, 113, 475–488. [Google Scholar] [CrossRef]
- Diniz, T.C.; Silva, J.C.; de Lima-Saraiva, S.R.; Ribeiro, F.P.; Pacheco, A.G.; de Freitas, R.M.; Quintans-Júnior, L.J.; Quintans, J.D.S.; Mendes, R.L.; Almeida, J.R. The role of flavonoids on oxidative stress in epilepsy. Oxid. Med. Cell. Longev. 2015, 2015, 171756. [Google Scholar] [CrossRef]
- Fahnenstich, H.; Scarpeci, T.E.; Valle, E.M.; Flügge, U.I.; Maurino, V.G. Generation of hydrogen peroxide in chloroplasts of Ara-bidopsis overexpressing glycolate oxidase as an inducible system to study oxidative stress. Plant Physiol. 2008, 148, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.L.; Graham, M.Y.; Subramanian, S.; Yu, O. RNAi Silencing of Genes for Elicitation or Biosynthesis of 5-Deoxyisoflavonoids Suppresses Race-Specific Resistance and Hypersensitive Cell Death in Phytophthora sojae Infected Tissues. Plant Physiol. 2007, 144, 728–740. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.-G.; Sheng, Z.-W.; Xu, W.-F.; Li, Y.-X.; Liu, Y.-G.; Xia, Y.-J.; Zhang, J.-H. Modulation of Anti-Oxidation Ability by Proanthocyanidins during Germination of Arabidopsis thaliana Seeds. Mol. Plant 2012, 5, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, M.; Sa-kakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of an-tioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Falcone, F.M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, J.K.; Wakatake, T.; Yoshida, S.; Takebayashi, Y.; Kasahara, H.; Wafula, E.; Depamphilis, C.W.; Namba, S.; Shirasu, K. Local Auxin Biosynthesis Mediated by a YUCCA Flavin Monooxygenase Regulates Haustorium Development in the Parasitic Plant Phtheirospermum japonicum. Plant Cell 2016, 28, 1795–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, E.; Qu, L.; Hu, J.; Huang, Y.; Qiu, L.; Lu, H.; Jiang, B.; Liu, C.; Peng, T.; Zhao, Y.; et al. Mechanisms of Soybean Roots’ Tol-erances to Salinity Revealed by Proteomic and Phosphoproteomic Comparisons Between Two Cultivars. Mol. Cell. Proteom. 2016, 15, 266–288. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.; Barah, P.; Suarez-Rodriguez, M.C.; Bressendorff, S.; Friis, P.; Costantino, P.; Bones, A.M.; Nielsen, H.B.; Mundy, J. Transcriptome Responses to Combinations of Stresses in Arabidopsis. Plant Physiol. 2013, 161, 1783–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.S.; Zhao, X.Q.; Li, M.; Huang, L.Y.; Xu, J.L.; Zhang, F.; Cui, Y.R.; Fu, B.Y.; Li, Z.K. Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J. Exp. Bot. 2016, 67, 405–419. [Google Scholar] [CrossRef] [Green Version]
- Brinker, M.; Brosché, M.; Vinocur, B.; Abo-Ogiala, A.; Fayyaz, P.; Janz, D.; Ottow, E.A.; Cullmann, A.D.; Saborowski, J.; Kangasjärvi, J.; et al. Linking the Salt Transcriptome with Physiological Responses of a Salt-Resistant Populus Species as a Strategy to Identify Genes Important for Stress Acclimation. Plant Physiol. 2010, 154, 1697–1709. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Peng, Z.; Mao, X.; Wang, J.; Li, C.; Chang, X.; Jing, R. Genetic insights into natural variation underlying salt tolerance in wheat. J. Exp. Bot. 2020, 500. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Y.; Jiao, B.; Chen, G.; Huang, S.; Guo, F.; Shen, Y.; Huang, Z.; Zhao, B. Overexpression of the wheat salt tolerance-related gene TaSC enhances salt tolerance in Arabidopsis. J. Exp. Bot. 2012, 63, 5463–5473. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Zhu, Y.; Zhao, Y.; Wang, Y.; Li, J.; Wang, Q.; Liu, Y. De novo transcriptome sequencing and analysis of salt-, alkali-, and drought-responsive genes in Sophora alopecuroides. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Rao, G.; Sui, J.; Zhang, J. Metabolomics reveals significant variations in metabolites and correlations regarding the maturation of walnuts (Juglans regia L.). Biol. Open 2016, 5, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heischmann, S.; Quinn, K.; Cruickshank-Quinn, C.; Liang, L.P.; Reisdorph, R.; Reisdorph, N.; Patel, M. Exploratory Metabo-lomics Profiling in the Kainic Acid Rat Model Reveals Depletion of 25-Hydroxyvitamin D3 during Epileptogenesis. Sci. Rep. 2016, 6, 31424. [Google Scholar] [CrossRef] [Green Version]
- Haspel, J.A.; Chettimada, S.; Shaik, R.S.; Chu, J.-H.; Raby, B.A.; Cernadas, M.; Carey, V.; Process, V.; Hunninghake, G.M.; Ifedigbo, E.; et al. Circadian rhythm reprogramming during lung inflammation. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Mao, C.; Shi, Z.; Kou, X. The Amino Acid Metabolic and Carbohydrate Metabolic Pathway Play Important Roles during Salt-Stress Response in Tomato. Front. Plant Sci. 2017, 8, 1231. [Google Scholar] [CrossRef] [Green Version]
- Diao, H.-Y.; Wang, A.-Z.; Yuan, F.-H.; Guan, D.-X.; Sun, Y.; Wu, J.-B. Applications of compound-specific isotope analysis in tree non-structural carbohydrates research: A review. Ying Yong Sheng Tai Xue Bao 2020, 31, 4291–4300. [Google Scholar]
- Kumar, S.; Li, G.; Yang, J.; Huang, X.; Ji, Q.; Zhou, K.; Khan, S.; Ke, W.; Hou, H. Investigation of an Antioxidative System for Salinity Tolerance in Oenanthe javanica. Antioxidants 2020, 9, 940. [Google Scholar] [CrossRef]
- Kerepesi, L.; Galiba, G. Osmotic and salt stress induced alteration in soluble carbohydratecontent in wheat seedlings. Crop Sci. 2000, 40, 482–487. [Google Scholar] [CrossRef]
- Zrig, A.; Ferreira, J.F.S.; Hamouda, F.; Tounekti, T.; Selim, S.; al Jaouni, S.; Khemira, H.; Abdel, G.H. The impact of foliar fertilizers on growth and biochemical responses of Thymus vulgaris to salinity stress. Arid. Land Res. Manag. 2019, 33, 297–320. [Google Scholar] [CrossRef]
- Zhu, Y.; Yin, J.; Liang, Y.; Liu, J.; Jia, J.; Huo, H.; Wu, Z.; Yang, R.; Gong, H. Transcriptomic dynamics provide an insight into the mechanism for silicon-mediated alleviation of salt stress in cucumber plants. Ecotoxicol. Environ. Saf. 2019, 174, 245–254. [Google Scholar] [CrossRef]
- Cheng, P.; Gao, J.; Feng, Y.; Zhang, Z.; Liu, Y.; Fang, W.; Chen, S.; Chen, F.; Jiang, J. The chrysanthemum leaf and root transcript profiling in response to salinity stress. Gene 2018, 674, 161–169. [Google Scholar] [CrossRef]
- Lv, P.-P.; Hu, J.; Chen, S.-L.; Shen, X.; Yin, W.-B.; Chen, Y.-H.; Sun, Y.-R.; Hu, Z.-M. Function of the putative Na+/H+ antiporter gene PeNhaD1 from salt-resistant Populus euphratica Oliv. Zhi wu sheng li yu fen zi sheng wu xue xue bao = J. Plant Physiol. Mol. Boil. 2007, 33, 173–178. [Google Scholar]
- Zhang, A.; Han, D.; Wang, Y.; Mu, H.; Zhang, T.; Yan, X.; Pang, Q. Transcriptomic and proteomic feature of salt stress-regulated network in Jerusalem artichoke (Helianthus tuberosus L.) root based on de novo assembly sequencing analysis. Planta 2017, 247, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Q.; Zhai, H.; Li, Y.; Wang, X.; Liu, Q.; He, S. Transcript profile analysis reveals important roles of jasmonic acid signalling pathway in the response of sweet potato to salt stress. Sci. Rep. 2017, 7, 40819. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Li, W.; Zhang, F.; Guo, W. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Skorupa, M.; Gołębiewski, M.; Domagalski, K.; Kurnik, K.; Abu, N.K.; Złoch, M.; Tretyn, A.; Tyburski, J. Transcriptomic profiling of the salt stress response in excised leaves of the halophyte Beta vulgaris ssp. maritima. Plant Sci. 2016, 243, 56–70. [Google Scholar] [CrossRef]
- Janz, D.; Behnke, K.; Schnitzler, J.-P.; Kanawati, B.; Schmitt-Kopplin, P.; Polle, A. Pathway analysis of the transcriptome and metabolome of salt sensitive and tolerant poplar species reveals evolutionary adaption of stress tolerance mechanisms. BMC Plant Biol. 2010, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Lee, H.; Song, Y.H.; Kim, H. Effect of salt stress on the growth, mineral contents, and metabolite profiles of spinach. J. Sci. Food Agric. 2021, 1. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, X.; Zhao, C.; Fang, Z.; Jiao, P. Transcriptomic and metabolomic analysis reveals the role of CoA in the salt tolerance of Zygophyllum spp. BMC Plant Biol. 2020, 20, 9–14. [Google Scholar] [CrossRef]
- Keller, C.; Maeda, J.; Jayaraman, D.; Chakraborty, S.; Sussman, M.R.; Harris, J.M.; Ané, J.M.; Li, L. Comparison of Vacuum MALDI and AP-MALDI Platforms for the Mass Spectrometry Imaging of Metabolites Involved in Salt Stress in Medicago trun-catula. Front. Plant Sci. 2018, 9, 1238. [Google Scholar] [CrossRef]
- Rouphael, Y.; Raimondi, G.; Lucini, L.; Carillo, P.; Kyriacou, M.C.; Colla, G.; Cirillo, V.; Pannico, A.; El-Nakhel, C.; de Pascale, S. Physiological and Metabolic Responses Triggered by Omeprazole Improve Tomato Plant Tolerance to NaCl Stress. Front. Plant Sci. 2018, 9, 249. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, M.R.; Ghabooli, M.; Khatabi, B.; Hajirezaei, M.R.; Schweizer, P.; Salekdeh, G.H. Metabolic and transcriptional response of central metabolism affected by root endophytic fungus Piriformospora indica under salinity in barley. Plant Mol. Biol. 2016, 90, 699–717. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, D.; Li, M.; Shi, L. Metabolic Profiles Reveal Changes in Wild and Cultivated Soybean Seedling Leaves under Salt Stress. PLoS ONE 2016, 11, e0159622. [Google Scholar] [CrossRef] [Green Version]
- Jorge, T.F.; Duro, N.; da Costa, M.; Florian, A.; Ramalho, J.C.; Ribeiro-Barros, A.I.; Fernie, A.R.; António, C. GC-TOF-MS analysis reveals salt stress-responsive primary metabolites in Casuarina glauca tissues. Metabolomics 2017, 13, 95. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Pieckenstain, F.L.; Escaray, F.; Erban, A.; Kraemer, U.; Udvardi, M.K.; Kopka, J. Comparative ionomics and metabolomics in extremophile and glycophytic Lotus species under salt stress challenge the metabolic pre-adaptation hypothesis. Plant Cell Environ. 2011, 34, 605–617. [Google Scholar] [CrossRef]
- Takahashi, F.; Shinozaki, K. Long distance signalingin plant stress response. Curr. Opin. Plant Biol. 2019, 47, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.; Chauhan, R.; Gill, T.; Swarnkar, M.K.; Sreenivasulu, Y.; Kumar, S.; Kumar, N.; Shankar, R.; Ahuja, P.S.; Singh, A.K. Expression of SOD and APX genes positively regulates secondary cell wall biosynthesis and promotes plant growth and yield in Arabidopsis under salt stress. Plant Mol. Biol. 2015, 87, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.Q.; Brown, C.W.; Pegler, J.L.; Eamens, A.L.; Grof, C.P.L. Molecular Manipulation of MicroRNA397 Abundance Influ-ences the Development and Salt Stress Response of Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 7879. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Mostafa, H.H.A.; Yang, W.; Wang, J.; Nuerawuti, M.; Wang, Y.; Song, J.; Zhang, X.; Ma, L.; Wang, H.; et al. Com-parative transcriptome profiling reveals that brassinosteroid-mediated lignification plays an important role in garlic adaption to salt stress. Plant Physiol. Biochem. 2021, 158, 34–42. [Google Scholar] [CrossRef]
- Duan, A.-Q.; Tao, J.-P.; Jia, L.-L.; Tan, G.-F.; Liu, J.-X.; Li, T.; Chen, L.-Z.; Su, X.-J.; Feng, K.; Xu, Z.-S.; et al. AgNAC1, a celery transcription factor, related to regulation on lignin biosynthesis and salt tolerance. Genome 2020, 112, 5254–5264. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-C.; Kim, Y.-H.; Jeong, J.C.; Kim, C.Y.; Lee, H.-S.; Bang, J.-W.; Kwak, S.-S. Sweet potato late embryogenesis abundant 14 (IbLEA14) gene influences lignification and increases osmotic- and salt stress-tolerance of Transgenic calli. Planta 2010, 233, 621–634. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, K.; Yang, C. BpNAC012 Positively Regulates Abiotic Stress Responses and Secondary Wall Biosynthesis. Plant Physiol. 2019, 179, 700–717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, S.; Li, C.; Zhang, P.; Zhang, P. Transcriptome sequencing of Antarctic moss under salt stress emphasizes the important roles of the ROS-scavenging system. Gene 2019, 696, 122–134. [Google Scholar] [CrossRef]
- Chen, K.; Song, M.; Guo, Y.; Liu, L.; Xue, H.; Dai, H.; Zhang, Z. MdMYB46 could enhance salt and osmotic stress tolerance in apple by directly activating stress-responsive signals. Plant Biotechnol. J. 2019, 17, 2341–2355. [Google Scholar] [CrossRef] [Green Version]
- Onohata, T.; Gomi, K. Overexpression of jasmonate-responsive OsbHLH034 in rice results in the induction of bacterial blight resistance via an increase in lignin biosynthesis. Plant Cell Rep. 2020, 39, 1175–1184. [Google Scholar] [CrossRef]
- Srivastava, S.; Vishwakarma, R.K.; Arafat, Y.A.; Gupta, S.K.; Khan, B.M. Abiotic stress induces change in Cinnamoyl CoA Re-ductase (CCR) protein abundance and lignin deposition in developing seedlings of Leucaena leucocephala. Physiol. Mol. Biol. Plants 2015, 21, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Pi, E.; Zhu, C.; Fan, W.; Huang, Y.; Qu, L.; Li, Y.; Zhao, Q.; Ding, F.; Qiu, L.; Wang, H.; et al. Quantitative Phosphoproteomic and Metabolomic Analyses Reveal GmMYB173 Optimizes Flavonoid Metabolism in Soybean under Salt Stress. Mol. Cell. Proteom. 2018, 17, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.; Li, W.; Niu, C.; Wei, W.; Hu, Y.; Han, J.; Lu, X.; Tao, J.; Jin, M.; Qin, H.; et al. A class B heat shock factor selected for during soybean domestication contributes to salt tolerance by promoting flavonoid biosynthesis. New Phytol. 2019, 225, 268–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, E.; Xu, J.; Li, H.; Fan, W.; Zhu, C.; Zhang, T.; Jiang, J.; He, L.; Lu, H.; Wang, H.; et al. Enhanced Salt Tolerance of Rhizobia-inoculated Soybean Correlates with Decreased Phosphorylation of the Transcription Factor GmMYB183 and Altered Flavonoid Biosynthesis. Mol. Cell. Proteom. 2019, 18, 2225–2243. [Google Scholar] [CrossRef]
- Wang, Q.; Ni, J.; Shah, F.; Liu, W.; Wang, D.; Yao, Y.; Hu, H.; Huang, S.; Hou, J.; Fu, S.; et al. Overexpression of the Stress-Inducible SsMAX2 Promotes Drought and Salt Resistance via the Regulation of Redox Homeostasis in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zhu, H.; Kong, W.; Peng, R.; Liu, Q.; Yao, Q. The Antirrhinum AmDEL gene enhances flavonoids accumulation and salt and drought tolerance in transgenic Arabidopsis. Planta 2016, 244, 59–73. [Google Scholar] [CrossRef]
- Kim, J.; Lee, W.J.; Vu, T.T.; Jeong, C.Y.; Hong, S.-W.; Lee, H. High accumulation of anthocyanins via the ectopic expression of AtDFR confers significant salt stress tolerance in Brassica napus L. Plant Cell Rep. 2017, 36, 1215–1224. [Google Scholar] [CrossRef]
- Zhan, X.; Shen, Q.; Chen, J.; Yang, P.; Wang, X.; Hong, Y. Rice sulfoquinovosyltransferase SQD2.1 mediates flavonoid glyco-sylation and enhances tolerance to osmotic stress. Plant Cell Environ. 2019, 42, 2215–2230. [Google Scholar] [CrossRef]
- Zhang, H.; Du, C.; Wang, Y.; Wang, J.; Zheng, L.; Wang, Y. The Reaumuria trigyna leucoanthocyanidin dioxygenase (RtLDOX) gene complements anthocyanidin synthesis and increases the salt tolerance potential of a transgenic Arabidopsis LDOX mutant. Plant Physiol. Biochem. 2016, 106, 278–287. [Google Scholar] [CrossRef]
- Chen, S.; Wu, F.; Li, Y.; Qian, Y.; Pan, X.; Li, F.; Wang, Y.; Wu, Z.; Fu, C.; Lin, H.; et al. NtMYB4 and NtCHS1 Are Critical Factors in the Regulation of Flavonoid Biosynthesis and Are Involved in Salinity Responsiveness. Front. Plant Sci. 2019, 10, 178. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2012, 8, 17–32. [Google Scholar] [CrossRef]
- Shivakrishna, P.; Reddy, K.A.; Rao, D.M. Effect of PEG-6000 imposed drought stress on RNA content, relative water content (RWC), and chlorophyll content in peanut leaves and roots. Saudi J. Biol. Sci. 2018, 25, 285–289. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in NR | 80,384 | 55.8 |
| Annotated in NT | 72,447 | 50.29 |
| Annotated in KO | 31,769 | 22.05 |
| Annotated in SwissProt | 59,963 | 41.62 |
| Annotated in PFAM | 61,906 | 42.97 |
| Annotated in GO | 61,901 | 42.97 |
| Annotated in KOG | 27,796 | 19.29 |
| Annotated in all Databases | 12,155 | 8.43 |
| Annotated in at least one Database | 105,318 | 73.11 |
| Total Unigenes | 144,051 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Wang, Q.; Wang, Y.; Xu, Y.; Li, J.; Zhao, S.; Wang, D.; Ma, Z.; Yan, F.; Liu, Y. Combined Transcriptomic and Metabolomic Analysis Reveals the Role of Phenylpropanoid Biosynthesis Pathway in the Salt Tolerance Process of Sophora alopecuroides. Int. J. Mol. Sci. 2021, 22, 2399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052399
Zhu Y, Wang Q, Wang Y, Xu Y, Li J, Zhao S, Wang D, Ma Z, Yan F, Liu Y. Combined Transcriptomic and Metabolomic Analysis Reveals the Role of Phenylpropanoid Biosynthesis Pathway in the Salt Tolerance Process of Sophora alopecuroides. International Journal of Molecular Sciences. 2021; 22(5):2399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052399
Chicago/Turabian StyleZhu, Youcheng, Qingyu Wang, Ying Wang, Yang Xu, Jingwen Li, Shihui Zhao, Doudou Wang, Zhipeng Ma, Fan Yan, and Yajing Liu. 2021. "Combined Transcriptomic and Metabolomic Analysis Reveals the Role of Phenylpropanoid Biosynthesis Pathway in the Salt Tolerance Process of Sophora alopecuroides" International Journal of Molecular Sciences 22, no. 5: 2399. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052399