
Mitochondrial Calcium Signaling in Pancreatic β-Cell


1
Nestlé Research—EPFL Innovation Park, CH-1015 Lausanne, Switzerland
2
Molecular Nutritional Medicine, Else Kröner Fresenius Center for Nutritional Medicine, Technische Universität München, 85354 Freising, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(5), 2515; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052515
Submission received: 1 February 2021
/
Revised: 22 February 2021
/
Accepted: 26 February 2021
/
Published: 3 March 2021
(This article belongs to the Special Issue Mitochondrial Calcium Signaling)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Accumulation of calcium in energized mitochondria of pancreatic β-cells is emerging as a crucial process for pancreatic β-cell function. β-cell mitochondria sense and shape calcium signals, linking the metabolism of glucose and other secretagogues to the generation of signals that promote insulin secretion during nutrient stimulation. Here, we describe the role of mitochondrial calcium signaling in pancreatic β-cell function. We report the latest pharmacological and genetic findings, including the first mitochondrial calcium-targeted intervention strategies developed to modulate pancreatic β-cell function and their potential relevance in the context of diabetes.
1. Introduction
Pancreatic β-cells constitute a small endocrine tissue organized, together with other endocrine cells, in the islets of Langerhans, scattered throughout the exocrine tissue of the pancreas [1]. β-cells sense glucose and secrete insulin in order to lower blood glucose levels after a meal. Defective insulin secretion underlies diabetes mellitus, which is a metabolic disorder characterized by elevated blood glucose levels [2,3]. The WHO’s first global report on diabetes indicates that the number of adults living with diabetes has almost quadrupled since 1980 to 422 million adults. This dramatic increase is largely due to the rise in Type 2 diabetes, whose driving factors include overweight and obesity. This disease develops when the β-cells of the endocrine pancreas fail to secrete sufficient hormones to compensate for the insulin resistance in the peripheral target tissues, liver, muscle and fat [4]. Diabetes is a non-communicable disease for which new approaches to prevention and treatment urgently need to be found. Targeting pancreatic β-cells is a promising strategy for the treatment of diabetes, due to the crucial role of the pancreatic β-cell in the pathogenesis of both Type 1 and Type 2 diabetes [5]. Therefore, preservation, expansion or improved function of β-cells are current approaches for targeting this cell type in the management of diabetes. Modulation of the biological pathways that regulate β-cell function represents the next stage of discovery in this field [5].
In this context, targeting mitochondrial Ca2+ represents an innovative approach to modulate β-cell function and to potentially promote beneficial effects for diabetic patients. Thus, dysregulation of Ca2+ signaling has been reported to have profound effects on β-cell performance and to increase the risk of developing diabetes [6,7]. Furthermore, modulation of dynamic cellular Ca2+ homeostasis has been proposed to prevent cytokine-mediated β-cell loss in diabetes [8]. In the pancreatic β-cell, Ca2+ homeostasis and β-cell function are substantially linked to mitochondrial function [9]. Therefore, mitochondria play a key role in β-cells during nutrient stimulation by linking the metabolism of glucose and other secretagogues to the generation of signals that promote insulin secretion [9]. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells [10]. Mitochondria are versatile intracellular organelles that are able to take up and release calcium [11,12]. Mitochondrial matrix Ca2+ is an activating signal for insulin secretion, and its requirements for signal-dependent hormone secretion have been highlighted [13]. Recently, the molecular identity of the mitochondrial Ca2+ uniporter (MCU), the transporter that mediates mitochondrial calcium uptake, has been revealed [14]. Genetic and pharmacological evidence has demonstrated the crucial role of mitochondrial Ca2+ in modulating pancreatic β-cell signal transduction, opening new perspectives for intervention [15,16,17].
Excellent reviews on the bioenergetic role of mitochondria and mitochondrial Ca2+ in metabolism–secretion coupling in the pancreatic β-cell are available [9,18,19], including the control of mitochondrial structure and function by calcium [20]. The role of mitochondrial ion channels in the pathophysiology of the pancreatic β-cell has also been described recently [21]. In this paper, we focus on the role of mitochondrial Ca2+ in pancreatic β-cell signal transduction. We report the latest pharmacological and genetic evidence, including the first intervention strategy targeting mitochondrial Ca2+ in the β-cell.
2. Pancreatic β-Cell Signal Transduction and Ca2+ Homeostasis
In the pancreatic β-cell, metabolism–secretion coupling describes the molecular mechanism linking nutrient sensing and signaling to insulin secretion. This process relates to the consensus model and additional coupling factors (including both triggering and amplifying pathways) of glucose-stimulated insulin secretion [22,23]. Glucose-stimulated insulin secretion is relatively well characterized and requires the sequential activation of several biological processes (Figure 1).
Glucose enters the β-cell by glucose-mediated transporters (GLUT). In the cytosol, it is metabolized by glycolysis to generate pyruvate, which is taken up by mitochondria. Mitochondrial pyruvate is metabolized by the tricarboxylic acid (TCA) cycle, which generates reducing equivalents NADH (reduced nicotinamide adenine dinucleotide) and FADH2 (reduced flavin adenine dinucleotide), which are substrates of the mitochondrial respiratory chain. Activation of mitochondrial respiration leads to mitochondrial ATP synthesis and thus to an increased cytosolic ATP-to-ADP ratio, which induces the closure of plasma membrane KATP channels and promotes plasma membrane depolarization. This opens voltage-gated plasma membrane Ca2+ channels, leading to an increase in cytosolic Ca2+ concentration, which finally triggers insulin exocytosis by activating Ca2+-sensitive granule-resident proteins (e.g., synaptotagmin-7 [24]). The amplifying pathways of metabolism–secretion coupling are contributed by additive coupling factors, and mitochondria have been characterized as a source of coupling factors [9,22].
The coupling between nutrient stimulation and hormone secretion is closely linked to Ca2+ homeostasis in the pancreatic β-cell [7,25,26,27]. Therefore, insulin secretion is driven by electrical activity and oscillations of intracellular Ca2+ concentrations. However, in addition to KATP channels and voltage-dependent Ca2+ channels [28], other plasma membrane channels and intracellular stores have been shown to be involved in insulin secretion in pancreatic β-cells [26,29]. In particular, store-operated Ca2+ channels, which are voltage-independent Ca2+ channels activated upon depletion of the endoplasmic reticulum Ca2+ stores, and transient receptor potential channel 1 (TRPC1), have been indicated to be involved in insulin secretion [30,31]. For store-operated Ca2+ channels, Orai1 has been identified as the main protein that conducts the previously described Ca2+ release-activated current (ICRAC). The activity of Orai1 channels is tightly controlled by the endoplasmic reticulum membrane protein stromal interacting molecule 1 (STIM1), which acts as an endoplasmic reticulum Ca2+ sensor and translocates, upon endoplasmic reticulum depletion, to endoplasmic reticulum/plasma membrane regions, where Orai1 is clustered [31]. In addition, mobilization of intracellular Ca2+ from the endoplasmic reticulum has been suggested to potentiate glucose-stimulated hormone secretion [32], and the Type 2 ryanodine receptor (RyR2) has been proposed to play a crucial role in regulating insulin secretion and glucose homeostasis [33]. Moreover, an atypical Ca2+ leak has been observed in the endoplasmic reticulum, specifically in pancreatic islets and β-cells. This continuous Ca2+ efflux from the endoplasmic reticulum was modulated by GSK3β-dependent phosphorylation of presenilin-1 and promoted mitochondrial activation [34,35]. Additional intracellular acidic compartments may contribute to the local modulation of β-cell Ca2+ homeostasis and thus β-cell function, including insulin granules and other acidic stores (e.g., lysosomes) [26].
The last player in pancreatic β-cell Ca2+ homeostasis and signal transduction is represented by the mitochondrial network. Therefore, mitochondria are intracellular organelles that take up and release Ca2+, promoting the sensing and shaping of cytosolic Ca2+ signals [36]. The role of mitochondrial Ca2+ signaling in energized mitochondria of the pancreatic β-cell is emerging as a biological process of critical importance to pancreatic β-cell function and is highlighted in the next section.
3. The Mitochondrial Calcium Uniporter and Its Existence in Pancreatic β-Cells
Despite the undisputed role of cytosolic Ca2+ elevation in triggering insulin secretion in the pancreatic β-cell, it is also accepted that such a rise in itself does not sustain insulin secretion [18,37,38]. Therefore, mitochondria have been demonstrated to contribute to robust insulin secretion by triggering additional regulatory factors, and it has been proposed that mitochondrial Ca2+ plays a crucial role as a receiver and generator of the signals essential for metabolism–secretion coupling [9,39]. The discovery and molecular definition of the MCU [14,40], the transporter which mediates the transport of Ca2+ in the mitochondrial matrix under physiological conditions, is shedding light on the role of mitochondrial Ca2+ elevation in different tissues, including the pancreatic β-cell (see Section 4).
The MCU complex is a low-affinity, high-capacity Ca2+ uniporter embedded in the inner mitochondrial membrane with an approximate in vitro Ca2+ binding affinity estimated to be between 10 and 70 µM [41,42,43,44] in different tissues. The transport of Ca2+ ions into the mitochondrial matrix is electrochemically driven by a strong electrical gradient (~180 mV) through the ion-impermeable inner mitochondrial membrane [45]. As reported for many other tissues, in the pancreatic β-cell, resting-state mitochondrial Ca2+ levels are also very close to cytosolic Ca2+ levels, indicating tight regulation of the MCU [18]. Permeabilized INS-1 cells (a cell-line model of pancreatic insulin-secreting cells) display significant mitochondrial Ca2+ uptake when perfused with buffers containing a Ca2+ concentration of only 150 nM [46], which is below the general threshold of MCU activation [47]. This study suggests a slightly higher affinity configuration of the β-cell MCU, compared with the MCU of other tissues. However, in permeabilized β-cells perfused with a range of different Ca2+ buffers, mitochondrial Ca2+ uptake was more efficient above 2 µM [48]. These data indicate that although some studies demonstrated a lower Ca2+ activation threshold, the MCU of β-cells also behaves as a low affinity, high capacity Ca2+ transport system, with similar properties to those reported in other tissues. In any case, the mitochondrial Ca2+ concentration in glucose-stimulated β-cells was reported to reach only 600–800 nM [13,18,49], mirroring cytosolic Ca2+ events. Purinergic activation and potassium-induced depolarization generate cytosolic Ca2+ transients around one micromolar, with the corresponding mitochondrial Ca2+ rises reaching nearly 5 µM [39]. These results indicate that, on average, β-cell mitochondria barely reach micromolar Ca2+ concentrations in the matrix during physiological activation.
The MCU complex consists of at least six subunits, each of which plays an individual role in orchestrating mitochondrial Ca2+ uptake [41]. The MCU subunit is a 40 kDa protein that, together with its paralog, MCUb [50], has been proposed to form a tetramerizing pore which penetrates the inner mitochondrial membrane [51]. In order to channel Ca2+ ions into the mitochondrial matrix, it requires a third subunit known as the essential MCU regulator (EMRE), a 10 kDa protein [50,52]. EMRE is a single-transmembrane protein whose transmembrane helix connects it to the MCU subunit [53]. The MCU and MCUb share 50% sequence similarity and have opposite effects on mitochondrial Ca2+ uptake [50]. While the MCU has a promoting effect on mitochondrial Ca2+ uptake, MCUb exerts a negative effect on mitochondrial Ca2+ uptake [54]. The ratio of MCU and MCUb varies among tissues and is potentially influenced by metabolic impairments, leading to the hypothesis that an altered MCU/MCUb ratio may affect β-cell function [55,56,57]. When Ca2+ in the intermembrane space exceeds ~0.6 µM, it initiates activation of the MCU complex via the MCU gatekeeper protein paralogs mitochondrial calcium uptake 1 and 2 (MICU1 and MICU2) [44,58]. MICU2 is a 50 kDa protein and shares approximately 25% sequence identity with the 54 kDa protein MICU1 [42]. Neither MICU1 nor MICU2 contain transmembrane domains that would link them to the MCU subunit, so it has been proposed that MICU1 is linked to the MCU via EMRE through electrostatic interaction [58]. However, a more recent study revealed an additional direct binding of MICU1 to the highly conserved DIME motif of the MCU [59,60]. In addition, another study showed an interaction between MICU1/MICU2 and cardiolipin [58]. Through cysteine residues, MICU2 forms an intermembrane space-facing heterodimer with MICU1 via disulfide bonds [51]. Both gatekeeper proteins contain EF-hand motifs (consisting of a helix E, a loop and another helix F), with a Ca2+ binding affinity of ~0.3 µM for MICU1 and a Ca2+ binding affinity of ~0.6 µM for the MICU1–MICU2 dimer [58].
Genetic ablation of MICU2 lowers the required Ca2+ concentration in the intermembrane space for MICU1-regulated MCU activation, resulting in increased mitochondrial Ca2+ uptake [52]. On the other hand, ablation of MICU1 prevents indirect binding of MICU2 to the MCU, resulting in unregulated Ca2+ entry into the mitochondria [61,62]. Studies with MICU1 knockout as well as with MICU1 knockdown have described an inverse relationship between extra-mitochondrial Ca2+ concentrations and mitochondrial Ca2+ uptake [62,63]. This suggests that the absence of MICU1 at low/resting extra-mitochondrial Ca2+ concentrations leads to increased mitochondrial Ca2+ uptake, while high extra-mitochondrial Ca2+ concentrations are associated with decreased mitochondrial Ca2+ uptake. The final subunit, which has been proposed to contribute to the structure of the MCU complex is a scaffolding factor called mitochondrial calcium uniporter regulator 1 (MCUR1), which can interact with the MCU but not with MICU1 [64]. However, there are two possible interpretations of the direct or indirect role of MCUR1 in modulating mitochondrial Ca2+ uptake. Several studies have demonstrated a regulatory effect of MCUR1 on MCU complex activity, noting reduced mitochondrial Ca2+ uptake in the absence of MCUR1 and a decrease in reducing equivalents as well as ATP generation [65,66,67]. Alternatively, MCUR1 has been suggested to have a potential role as a Complex IV assembly factor in the mitochondrial respiratory chain [68]. Interestingly, the relative abundance of distinct MCU components has been reported to be different in distinct cell types or tissues [35], and it has been proposed that the specific stoichiometry of the MCU subunits may define the functional characteristics of the channel, including Ca2+ permeation across the pore, the activation threshold and the cooperativity. Consistent with this hypothesis, it has been reported that in parallel with the distinct relative abundance of MCU components [35], MCU activity also varies greatly among cell types [69]. Although the stoichiometry of the different MCU subunits and their relative abundance in different tissues, including in the pancreatic β-cell, are largely unknown, we tentatively speculate that these cells may have a specific configuration of the MCU that reduces the Ca2+ activation threshold.
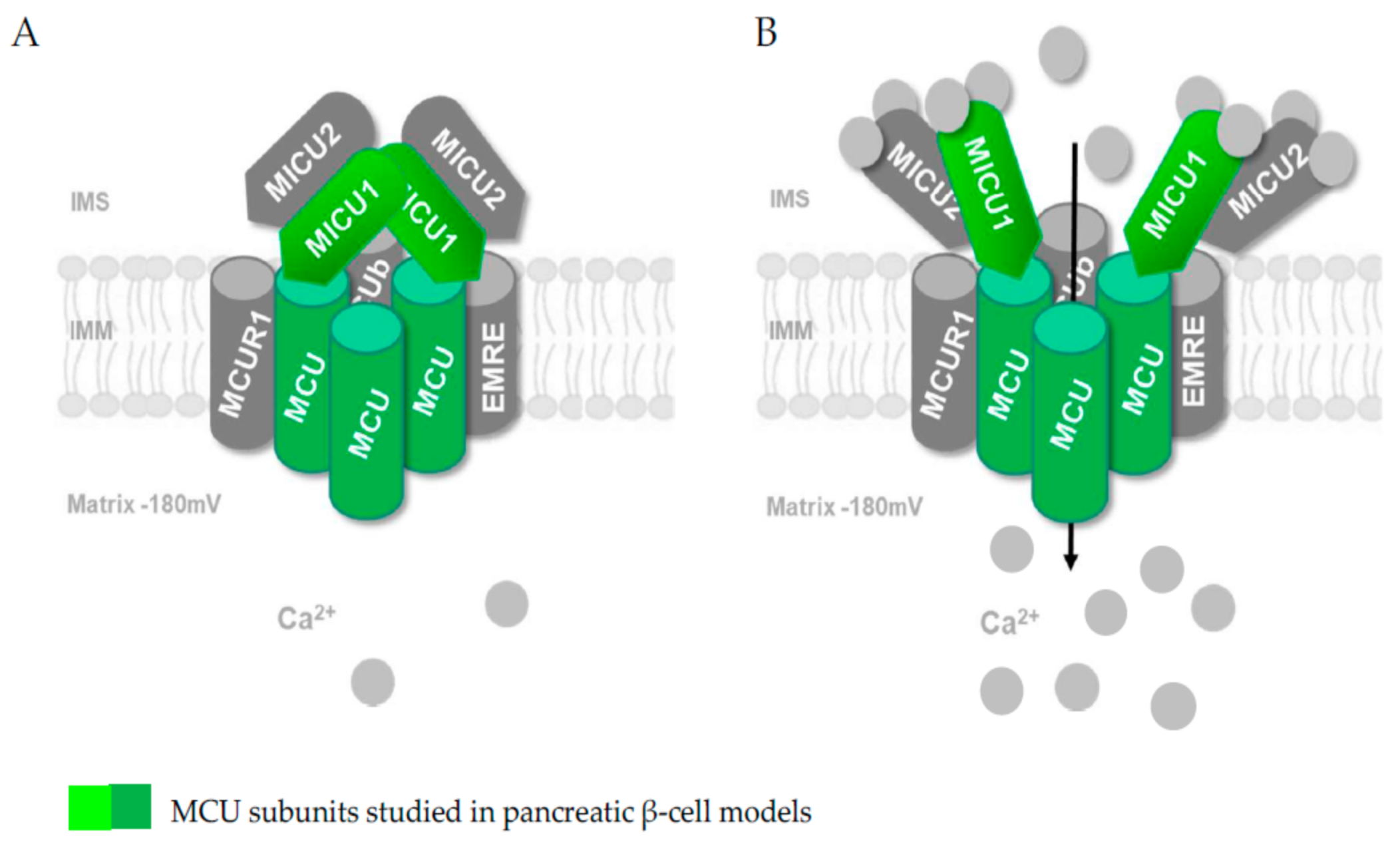
Pancreatic β-cells have been demonstrated to express a functional MCU, and some studies have investigated the role of MCU subunits on insulin secretion [15,16,17] (Figure 2).
Not surprisingly, the existence of MCU and MICU1 subunits in pancreatic β-cell models has been reported and functionally validated (see Section 4) [15,16,17,55]. In addition, a study recently published as a preprint in bioRxiv but not yet peer-reviewed proposes the existence of MICU2 subunits in rat and human insulin-secreting cell lines as well as in mouse islets (Mitochondrial Clearance of Ca2+ Controls Insulin Secretion; https://0-doi-org.brum.beds.ac.uk/10.1101/830323 (accessed on 1 February 2021); Vishnu, Hamilton, Bagge, Wernersson, Cowan, Barnard, Sancak, Kamer, Spégel, Fex, Tengholm, Mootha, Nicholls and Mulder). Further studies are expected to consolidate the specific role of the other subunits of the MCU in the pancreatic β-cell (Figure 2) and fully define the structure of this transporter. However, the crucial role of the MCU complex in the mitochondrial Ca2+ transport of the pancreatic β-cell and in the pancreatic β-cell metabolism–secretion coupling is already well established and will be discussed in the next section.
4. Role of Mitochondrial Ca2+ Uptake in the Pancreatic β-Cell
The importance of mitochondrial Ca2+ in pancreatic β-cell function was efficiently highlighted by modulating mitochondrial Ca2+ via increased matrix Ca2+ buffering capacity, achieved by targeting exogenous Ca2+-binding proteins into the matrix [13] or by loading the cells with the Ca2+ chelator BAPTA (1, 2-bis(2-aminophenoxy) ethane-N, N, N′, N′-tetraacetic acid) [70]. The impaired bioenergetic response to elevated glucose recorded in these two studies revealed the physiological importance of mitochondrial Ca2+ in energy metabolism required for signal-dependent hormone secretion. The discovery of the molecular nature of the mitochondrial Ca2+ uniporter [14,40] then enabled clarification of the role of mitochondrial Ca2+ in pancreatic β-cell function and opened new perspectives for pharmacological and nutritional interventions. In murine pancreatic β-cells, knockdown of MCU showed a strong reduction in mitochondrial Ca2+ uptake, accompanied by impaired ATP production [17], consistent with a role of mitochondrial Ca2+ in metabolism–secretion coupling. Similar results were obtained in pancreatic β-cell lines (INS-1 and INS-1E cells). Genetic depletion of two subunits of the MCU complex (MCU and MICU1) was characterized by reduced hyperpolarization of the inner mitochondrial membrane, diminished mitochondrial Ca2+ transients, impaired respiration rate and ATP levels, and reduced insulin secretion during glucose stimulation [15,16]. Taken together, these results demonstrate the importance of mitochondrial Ca2+ for signal transduction in β-cells and the impact of the MCU on signal-dependent hormone secretion.
The crucial role of mitochondrial Ca2+ in glucose-induced insulin secretion was finally validated by Georgiadou and collaborators in a mouse model, in which MCU was highly selectively deleted in pancreatic β-cells [55]. In this paper, the authors showed that mitochondrial Ca2+ uptake, glucose-induced ATP production and insulin secretion were substantially impaired in their animal model in vitro. In the living β-cell-specific MCU-null mice, the first phase of insulin release was also impaired, despite paradoxical improvements in systemic glucose tolerance. Despite the apparent compensatory mechanisms proposed to explain the maintained glucose tolerance in β-cell-specific MCU-null mice (which remain to be established), this study indicated that agents affecting mitochondrial Ca2+ uptake in the pancreatic β-cell may alter insulin secretion and diabetes risk.
The molecular characterization of the MCU is shedding light on the role of this transporter in pancreatic β-cell function; however, the precise mechanism and contribution of mitochondrial Ca2+ to the regulation of insulin secretion is not fully understood. Consistent with the universal role of mitochondrial Ca2+ in the regulation of cellular energetics [12,71], an essential role of mitochondrial Ca2+ for effective ATP generation in pancreatic β-cells is well established [18]. Mitochondrial Ca2+ is required for the activation of matrix dehydrogenases involved in pyruvate metabolism and the TCA cycle [18,72]. Mitochondrial Ca2+-activated dehydrogenases form reducing equivalents in the mitochondrial matrix, promoting the reduction of mitochondrial redox pairs. Most notably, studies in Bristol in the 1960s and 1970s led to the recognition that mitochondrial Ca2+ promotes the supply of reducing equivalents in the form of NADH or FADH2 [73,74,75]. Four Ca2+-activated mitochondrial dehydrogenases were specifically described: FAD-glycerol phosphate dehydrogenase (located on the outer surface of the inner mitochondrial membrane and influenced by changes in cytoplasmic Ca2+ concentration), pyruvate dehydrogenase, NAD-isocitrate dehydrogenase and oxoglutarate dehydrogenase (the latter three are localized within the mitochondria and are regulated by changes in mitochondrial matrix Ca2+ concentration). Following early studies with isolated mitochondria, the effects on Ca2+ regulation of mitochondrial metabolism were confirmed in situ [71]. In the pancreatic β-cell, the mitochondrial Ca2+ level reached during glucose-induced cell activation (see Section 3) was sufficient to stimulate matrix dehydrogenases. Therefore, the K0.5 of pyruvate dehydrogenase for Ca2+ activation is about 1 µM and the K0.5 of oxoglutarate dehydrogenase is in the range of 0.2 to 2 µM, depending on the ATP/ADP levels [76]. In this context, we proposed to extend this bioenergetics and dehydrogenase-dependent role of mitochondrial Ca2+ by discovering the significant contribution of the ATP-synthase-dependent mitochondrial respiration [77]. According to our model, in the pancreatic β-cell, the cytosolic Ca2+ rise during glucose stimulation affects mitochondrial activity. Mitochondria take up and release Ca2+ ions, leading to a transient increase in matrix Ca2+. These mitochondrial Ca2+ signals accelerate oxidative metabolism and simultaneously stimulate ATP-synthase-dependent respiration. Coordinated activation of these two processes allows the respiratory rate to change several-fold with only small alterations in the NAD(P)H:NAD(P)+ ratio (the ratio between the reduced and oxidized form of the nicotinamide adenine dinucleotide, or of the nicotinamide adenine dinucleotide phosphate), promoting robust insulin secretion [77]. Consistent with the proposition that mitochondrial Ca2+ uptake is a critical event in cellular bioenergetics and thus in the metabolic coupling of insulin, specific buffering or suppression of the mitochondrial Ca2+ increase lowers glucose-induced respiration and ATP synthesis, and impairs second-phase insulin secretion [13,17].
An additional mechanism linking mitochondrial Ca2+ to signal transduction in β-cells is related to the ability of mitochondria to take up and buffer intracellular Ca2+ and to affect the tone and frequency of cytosolic Ca2+ oscillations [18]. This regulation may contribute to control the pulsatility of insulin release [78]. The ability of mitochondria to buffer cytosolic Ca2+ has also recently been linked to the regulation of the insulin secretion through local Ca2+ buffering at the sub-plasma membrane level [79]. In this study, linking mitochondrial localization with mitochondrial Ca2+ and β-cell function, the authors proposed that changes in mitochondrial distribution may be important for the generation of the Ca2+ microdomains required for efficient insulin granule release. The precise contribution of mitochondrial Ca2+ rise to the shaping of cytosolic Ca2+ signaling is not completely established in β-cells. However, the aforementioned preprint in bioRxiv, which has not yet been peer-reviewed, (Mitochondrial Clearance of Ca2+ Controls Insulin Secretion; https://0-doi-org.brum.beds.ac.uk/10.1101/830323; Vishnu, Hamilton, Bagge, Wernersson, Cowan, Barnard, Sancak, Kamer, Spégel, Fex, Tengholm, Mootha, Nicholls and Mulder), seems to shed some light on this function. Thus, by ablating MICU2 subunits of the MCU complex in insulin-secreting rat and human cell lines as well as in mouse islets, the authors reported reduced glucose-induced mitochondrial Ca2+ elevation and impaired bioenergetics and insulin secretion. In MICU2-deficient cells, although KCl-evoked sub-plasmalemmal Ca2+ increases were more pronounced, the global cytosolic Ca2+ response was surprisingly reduced. The authors concluded that MCU plays a role in stimulated β-cells by regulating net Ca2+ entry across the plasma membrane. They proposed that this was linked to the clearing of sub-plasmalemmal Ca2+ levels by mitochondria located near the plasma membrane.
In addition to the regulation of bioenergetics and the modulation of cytosolic Ca2+, mitochondrial Ca2+ has also been proposed to modulate β-cell function, linked to the activation of the mitochondrial permeability transition pore (PTP) [80]. The PTP is a large mitochondrial inner membrane channel responsible for the so-called mitochondrial permeability transition, which is a mitochondrial Ca2+-dependent, redox-dependent and cyclosporine-A-inhibited permeabilization of the inner mitochondrial membrane [81,82]. The opening of the PTP plays an important role in the physiopathology of several tissues, including pancreatic β-cells [83,84,85,86,87]. Given the crucial role of the PTP in cellular physiopathology and its mitochondrial Ca2+-dependence [88], the modulation of this channel provides an additional link between mitochondrial Ca2+ signaling and the function and fate of pancreatic β-cells.
The molecular identity of the PTP is still a matter of debate [81,89,90,91]; however, the protein cyclophilin D has been demonstrated to be a regulator of the PTP [92,93]. Therefore, the PTP inhibitor cyclosporine-A prevented PTP activation in wild-type mice but not in cyclophilin D knockout animals, demonstrating that cyclophilin D is a regulatory subunit of the PTP and represents the target for PTP inhibition by cyclosporine A. Although the specificity of cyclosporine A has been criticized, several pharmacological cyclosporine A-based studies in pancreatic β-cells proposed the existence of the mitochondrial permeability transition in this cell type [85,87,94]. These manuscripts emphasized the importance of the PTP for the secretory function of β-cells [83,85], and as a common effector of both apoptosis and necrosis [94,95].
In two semi-permeabilized pancreatic β-cell lines (MIN6 and INS-1), the existence of both a mitochondrial Ca2+-induced and thiol cross-linking-dependent, and a mitochondrial Ca2+-independent mitochondrial permeability transition has been reported [85]. Inhibition of PTP opening with cyclosporine A suppressed glucose-induced insulin secretion [83,85]. In another study, cyclosporine A decreased Pdx1 deficiency-induced cell death in mouse insulinoma MIN6 cells by preventing PTP opening [84]. These results were confirmed in a genetic mouse model after ablation of Ppif, the gene encoding cyclophilin D. Disruption of this gene restored β-cell mass, reduced β-cell death, and normalized fasting blood glucose and glucose and insulin responses to an acute glucose challenge in adult mice previously kept on a high-fat diet [84]. The authors concluded that the PTP is a critical regulator of pancreatic β-cell death. Another study showed that cyclosporine A prevented PTP opening and protected against both high-dose glucose- and fructose-induced cell death [96]. In addition, it has been suggested that pharmacological inhibition of the PTP during islet transplantation may improve islet cell survival and graft success [86].
In summary, the mitochondrial Ca2+-induced PTP may play two distinct roles in the physiology and pathology of the pancreatic β-cell. While activation of the PTP is required to promote insulin secretion upon glucose stimulation [83,85], PTP inhibition protects against glucotoxicity as well as hypoxia and substrate deficiency during islet transplantation [86,96]. Given the dependence of the PTP on mitochondrial Ca2+ levels, MCU activators are expected to promote insulin secretion; conversely, MCU inhibitors are potential agents to protect β-cells during islet transplantation or nutrient stress in vivo [97]. Importantly, a large number of inhibitors of mitochondrial Ca2+ transport have been synthesized recently [97,98,99,100]. However, MCU inhibitors can non-specifically affect not only different cellular transport systems of Ca2+ but also various systems in the mitochondria. For instance, the new penetrating inhibitor of the Ca2+ uniporter, DS16570511, has multiple effects on the mitochondria of cerebral cortex cells, while stimulating cellular survival during glutamate overload [101]. Thus, a comprehensive analysis of the mechanisms of action of mitochondrial Ca2+ transport modulators is crucial to conclusively establish the potential effects of these compounds on pancreatic β-cell function.
5. Mitochondrial Ca2+ Extrusion in the Pancreatic β-Cell
During cell stimulation, the amplitude and duration of mitochondrial Ca2+ elevation reflect the balance between uptake and release mechanisms [11,102,103]. Many studies have focused on MCU-mediated mitochondrial Ca2+ uptake in different systems, including the pancreatic β-cell. However, mitochondrial Ca2+ extrusion has also been demonstrated to play a key role in cellular physiopathology and signaling [64,102,104]. Therefore, prolonged (pathological) accumulation of Ca2+ in the matrix space can lead to mitochondrial Ca2+ overload, followed by opening of the mitochondrial permeability transition pore [88,89], resulting in the activation of cell death signals. To avoid this transition from the stimulatory to detrimental effects of Ca2+, mitochondria possess two membrane systems to extrude Ca2+: the mitochondrial Na+/Ca2+ exchanger and the mitochondrial H+/Ca2+ exchanger [64,102,104,105].
The molecular identity of the mitochondrial Na+/Ca2+ exchanger was revealed in 2010 and attributed to the protein NCLX [104]; subsequently, the impact of NCLX on pancreatic β-cell function was investigated [106]. The existence and role of a mitochondrial Ca2+/H+ exchanger in the pancreatic β-cell is a more complicated topic. Therefore, the protein LETM1 was proposed as a high-affinity mitochondrial Ca2+/H+ exchanger, capable of driving both the extrusion and uptake of Ca2+ into energized mitochondria at sub-micromolar Ca2+ concentrations [107,108]. Interestingly, LETM1 is expressed in the β-cell line INS-1 and it has been proposed to contribute to mitochondrial Ca2+ sequestration (but not extrusion), depending on the source and mode of mobilized Ca2+ [15]. Therefore, mitochondrial Ca2+ sequestration of Ca2+ that entered the INS-1 cells via depolarization-activated L-type Ca2+ channels of the plasma membrane was blunted by diminution of LETM1 expression [15]. However, silencing LETM1 in INS-1 cells did not attenuate mitochondrial sequestration of intracellularly released Ca2+. The authors concluded that these data point to LETM1 as a high-affinity Ca2+ carrier [15]. However, LETM1 was previously indicated to mediate K+/H+ exchange in the mitochondrial inner membrane [109,110]. In addition, exogenous LETM1 expression led to a direct increase in K+-induced proton extrusion, whereas mitochondrial Ca2+ efflux was not altered [64]. Other studies support a role of LETM1 as a K+/H+ exchanger [111,112]. Moreover, they indicate a key role of LETM1 in monovalent cation homeostasis and suggest an indirect effect of LETM1 on the modulation of mitochondrial transmembrane Ca2+ fluxes, which can reflect the effects on Na+/H+ exchange activity [111]. In summary, the existence and role of a mitochondrial Ca2+/H+ exchanger in the pancreatic β-cell remains to be elucidated, and additional studies are needed to reach a conclusion.
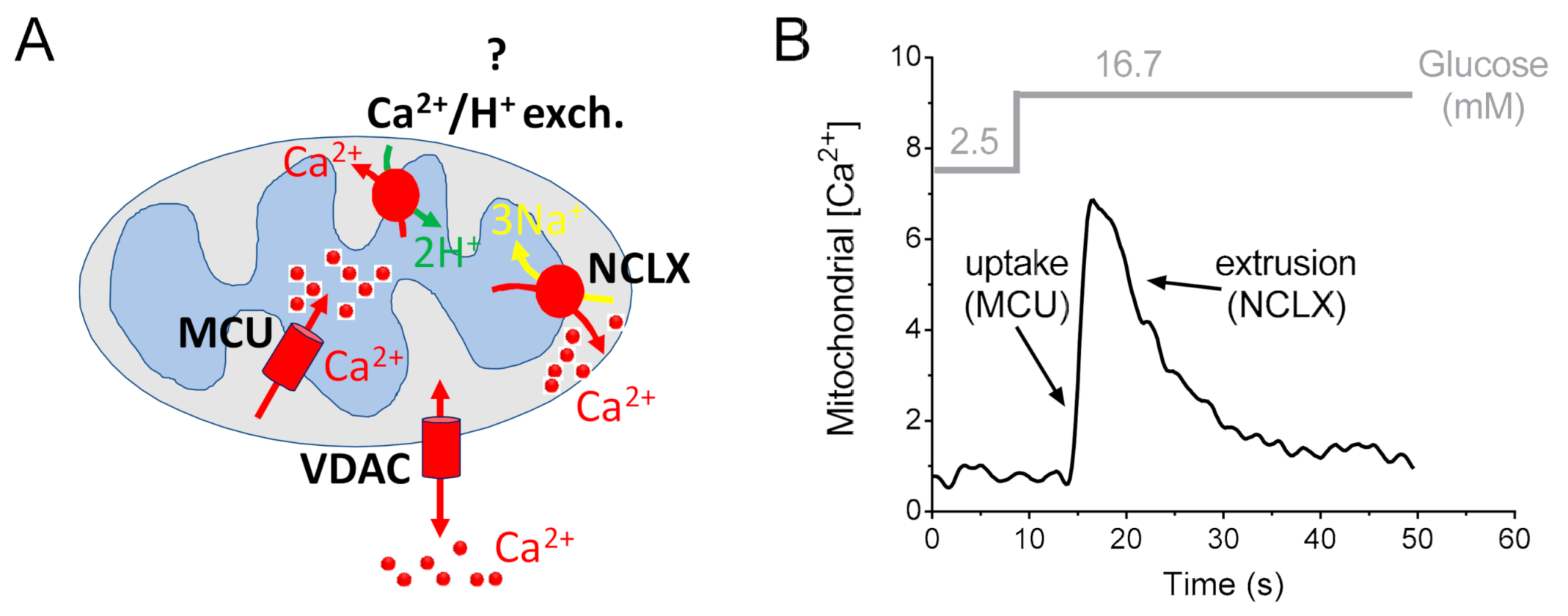
Conversely, convincing data on the molecular identity and function of the mitochondrial Na+/Ca2+ exchanger in the pancreatic β-cell have been reported (Figure 3).
Prior to the discovery of the molecular identity of the mitochondrial Na+/Ca2+ exchanger NCLX, previous studies used the mitochondrial Ca2+ exchanger inhibitor CGP-37157 to inhibit this transporter [114]. This treatment resulted in enhanced mitochondrial oxidative metabolism, ATP production and insulin secretion in rat pancreatic islets [114]. In contrast, other observations suggested that the use of the inhibitor CGP-37157 did not lead to enhanced glucose-dependent ATP production [115]. The major complication is that the CGP-37157 inhibitor is not specific and may interact with other Ca2+ transport pathways in the β-cell, including cytosolic Ca2+ signaling (by blocking the L-type Ca2+ channels, LTCC) [116], sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA pumps) and ryanodine receptors (RyR) [117].
The molecular discovery of NCLX allowed us to highlight the importance of mitochondrial extrusion in different tissues [64,104,105], including the pancreatic β-cell [106,118]. NCLX is expressed in the mitochondria of pancreatic β-cells, where it mediates mitochondrial Ca2+ extrusion [106,118]. By silencing NCLX using small interfering RNA, Nita and collaborators demonstrated that NCLX promotes mitochondrial Ca2+ extrusion from β-cells in MIN6 and in primary β-cells after glucose stimulation, and modulates both basal mitochondrial membrane potential and resting calcium levels. In addition, NCLX activity plays a major role in controlling both the rate and amplitude of cytosolic Ca2+ responses [106]. Surprisingly, NCLX had a small effect on high glucose-dependent ATP production but primarily regulated the rate of glucose-dependent insulin secretion, particularly during the first phase of insulin secretion [106]. Thus, the authors argued against a major energetic role for the exchanger in Ca2+ signaling linked to insulin secretion. They proposed that the Ca2+ transport activity mediated by NCLX and its strong effect on increasing cytosolic Ca2+ responses are the primary roles of NCLX in the context of insulin secretion. They suggested a model of mitochondrial Ca2+ efflux that modulates pancreatic β-cell function via NCLX-dependent shaping of the cytosolic glucose-dependent Ca2+ response, which may regulate the rate of insulin secretion. However, further in vivo studies with transgenic NCLX knockout mice are required to determine the precise contribution of mitochondrial Ca2+ extrusion to β-cell bioenergetics, Ca2+ homeostasis and islet physiology.
6. Mitochondrial Ca2+-Targeted Intervention Strategies to Modulate Pancreatic β-Cell Function
Given the importance of mitochondrial Ca2+ for pancreatic β-cell signal transduction and the possibility of modulating mitochondrial Ca2+ with natural bioactives [119], we recently investigated the effects of a mitochondrial Ca2+-targeted nutritional intervention strategy on metabolism–secretion coupling in a model of pancreatic insulin-secreting cells (INS-1E) [45]. We discovered that acute treatment of pancreatic INS-1E cells with the natural plant flavonoid and MCU activator kaempferol [119] increased glucose-stimulated mitochondrial Ca2+ elevation, which potentiated insulin secretion. Conversely, the MCU inhibitor mitoxantrone inhibited mitochondrial Ca2+ uptake and prevented both glucose-induced insulin secretion and kaempferol-potentiated effects. Kaempferol-dependent potentiation of insulin secretion was also validated in a model of standardized human pancreatic islets. We concluded that a mitochondrial Ca2+-targeted nutritional intervention activated metabolism–secretion coupling in insulin-secreting cells by modulating mitochondrial Ca2+ uptake.
Although pharmacological inhibition of the kaempferol-induced effect obtained by the mitochondrial Ca2+ inhibitor mitoxantrone [97] indicates a certain level of causality between mitochondrial Ca2+ modulation and β-cell function, it has not yet been definitively proven whether the modulation of mitochondrial Ca2+ by kaempferol is a direct or indirect effect of this polyphenol on the MCU complex.
In another intervention study, we demonstrated that the natural bioactive quinic acid increases glucose-stimulated mitochondrial Ca2+ rise, which enhanced insulin secretion in both INS-1E cells and mouse islets, and improved glucose tolerance in mice [120]. In this study, the naturally occurring polyol quinic acid was not a direct activator of MCU, but it enhanced the release of Ca2+ from the endoplasmic reticulum, thereby improving Ca2+ transfer between the endoplasmic reticulum and mitochondria. This transient mitochondrial Ca2+ increase was accompanied by the activation of two mitochondrial Ca2+-dependent processes (oxidative metabolism and ATP synthase-dependent respiration), which coordinately promoted sustained insulin secretion in the pancreatic β-cell [77].
Although preclinical and clinical evidence, including interventions in genetic MCU-ablated models, is still needed to validate the efficacy and safety of these phytochemical-based interventions, these studies suggest that bioactive agents that increase mitochondrial Ca2+ in pancreatic β-cells could be used to treat diabetes.
7. Conclusions
β-cell mitochondria contain a variety of ion channels in both the inner and the outer mitochondrial membrane, with important roles in stimulus–secretion coupling and cell viability [21]. The role of mitochondrial ion channels, including the MCU, in the β-cell has only partly been elucidated and is nowadays underestimated. The molecular definition of the MCU and its role in pancreatic β-cell signal transduction opens the possibility of developing a mitochondrial Ca2+-targeted intervention strategy for β-cell health and, prospectively, for diabetes treatment. Stimulation of the MCU has been demonstrated to improve β-cell energy metabolism during nutrient stimulation. Such a mechanism could explain how MCU activators might have a beneficial effect on stimulus–secretion coupling in β-cells, leading to improved glucose homeostasis. Encouraging results with the natural bioactive MCU activator kaempferol indicate the potential beneficial effects of the MCU-targeted strategy on pancreatic β-cell function. However, kaempferol is a non-specific activator of mitochondrial Ca2+ rise in pancreatic β-cells. A more specific MCU-targeted pharmacology is expected to improve MCU activation of the pancreatic β-cell, promoting beneficial effects in the context of diabetes treatment.
New studies targeting the proteins that control mitochondrial Ca2+ uptake should reveal whether altered insulin secretion is causally related to diabetes progression and could potentially expand the repertoire of therapeutic tools to treat this disease.
Author Contributions
Writing-original draft preparation, A.W., J.N.F. and U.D.M.; writing-review and editing A.W., J.N.F. and U.D.M.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors are employees of Nestlé Research, which is part of the Société des Produits Nestlé SA.
References
- Rahier, J.; Goebbels, R.M.; Henquin, J.-C. Cellular composition of the human diabetic pancreas. Diabetologia 1983, 24, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53, S16–S21. [Google Scholar] [CrossRef] [Green Version]
- Vetere, A.; Choudhary, A.; Burns, S.M.; Wagner, B.K. Targeting the pancreatic beta-cell to treat diabetes. Nat. Rev. Drug. Discov. 2014, 13, 278–289. [Google Scholar] [CrossRef]
- Gilon, P.; Chae, H.Y.; Rutter, G.A.; Ravier, M.A. Calcium signaling in pancreatic beta-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Kanekura, K.; Lavagnino, Z.; Spears, L.D.; Abreu, D.; Mahadevan, J.; Yagi, T.; Semenkovich, C.F.; Piston, D.W.; Uranoet, F. Targeting cellular calcium homeostasis to prevent cytokine-mediated beta cell death. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell Endocrinol. 2012, 353, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Wiederkehr, A.; De Marchi, U. Modulation of the matrix redox signaling by mitochondrial Ca2+. World J. Biol. Chem. 2015, 6, 310–323. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Szanda, G.; Akhmedov, D.; Mataki, C.; Heizmann, C.W.; Schoonjans, K.; Pozzan, T.; Spat, A.; Wollheim, C.B. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011, 13, 601–611. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Nguyen, T.T.; Choi, S.K.; Xu, S.; Das, R.; Cha, S.K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.I.; Semplici, F.; Ravier, M.A.; Bellomo, E.A.; Pullen, T.J.; Gilon, P.; Sekler, I.; Rizzuto, R.; Rutter, G.A. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic beta-cells. PLoS ONE 2012, 7, e39722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium 2008, 44, 64–76. [Google Scholar] [CrossRef]
- Nicholls, D.G. The Pancreatic beta-cell: A bioenergetic perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef] [Green Version]
- Georgiadou, E.; Rutter, G.A. Control by Ca(2+) of mitochondrial structure and function in pancreatic beta-cells. Cell Calcium 2020, 91, 102282. [Google Scholar] [CrossRef]
- De Marchi, U.; Fernandez-Martinez, S.; de la Fuente, S.; Wiederkehr, A.; Santo-Domingo, J. Mitochondrial ion channels in pancreatic beta-cells: Novel pharmacological targets for the treatment of Type 2 diabetes. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maechler, P. Mitochondrial function and insulin secretion. Mol. Cell. Endocrinol. 2013, 379, 12–18. [Google Scholar] [CrossRef]
- Henquin, J.-C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000, 49, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, B.R.; Wollheim, C.B. Synaptotagmins bind calcium to release insulin. Am. J. Physiol. Metab. 2008, 295, E1279–E1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idevall-Hagren, O.; Tengholm, A. Metabolic regulation of calcium signaling in beta cells. Semin. Cell Dev. Biol. 2020, 103, 20–30. [Google Scholar] [CrossRef]
- Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium signaling in ß-cell physiology and pathology: A revisit. Int. J. Mol. Sci. 2019, 20, 6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabourin, J.; Allagnat, F. Store-operated Ca2+ entry: A key component of the insulin secretion machinery. J. Mol. Endocrinol. 2016, 57, F35–F39. [Google Scholar] [CrossRef]
- Miura, Y.; Henquin, J.C.; Gilon, P. Emptying of intracellular Ca2+ stores stimulates Ca2+ entry in mouse pancreatic beta-cells by both direct and indirect mechanisms. J. Physiol. 1997, 503, 387–398. [Google Scholar] [CrossRef]
- Sabourin, J.; Le Gal, L.; Saurwein, L.; Haefliger, J.A.; Raddatz, E.; Allagnat, F. Store-operated Ca2+ entry mediated by orai1 and TRPC1 participates to insulin secretion in rat beta. Cells J. Biol. Chem. 2015, 290, 30530–30539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentki, M.; Janjic, D.; Biden, T.J.; Blondel, B.; Wollheim, C.B. Regulation of Ca2+ transport by isolated organelles of a rat insulinoma: Studies with endoplasmic reticulum and secretory granules. J. Biol. Chem. 1984, 259, 10118–10123. [Google Scholar] [CrossRef]
- Santulli, G.; Pagano, G.; Sardu, C.; Xie, W.; Reiken, S.; D’Ascia, S.L.; Cannone, M.; Marziliano, N.; Trimarco, B.; Guise, T.A.; et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Investig. 2015, 125, 1968–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klec, C.; Madreiter-Sokolowski, C.T.; Ziomek, G.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; et al. Presenilin-1 established ER-Ca(2+) leak: A Follow up on its importance for the initial insulin secretion in pancreatic islets and beta-cells upon elevated glucose. Cell. Physiol. Biochem. 2019, 53, 573–586. [Google Scholar] [PubMed] [Green Version]
- Klec, C.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Sachdev, V.; Duta-Mare, M.; Gottschalk, B.; Depaoli, M.R.; Rost, R.; Hay, J.; Waldeck-Weiermair, M.; et al. Glycogen synthase kinase 3 beta controls presenilin-1-mediated endoplasmic reticulum Ca2+ leak directed to mitochondria in pancreatic islets and beta-cells. Cell. Physiol. Biochem. 2019, 52, 57–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouriou, Y.; Demaurex, N.; Bijlenga, P.; De Marchi, U. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie 2011, 93, 2060–2067. [Google Scholar] [CrossRef] [Green Version]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.Y.; Henquin, J.C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Jonas, J.C.; Li, G.; Palmer, M.; Weller, U.; Wollheim, C.B. Dynamics of Ca2+ and guanosine 5′-[gamma-thio]triphosphate action on insulin secretion from alpha-toxin-permeabilized HIT-T15 cells. Biochem. J. 1994, 301, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.; Theler, J.M.; Murgia, M.; Wollheim, C.B.; Pozzan, T.; Rizzuto, R. Stimulated Ca2+ influx raises mitochondrial free Ca2+ to supramicromolar levels in a pancreatic beta-cell line: Possible role in glucose and agonist-induced insulin secretion. J. Biol. Chem. 1993, 268, 22385–22390. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nat. Cell Biol. 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 2006–2011. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The mitochondrial Ca2+ uniporter: Structure, function, and pharmacology. Handb. Exp. Pharmacol. 2017, 240, 129–156. [Google Scholar]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foskett, J.K.; Madesh, M. Regulation of the mitochondrial Ca2+ uniporter by MICU1 and micubiochem. Biophys. Res. Commun. 2014, 449, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.; Feige, J.N.; De Marchi, U. Targeting mitochondrial calcium uptake with the natural flavonol kaempferol, to promote metabolism/secretion coupling in pancreatic β-cells. Nutrients 2020, 12, 538. [Google Scholar] [CrossRef] [Green Version]
- Pitter, J.; Maechler, P.; Wollheim, C.; Spät, A. Mitochondria respond to Ca2+ already in the submicromolar range: Correlation with redox state. Cell Calcium 2002, 31, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesada, I.; Villalobos, C.; Nunez, L.; Chamero, P.; Alonso, M.T.; Nadal, A.; Garcia-Sancho, J. Glucose induces synchronous mitochondrial calcium oscillations in intact pancreatic islets. Cell Calcium 2008, 43, 39–47. [Google Scholar] [CrossRef]
- Kennedy, E.D.; Rizzuto, R.; Theler, J.M.; Pralong, W.F.; Bastianutto, C.; Pozzan, T.; Wollheim, C.B. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressing INS-1 cells. J. Clin. Investig. 1996, 98, 2524–2538. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Fan, M. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.B.; Tsai, C.W.; Tsai, M.F. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. eLife 2019, 8. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef] [Green Version]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; da Silva Xavier, G.; Davis, S.P.X.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca(2+) uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.L.; Murphy, E. The mitochondrial calcium uniporter: Mice can live and die without it. J. Mol. Cell. Cardiol. 2015, 78, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Csordás, G.; Huang, K.-T.; Várnai, P.; Joseph, S.K.; Hajnóczky, G. MICU1 interacts with the D-ring of the MCU pore to control its Ca(2+) flux and sensitivity to Ru. Mol. Cell 2018, 72, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.-F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.-W.; Wu, Y.; Willliams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. eLife 2016, 5, e15545. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 serves as a molecular gatekeeper to prevent In Vivo mitochondrial calcium overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, S.; Matesanz-Isabel, J.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. Biochem. J. 2014, 458, 33–40. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Min, C.K.; Kim, T.G.; Song, H.K.; Lim, Y.; Kim, D.; Shin, K.; Kang, M.; Kang, J.Y.; Youn, H.S.; et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 2015, 16, 1318–1333. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [Green Version]
- Enslow, B.T.; Madaris, T.; Ravichandran, J.; Jacob, R.; Srikantan, S.; Stathopulos, P.; Muniswamy, M. Identification of critical MCUR1 domains in the mitochondrial calcium uniporter complex that regulates cellular metabolism. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Paupe, V.; Prudent, J.; Dassa, E.P.; Rendon, O.Z.; Shoubridge, E.A. CCDC90A (MCUR1) is a cytochrome C Oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015, 21, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Nagashima, K.; Inagaki, N.; Kioka, H.; Takashima, S.; Fukuoka, H.; Noji, H.; Kakizuka, A.; Imamura, H. Glucose-stimulated single pancreatic islets sustain increased cytosolic ATP levels during initial Ca2+ influx and subsequent Ca2+ oscillations. J. Biol. Chem. 2014, 289, 2205–2216. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M.; McCormack, J.G. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium 1986, 7, 377–386. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Thevenet, J.; Hermant, A.; Dioum, E.; Wiederkehr, A. Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J. Biol. Chem. 2014, 289, 9182–9194. [Google Scholar] [CrossRef] [Green Version]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [CrossRef]
- Griesche, N.; Sanchez, G.; Hermans, C.; Idevall-Hagren, O. Cortical mitochondria regulate insulin secretion by local Ca(2+) buffering in rodent beta cells. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes mellitus, mitochondrial dysfunction and Ca(2+)-dependent permeability transition pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Gutierrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef]
- Dufer, M.; Krippeit-Drews, P.; Lembert, N.; Idahl, L.A.; Drews, G. Diabetogenic effect of cyclosporin A is mediated by interference with mitochondrial function of pancreatic B-cells. Mol. Pharmacol. 2001, 60, 873–879. [Google Scholar] [PubMed]
- Fujimoto, K.; Chen, Y.; Polonsky, K.S.; Dorn, G.W. Targeting cyclophilin D and the mitochondrial permeability transition enhances beta-cell survival and prevents diabetes in Pdx1 deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 10214–10219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells: Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lablanche, S.; Cottet-Rousselle, C.; Argaud, L.; Laporte, C.; Lamarche, F.; Richard, M.J.; Berney, T.; Benhamou, P.Y.; Fontaine, E. Respective effects of oxygen and energy substrate deprivation on beta cell viability. Biochim. Biophys. Acta 2015, 1847, 629–639. [Google Scholar] [CrossRef]
- Lu, A.; Chu, C.; Mulvihill, E.; Wang, R.; Liang, W. ATP-sensitive K(+) channels and mitochondrial permeability transition pore mediate effects of hydrogen sulfide on cytosolic Ca(2+) homeostasis and insulin secretion in beta-cells. Pflugers Arch. 2019, 471, 1551–1564. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabo, I.; De Marchi, U. Mitochondrial permeability transitions: How many doors to the house? Biochim. Biophys. Acta 2005, 1706, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. Why F-ATP synthase remains a strong candidate as the mitochondrial permeability transition pore. Front. Physiol. 2018, 9, 1543. [Google Scholar] [CrossRef] [Green Version]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The unique cysteine of F-ATP synthase OSCP subunit participates in modulation of the permeability transition pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef] [PubMed]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D.J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Basso, E.; Szabo, I.; Zoratti, M. Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol. Membr. Biol. 2006, 23, 521–530. [Google Scholar] [CrossRef]
- Barbu, A.; Welsh, N.; Saldeen, J. Cytokine-induced apoptosis and necrosis are preceded by disruption of the mitochondrial membrane potential (Deltapsi(m)) in pancreatic RINm5F cells: Prevention by Bcl. Mol. Cell Endocrinol. 2002, 190, 75–82. [Google Scholar] [CrossRef]
- Contreras, J.L.; Smyth, C.A.; Bilbao, G.; Young, C.J.; Thompson, J.A.; Eckhoff, D.E. 17beta-Estradiol protects isolated human pancreatic islets against proinflammatory cytokine-induced cell death: Molecular mechanisms and islet functionality. Transplantation 2002, 74, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 beta-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol. Cell 2017, 67, 711–723. [Google Scholar] [CrossRef]
- Woods, J.J.; Lovett, J.; Lai, B.; Harris, H.H.; Wilson, J.J. Redox stability controls the cellular uptake and activity of ruthenium-based inhibitors of the mitochondrial calcium uniporter (MCU). Angew. Chem. Int. Ed. Engl. 2020, 59, 6482–6491. [Google Scholar] [CrossRef]
- Woods, J.J.; Wilson, J.J. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Cur. Opin. Chem. Biol. 2020, 55, 9–18. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The effect of DS16570511, a new inhibitor of mitochondrial calcium uniporter, on calcium homeostasis, metabolism, and functional state of cultured cortical neurons and isolated brain mitochondria. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.; Trebak, M. Mitochondrial Ca(2+) signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assali, E.A.; Jones, A.E.; Veliova, M.; Acin-Perez, R.; Taha, M.; Miller, N.; Shum, M.; Oliveira, M.F.; Las, G.; Liesa, M.; et al. NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun. 2020, 11, 3347. [Google Scholar] [CrossRef]
- Nita, I.I.; Hershfinkel, M.; Fishman, D.; Ozeri, E.; Rutter, G.A.; Sensi, S.L.; Khananshvili, D.; Lewis, E.C.; Sekler, I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE 2012, 7, e46649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clish, C.B.; Clapham, D.E. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, E2249–E2254. [Google Scholar] [CrossRef] [Green Version]
- Dimmer, K.S.; Navoni, F.; Casarin, A.; Trevisson, E.; Endele, S.; Winterpacht, A.; Salviati, L.; Scorrano, L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 2008, 17, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Nowikovsky, K.; Froschauer, E.M.; Zsurka, G.; Samaj, J.; Reipert, S.; Kolisek, M.; Wiesenberger, G.; Schweyen, R.J. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J. Biol. Chem. 2004, 279, 30307–30315. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; Tavakoli, M.; Pfeiffer, C.; Seifert, J.; Mattarei, A.; De Stefani, D.; Zoratti, M.; Nowikovsky, K. LETM1-Mediated K(+) and Na(+) homeostasis regulates mitochondrial Ca(2+) efflux. Front. Physiol. 2017, 8, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimi, H.; McDonald, L.; Stribrna, E.; Lukes, J. Trypanosome Letm1 protein is essential for mitochondrial potassium homeostasis. J. Biol. Chem. 2013, 288, 26914–26925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Miles, P.D.; Vargas, L.; Luan, P.; Glasco, S.; Kushnareva, Y.; Kornbrust, E.S.; Grako, K.A.; Wollheim, C.B.; Maechler, P.; et al. Inhibition of mitochondrial Na+-Ca2+ exchanger increases mitochondrial metabolism and potentiates glucose-stimulated insulin secretion in rat pancreatic islets. Diabetes 2003, 52, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Luciani, D.S.; Ao, P.; Hu, X.; Warnock, G.L.; Johnson, J.D. Voltage-gated Ca(2+) influx and insulin secretion in human and mouse beta-cells are impaired by the mitochondrial Na(+)/Ca(2+) exchange inhibitor CGP. Eur. J. Pharmacol. 2007, 576, 18–25. [Google Scholar]
- Thu, T.; Ahn, J.R.; Woo, S.H. Inhibition of L-type Ca2+ channel by mitochondrial Na+-Ca2+ exchange inhibitor CGP-37157 in rat atrial myocytes. Eur. J. Pharmacol. 2006, 552, 15–19. [Google Scholar] [CrossRef]
- Neumann, J.T.; Diaz-Sylvester, P.L.; Fleischer, S.; Copello, J.A. CGP-37157 inhibits the sarcoplasmic reticulum Ca(2)+ ATPase and activates ryanodine receptor channels in striated muscle. Mol. Pharmacol. 2011, 79, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Nita, I.I.; Hershfinkel, M.; Kantor, C.; Rutter, G.A.; Lewis, E.C.; Sekler, I. Pancreatic beta-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 2014, 28, 3301–3312. [Google Scholar] [CrossRef] [PubMed]
- Montero, M.; Lobaton, C.D.; Hernandez-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Heikkila, E.; Hermant, A.; Thevenet, J.; Bermont, F.; Kulkarni, S.S.; Ratajczak, J.; Santo-Domingo, J.; Dioum, E.H.; Canto, C.; Barron, D.; et al. The plant product quinic acid activates Ca(2+) -dependent mitochondrial function and promotes insulin secretion from pancreatic beta cells. Br. J. Pharmacol. 2019, 176, 3250–3263. [Google Scholar] [CrossRef]
Figure 1.
Consensus model of the signal transduction pathway of pancreatic β-cells. Metabolism–secretion coupling of β-cells requires the sequential activation of glycolysis, mitochondrial oxidative metabolism and Ca2+ entry through the plasma membrane. Glucose stimulates glycolysis and pyruvate production. Pyruvate triggers mitochondrial metabolism and the formation of the reduced form of nicotinamide adenine dinucleotide NADH (by the TCA cycle), which is the fuel for the respiratory complexes (1,2,3,4), enabling ATP production by ATP-synthase (5). ATP inhibits the KATP channel, inducing membrane depolarization (ΔΨ↓) and Ca2+ entry through voltage-gated Ca2+ channels, promoting insulin secretion. Ca2+ is taken up in parallel by mitochondria via the mitochondrial Ca2+ uniporter (MCU) and facilitates sustained insulin secretion. The amplifying pathway of metabolism–secretion coupling is co-generated by additive coupling factors. Additional players contributing to Ca2+ homeostasis are mentioned in the main text.
Figure 1.
Consensus model of the signal transduction pathway of pancreatic β-cells. Metabolism–secretion coupling of β-cells requires the sequential activation of glycolysis, mitochondrial oxidative metabolism and Ca2+ entry through the plasma membrane. Glucose stimulates glycolysis and pyruvate production. Pyruvate triggers mitochondrial metabolism and the formation of the reduced form of nicotinamide adenine dinucleotide NADH (by the TCA cycle), which is the fuel for the respiratory complexes (1,2,3,4), enabling ATP production by ATP-synthase (5). ATP inhibits the KATP channel, inducing membrane depolarization (ΔΨ↓) and Ca2+ entry through voltage-gated Ca2+ channels, promoting insulin secretion. Ca2+ is taken up in parallel by mitochondria via the mitochondrial Ca2+ uniporter (MCU) and facilitates sustained insulin secretion. The amplifying pathway of metabolism–secretion coupling is co-generated by additive coupling factors. Additional players contributing to Ca2+ homeostasis are mentioned in the main text.

Figure 2.
The mitochondrial calcium uniporter (MCU) comprises distinct pore-forming (MCU/MCUb) and associated proteins (essential MCU regulator (EMRE), mitochondrial calcium uptake (MICU) 1, MICU2, MICU3 and mitochondrial calcium uniporter regulator 1 (MCUR1)). The subunits that have already been studied in models of the pancreatic β-cell are rendered in green. (A) Model of the closed conformation of MCU. (B) Following glucose stimulation, the entry of Ca2+ into the matrix space is mediated by MCU. IMM, inner mitochondrial membrane; IMS, intermembrane space.
Figure 2.
The mitochondrial calcium uniporter (MCU) comprises distinct pore-forming (MCU/MCUb) and associated proteins (essential MCU regulator (EMRE), mitochondrial calcium uptake (MICU) 1, MICU2, MICU3 and mitochondrial calcium uniporter regulator 1 (MCUR1)). The subunits that have already been studied in models of the pancreatic β-cell are rendered in green. (A) Model of the closed conformation of MCU. (B) Following glucose stimulation, the entry of Ca2+ into the matrix space is mediated by MCU. IMM, inner mitochondrial membrane; IMS, intermembrane space.

Figure 3.
Contribution of mitochondrial Ca2+ uptake and mitochondrial Ca2+ extrusion to mitochondrial Ca2+ signaling in the pancreatic β-cell. (A) Ca2+ transport proteins of pancreatic β-cell mitochondria. In the outer mitochondrial membrane, the voltage-dependent anion channel VDAC facilitates the diffusion of Ca2+, ions and other small solutes. However, mitochondrial Ca2+ uptake is mainly regulated at the level of the inner membrane. Therefore, in β-cell mitochondria, the influx of Ca2+ into the matrix is mediated by the Ca2+-selective channel MCU complex; Ca2+ is then extruded by the Na+/Ca2+ exchanger NCLX. The existence and the molecular identity of a pancreatic β-cell mitochondrial Ca2+/H+ exchanger has not yet been proven and is here highlighted with a question mark (?) (see text for details). (B) Example of a calibrated trace of mitochondrial Ca2+ elevation in the pancreatic β-cell line INS-1E stimulated with glucose, as indicated (gray). The uptake and release phases of the mitochondrial Ca2+ are highlighted. The transporters which have been demonstrated to substantially contribute to the uptake and the release are MCU and NCLX, respectively. To calibrate mitochondrial Ca2+, INS-1E cells were transfected with the genetically encoded mitochondrial luminescence Ca2+ sensor aequorin, which is reported to overestimate the average Ca2+ rise in cellular compartments [113].
Figure 3.
Contribution of mitochondrial Ca2+ uptake and mitochondrial Ca2+ extrusion to mitochondrial Ca2+ signaling in the pancreatic β-cell. (A) Ca2+ transport proteins of pancreatic β-cell mitochondria. In the outer mitochondrial membrane, the voltage-dependent anion channel VDAC facilitates the diffusion of Ca2+, ions and other small solutes. However, mitochondrial Ca2+ uptake is mainly regulated at the level of the inner membrane. Therefore, in β-cell mitochondria, the influx of Ca2+ into the matrix is mediated by the Ca2+-selective channel MCU complex; Ca2+ is then extruded by the Na+/Ca2+ exchanger NCLX. The existence and the molecular identity of a pancreatic β-cell mitochondrial Ca2+/H+ exchanger has not yet been proven and is here highlighted with a question mark (?) (see text for details). (B) Example of a calibrated trace of mitochondrial Ca2+ elevation in the pancreatic β-cell line INS-1E stimulated with glucose, as indicated (gray). The uptake and release phases of the mitochondrial Ca2+ are highlighted. The transporters which have been demonstrated to substantially contribute to the uptake and the release are MCU and NCLX, respectively. To calibrate mitochondrial Ca2+, INS-1E cells were transfected with the genetically encoded mitochondrial luminescence Ca2+ sensor aequorin, which is reported to overestimate the average Ca2+ rise in cellular compartments [113].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weiser, A.; Feige, J.N.; De Marchi, U. Mitochondrial Calcium Signaling in Pancreatic β-Cell. Int. J. Mol. Sci. 2021, 22, 2515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052515
AMA Style
Weiser A, Feige JN, De Marchi U. Mitochondrial Calcium Signaling in Pancreatic β-Cell. International Journal of Molecular Sciences. 2021; 22(5):2515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052515
Chicago/Turabian StyleWeiser, Anna, Jerome N. Feige, and Umberto De Marchi. 2021. "Mitochondrial Calcium Signaling in Pancreatic β-Cell" International Journal of Molecular Sciences 22, no. 5: 2515. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052515
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.