
Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
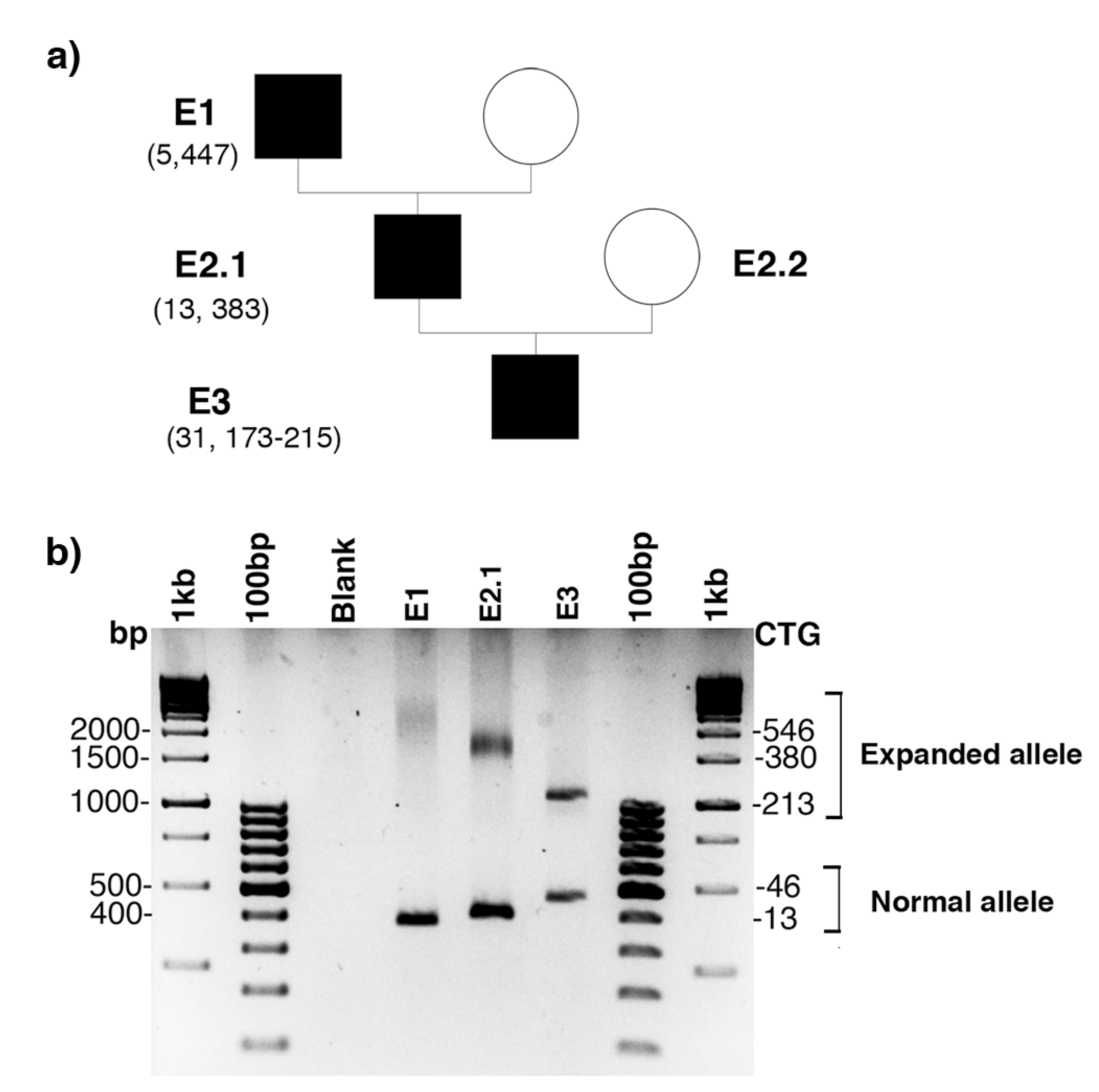
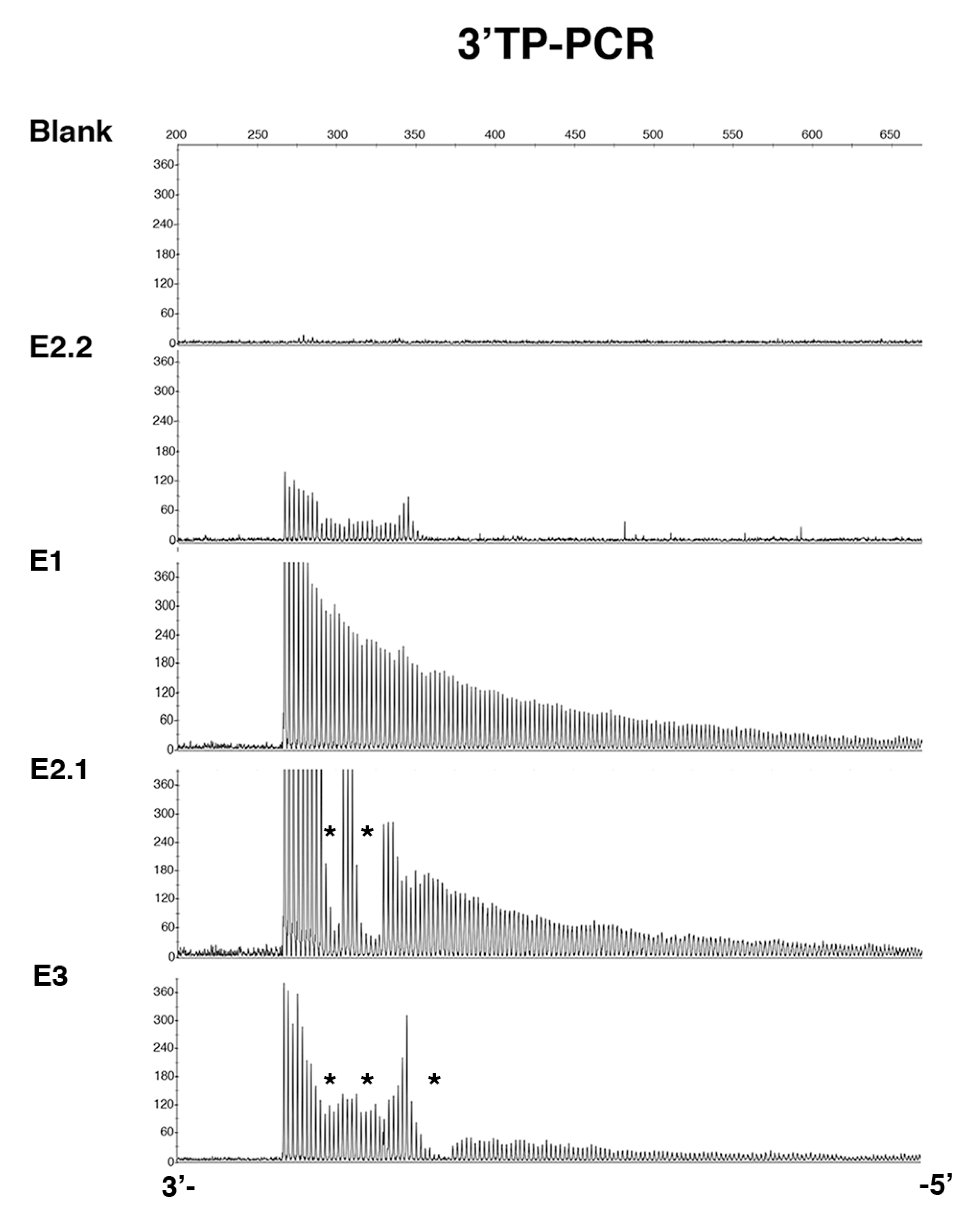
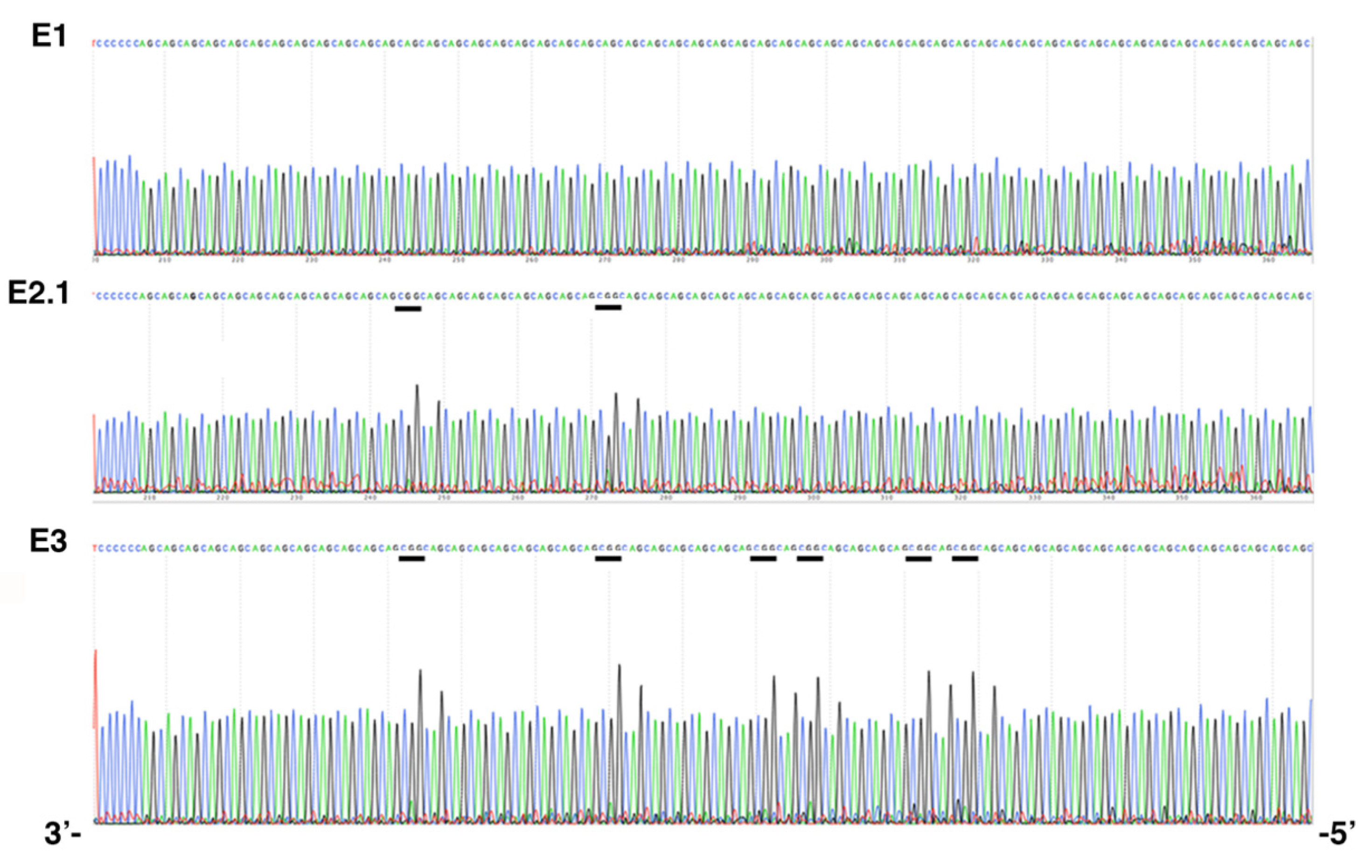
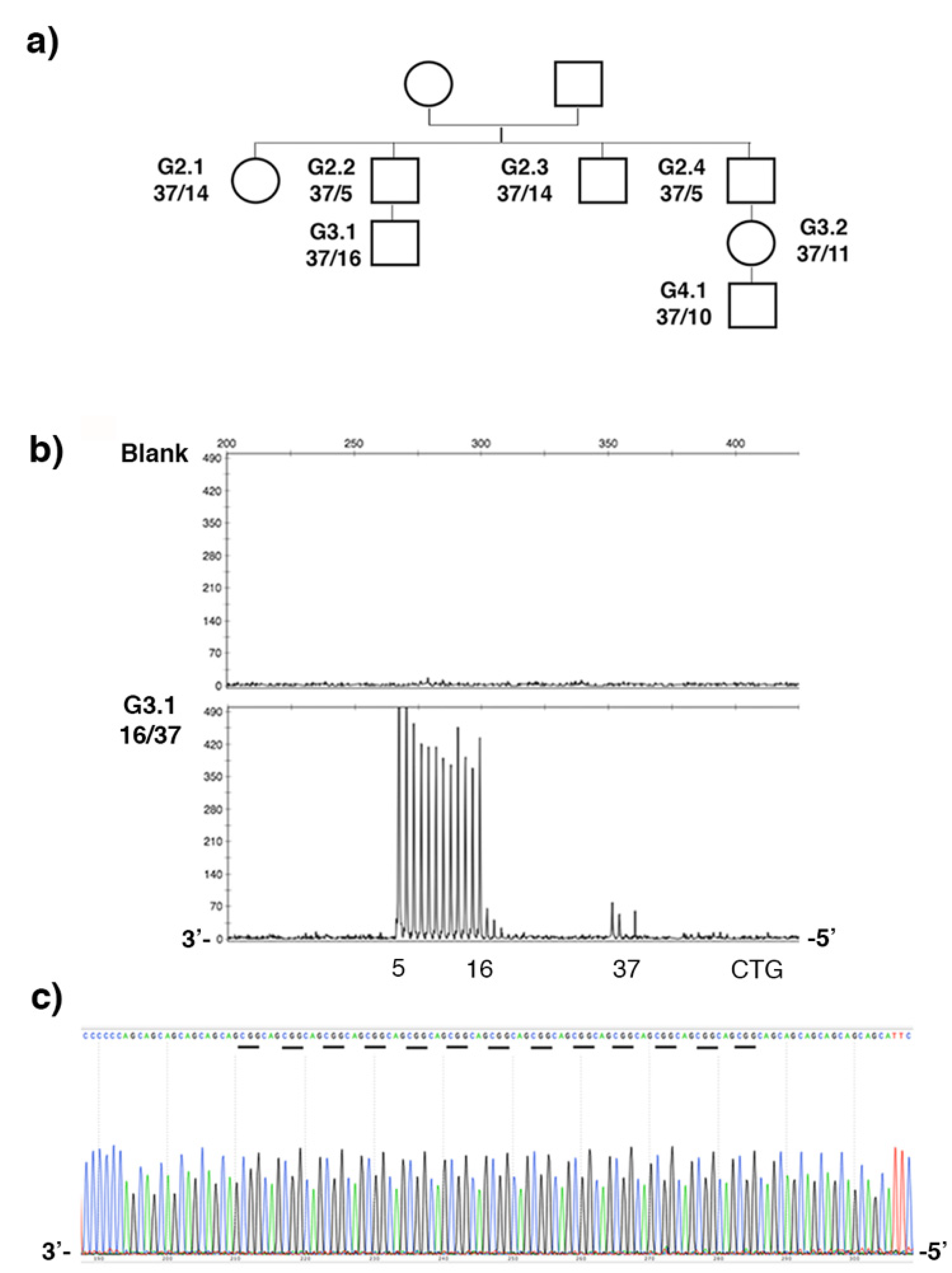
2.1. De Novo CCG Interruption in New DM1 Family Identified with the Sequel II System
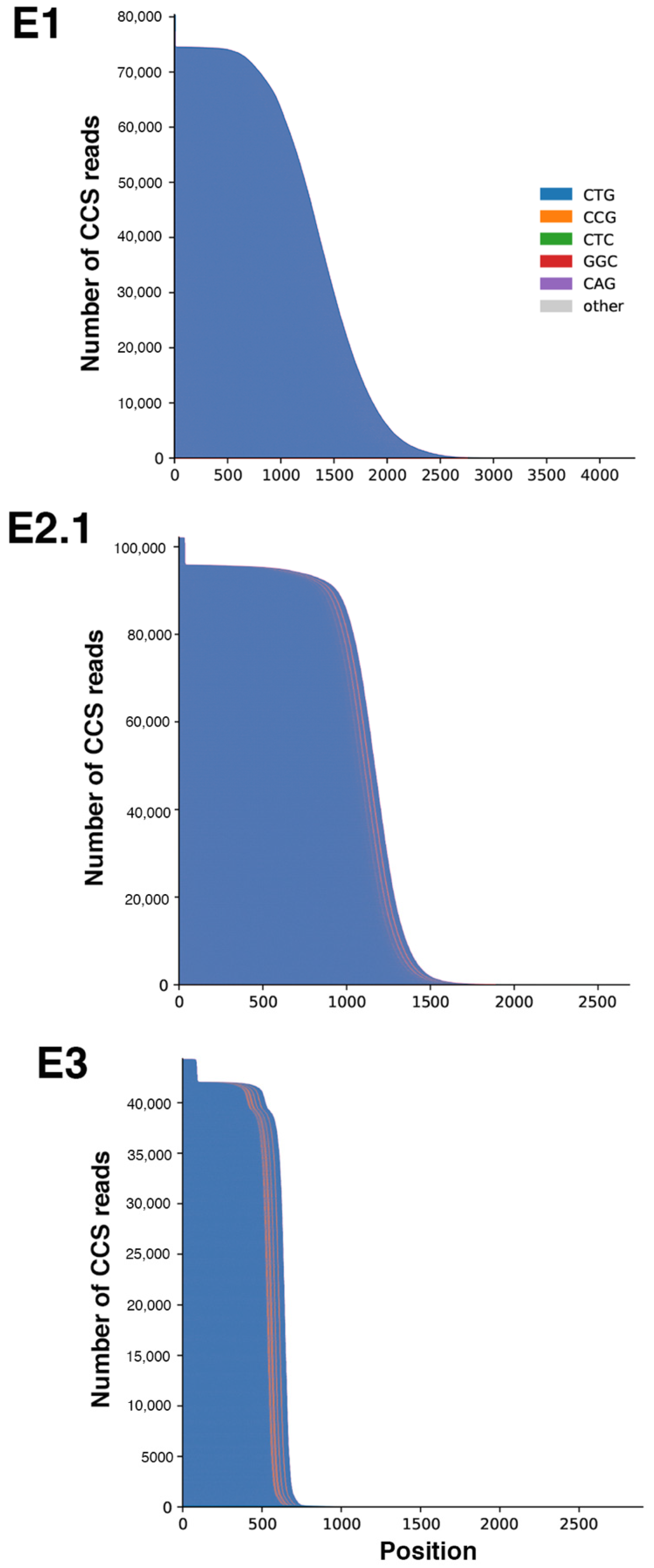
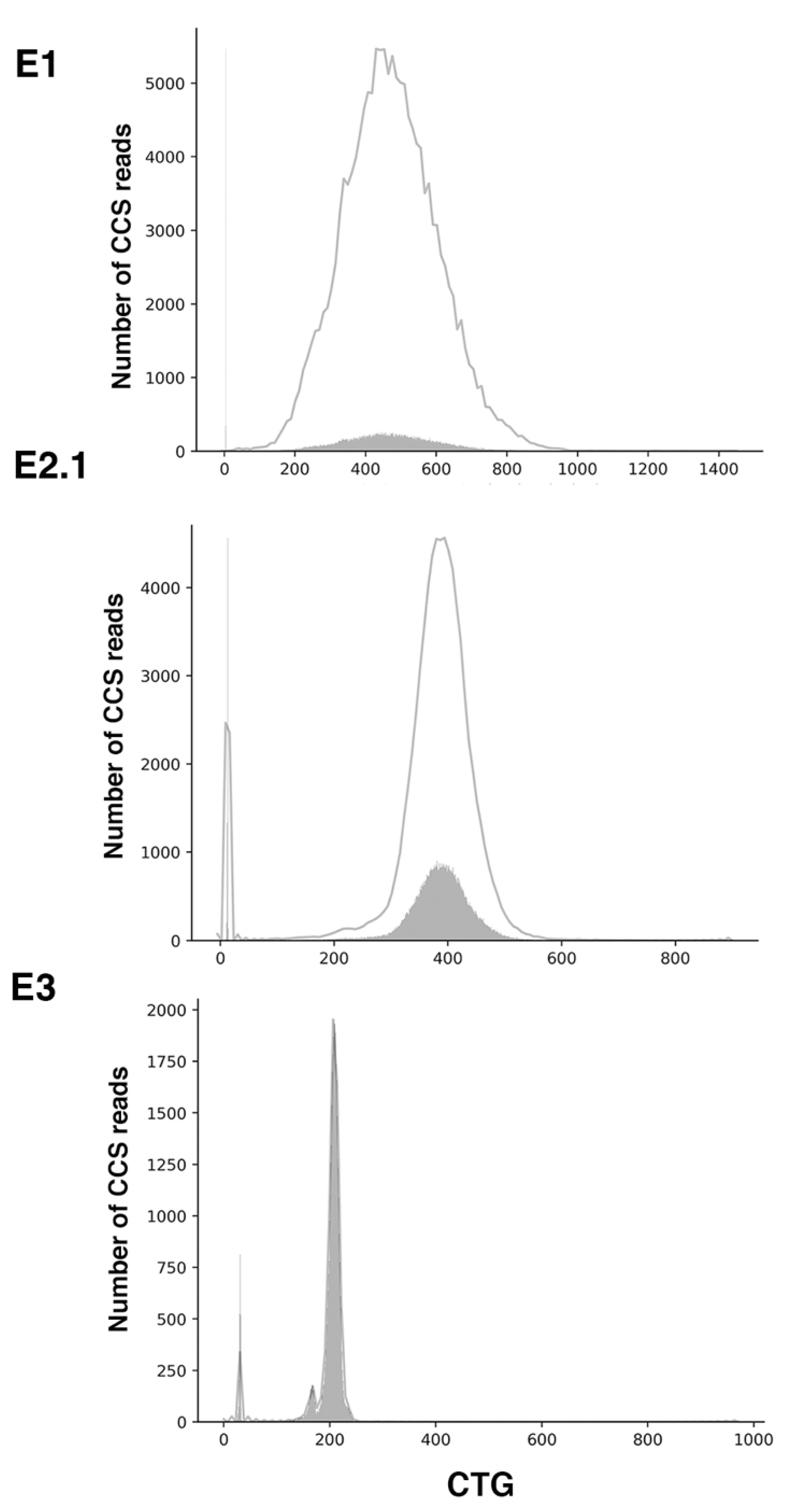
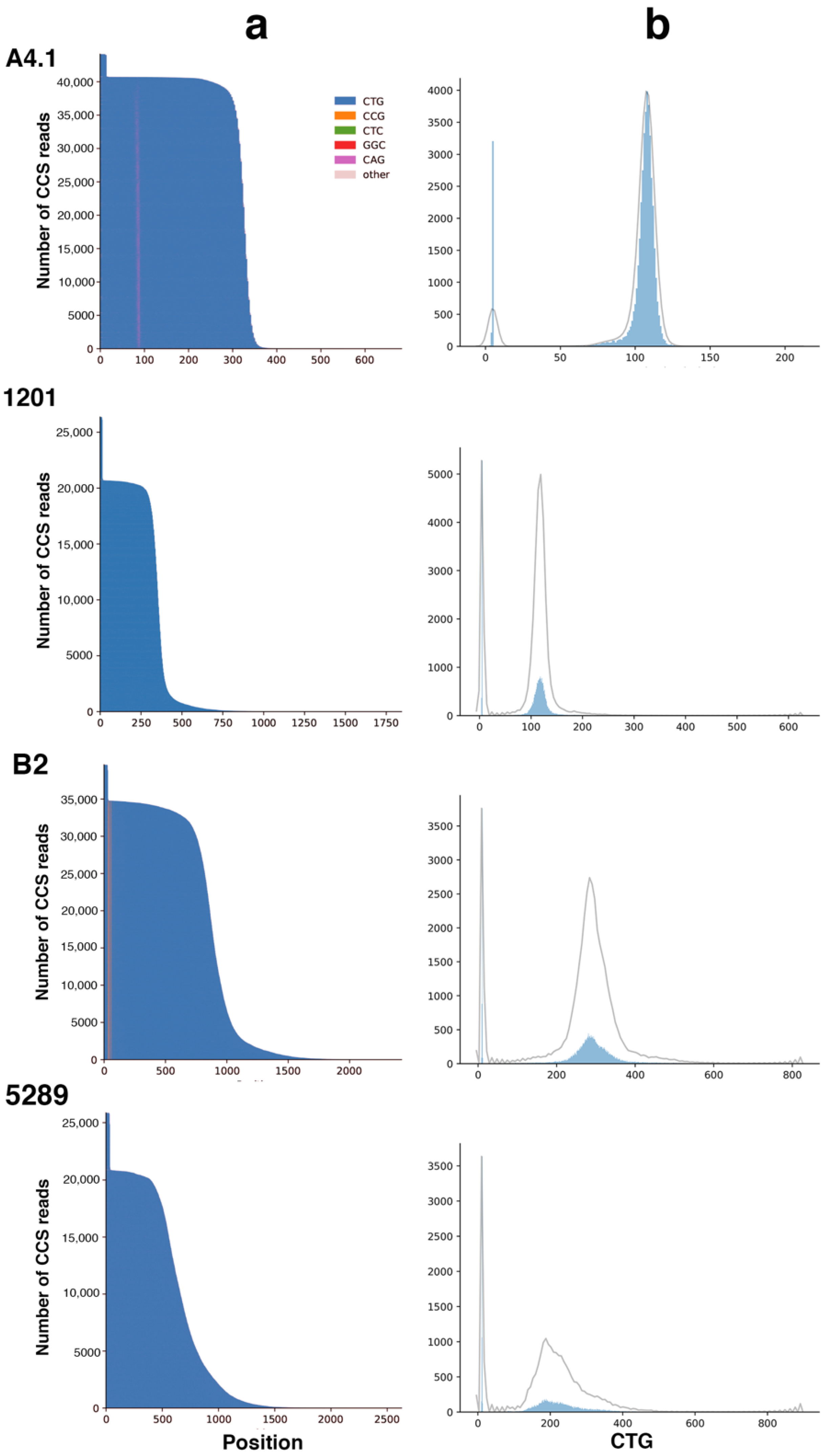
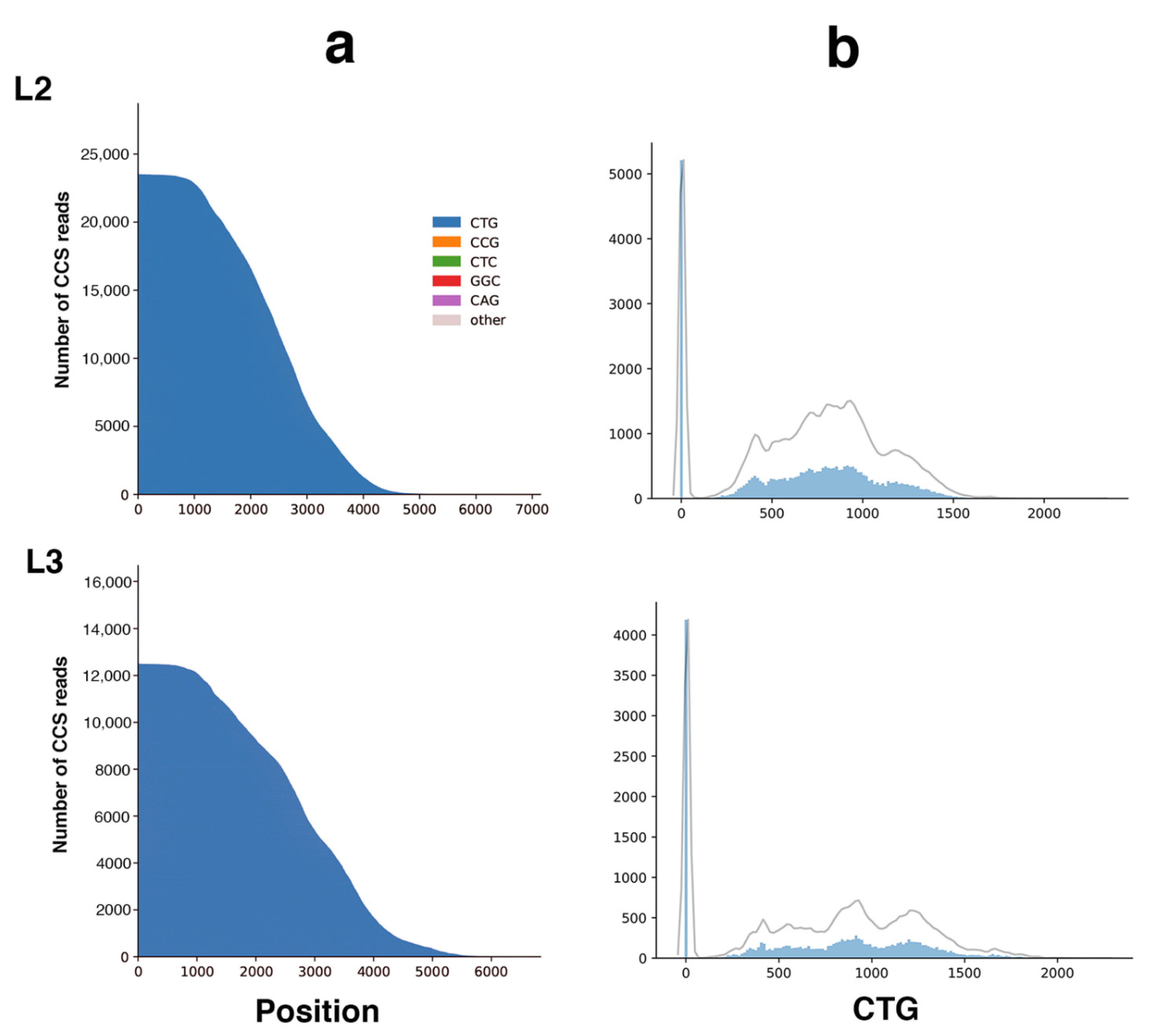
2.2. Sequel II System Makes It Possible to Estimate the CTG Repeat Length and the Interruptions as Well as the Somatic Mosaicism in DM1 Patients with at Least 1000 CTG Repeats
2.3. Stable CCG-Interrupted Allele with 37 Repeats Is Associated to DM1 Haplotype in a Large Family
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment
4.2. CTG Repeat Amplification
4.3. 3′Triplet-Primed PCR (TP-PCR)
4.4. CTG Repeat Sequencing
4.5. Sequel II System from PacBio (PacBio and Sequel Are Trademarks of Pacific Biosciences)
4.6. Haplotype Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paulson, H. Repeat expansion diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 147, pp. 105–123. ISBN 978-0-444-63233-3. [Google Scholar]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular Basis of Myotonic Dystrophy: Expansion of a Trinucleotide (CTG) Repeat at the 3′ End of a Transcript Encoding a Protein Kinase Family Member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Tomé, S.; Gourdon, G. DM1 Phenotype Variability and Triplet Repeat Instability: Challenges in the Development of New Therapies. Int. J. Mol. Sci. 2020, 21, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G. Unravelling the Myotonic Dystrophy Type 1 Clinical Spectrum: A Systematic Registry-Based Study with Implications for Disease Classification. Rev. Neurol. 2016, 172, 572–580. [Google Scholar] [CrossRef]
- Dogan, C.; De Antonio, M.; Hamroun, D.; Varet, H.; Fabbro, M.; Rougier, F.; Amarof, K.; Arne Bes, M.-C.; Bedat-Millet, A.-L.; Behin, A.; et al. Gender as a Modifying Factor Influencing Myotonic Dystrophy Type 1 Phenotype Severity and Mortality: A Nationwide Multiple Databases Cross-Sectional Observational Study. Plos ONE 2016, 11, e0148264. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Couto, J.M.; Higham, C.F.; Hogg, G.; Cuenca, P.; Braida, C.; Wilson, R.H.; Adam, B.; del Valle, G.; Brian, R.; et al. Somatic Instability of the Expanded CTG Triplet Repeat in Myotonic Dystrophy Type 1 Is a Heritable Quantitative Trait and Modifier of Disease Severity. Hum. Mol. Genet. 2012, 21, 3558–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-M.; Correia, K.; Loupe, J.; Kim, K.-H.; Barker, D.; Hong, E.P.; Chao, M.J.; Long, J.D.; Lucente, D.; Vonsattel, J.P.G.; et al. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, F.; Vásquez, M.; Corrales, E.; Vindas-Smith, R.; Santamaría-Ulloa, C.; Zhang, B.; Sirito, M.; Estecio, M.R.; Krahe, R.; Monckton, D.G. Longitudinal Increases in Somatic Mosaicism of the Expanded CTG Repeat in Myotonic Dystrophy Type 1 Are Associated with Variation in Age-at-Onset. Hum. Mol. Genet. 2020, 29, 2496–2507. [Google Scholar] [CrossRef]
- Overend, G.; Légaré, C.; Mathieu, J.; Bouchard, L.; Gagnon, C.; Monckton, D.G. Allele Length of the DMPK CTG Repeat Is a Predictor of Progressive Myotonic Dystrophy Type 1 Phenotypes. Hum. Mol. Genet. 2019, 28, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Jimenez-Moreno, C.; Okkersen, K.; Wenninger, S.; Daidj, F.; Hogarth, F.; Littleford, R.; Gorman, G.; Bassez, G.; Schoser, B.; et al. Genetic Determinants of Disease Severity in the Myotonic Dystrophy Type 1 OPTIMISTIC Cohort. Neurology 2019, 93, e995–e1009. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.; Vásquez, M.; Santamaría, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A Polymorphism in the MSH3 Mismatch Repair Gene Is Associated with the Levels of Somatic Instability of the Expanded CTG Repeat in the Blood DNA of Myotonic Dystrophy Type 1 Patients. DNA Repair 2016, 40, 57–66. [Google Scholar] [CrossRef]
- Flower, M.; Lomeikaite, V.; Ciosi, M.; Cumming, S.; Morales, F.; Lo, K.; Hensman Moss, D.; Jones, L.; Holmans, P.; Monckton, D.G.; et al. MSH3 Modifies Somatic Instability and Disease Severity in Huntington’s and Myotonic Dystrophy Type 1. Brain 2019, 142, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Latham, G.J.; Coppinger, J.; Hadd, A.G.; Nolin, S.L. The Role of AGG Interruptions in Fragile X Repeat Expansions: A Twenty-Year Perspective. Front. Genet. 2014, 5, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, Z.; Izumi, Y.; Kameyama, M.; Kawakami, H.; Nakamura, S. The EVect of CAT Trinucleotide Interruptions on the Age at Onset of Spinocerebellar Ataxia Type 1. J. Med. Genet. 1999, 36, 546–548. [Google Scholar] [PubMed]
- Kraus-Perrotta, C.; Lagalwar, S. Expansion, Mosaicism and Interruption: Mechanisms of the CAG Repeat Mutation in Spinocerebellar Ataxia Type 1. Cerebellum Ataxias 2016, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomé, S.; Dandelot, E.; Dogan, C.; Bertrand, A.; Geneviève, D.; Péréon, Y.; Simon, M.; Bonnefont, J.-P.; Bassez, G.; Gourdon, G. Unusual Association of a Unique CAG Interruption in 5′ of DM1 CTG Repeats with Intergenerational Contractions and Low Somatic Mosaicism. Hum. Mutat. 2018, 39, 970–982. [Google Scholar] [CrossRef]
- Braida, C.; Stefanatos, R.K.A.; Adam, B.; Mahajan, N.; Smeets, H.J.M.; Niel, F.; Goizet, C.; Arveiler, B.; Koenig, M.; Lagier-Tourenne, C.; et al. Variant CCG and GGC Repeats within the CTG Expansion Dramatically Modify Mutational Dynamics and Likely Contribute toward Unusual Symptoms in Some Myotonic Dystrophy Type 1 Patients. Hum. Mol. Genet. 2010, 19, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- The Scottish Myotonic Dystrophy Consortium; Cumming, S.A.; Hamilton, M.J.; Robb, Y.; Gregory, H.; McWilliam, C.; Cooper, A.; Adam, B.; McGhie, J.; Hamilton, G.; et al. De Novo Repeat Interruptions Are Associated with Reduced Somatic Instability and Mild or Absent Clinical Features in Myotonic Dystrophy Type 1. Eur. J. Hum. Genet. 2018, 26, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Masciullo, M.; Silvestri, G.; Novelli, G.; Botta, A. Myotonic Dystrophy Type 1: Role of CCG, CTC and CGG Interruptions within DMPK Alleles in the Pathogenesis and Molecular Diagnosis: Variant Interruptions in Pathogenesis and Molecular Diagnosis of DM1. Clin. Genet. 2017, 92, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Masciullo, M.; Pietrobono, R.; Conte, G.; Modoni, A.; Bianchi, M.L.E.; Rizzo, V.; Pomponi, M.G.; Tasca, G.; Neri, G.; et al. Molecular, Clinical, and Muscle Studies in Myotonic Dystrophy Type 1 (DM1) Associated with Novel Variant CCG Expansions. J. Neurol. 2013, 260, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Ballester-Lopez, A.; Koehorst, E.; Almendrote, M.; Martínez-Piñeiro, A.; Lucente, G.; Linares-Pardo, I.; Núñez-Manchón, J.; Guanyabens, N.; Cano, A.; Lucia, A.; et al. A DM1 Family with Interruptions Associated with Atypical Symptoms and Late Onset but Not with a Milder Phenotype. Hum. Mutat. 2020, 41, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakočević-Stojanović, V.; Savić-Pavićević, D. Repeat Interruptions Modify Age at Onset in Myotonic Dystrophy Type 1 by Stabilizing DMPK Expansions in Somatic Cells. Front. Genet. 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.N.; van der Plas, E.; Hamilton, M.; Koscik, T.R.; Gutmann, L.; Cumming, S.A.; Monckton, D.G.; Nopoulos, P.C. Variant Repeats within the DMPK CTG Expansion Protect Function in Myotonic Dystrophy Type 1. Neurol. Genet. 2020, 6, e504. [Google Scholar] [CrossRef] [PubMed]
- Radvansky, J.; Ficek, A.; Minarik, G.; Palffy, R.; Kadasi, L. Effect of Unexpected Sequence Interruptions to Conventional PCR and Repeat Primed PCR in Myotonic Dystrophy Type 1 Testing. Diagn. Mol. Pathol. 2011, 20, 48–51. [Google Scholar] [CrossRef] [PubMed]
- De Siena, C.; Cardani, R.; Brigonzi, E.; Bosè, F.; Fossati, B.; Meola, G.; Costa, E.; Valaperta, R. Incidence of Amplification Failure in DMPK Allele Due to Allelic Dropout Event in a Diagnostic Laboratory. Clin. Chim. Acta 2018, 484, 111–116. [Google Scholar] [CrossRef]
- Botta, A.; Rossi, G.; Marcaurelio, M.; Fontana, L.; D’Apice, M.R.; Brancati, F.; Massa, R.; G Monckton, D.; Sangiuolo, F.; Novelli, G. Identification and Characterization of 5′ CCG Interruptions in Complex DMPK Expanded Alleles. Eur. J. Hum. Genet. 2017, 25, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Fontana, L.; Masciullo, M.; Bianchi, M.L.E.; Rossi, S.; Leoncini, E.; Novelli, G.; Botta, A.; Silvestri, G. Expansion Size and Presence of CCG/CTC/CGG Sequence Interruptions in the Expanded CTG Array Are Independently Associated to Hypermethylation at the DMPK Locus in Myotonic Dystrophy Type 1 (DM1). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.; Serrenti, M.; Meloni, C.; Cau, M.; Melis, M.A. Triplet-Primed PCR Is More Sensitive than Southern Blotting–Long PCR for the Diagnosis of Myotonic Dystrophy Type1. Genet. Test. Mol. Biomark. 2012, 16, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Santoro, M.; D’Apice, M.R.; Gori, G.; Morrone, A.; Novelli, G.; Botta, A. Identification, Molecular Characterization and Segregation Analysis of a Variant DMPK Pre-Mutation Allele in a Three-Generation Italian Family. Acta. Myol. 2020, 39, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Lian, M.; Law, H.-Y.; Lee, C.G.; Chong, S.S. Defining the Performance Parameters of a Rapid Screening Tool for Myotonic Dystrophy Type 1 Based on Triplet-Primed PCR and Melt Curve Analysis. Expert Rev. Mol. Diagn. 2016, 16, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- P. Leeflang, E.; Arnhelm, N. A Novel Repeat Structure at the Myotonic Dystrophy Locus in a 37 Repeat Allele with Unexpectedly High Stability. Hum. Mol. Genet. 1995, 4, 135–136. [Google Scholar] [CrossRef]
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly Unstable Sequence Interruptions of the CTG Repeat in the Myotonic Dystrophy Gene. Am. J. Med. Genet. 2009, 149A, 1365–1374. [Google Scholar] [CrossRef]
- Tomé, S.; Gourdon, G. Fast Assays to Detect Interruptions in CTG.CAG Repeat Expansions. In Trinucleotide Repeats; Richard, G.-F., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2056, pp. 11–23. ISBN 978-1-4939-9783-1. [Google Scholar]
- Warner, J.P.; Barron, L.H.; Goudie, D.; Kelly, K.; Dow, D.; Fitzpatrick, D.R.; Brock, D.J. A General Method for the Detection of Large CAG Repeat Expansions by Fluorescent PCR. J. Med. Genet. 1996, 33, 1022–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomis, E.W.; Eid, J.S.; Peluso, P.; Yin, J.; Hickey, L.; Rank, D.; McCalmon, S.; Hagerman, R.J.; Tassone, F.; Hagerman, P.J. Sequencing the Unsequenceable: Expanded CGG-Repeat Alleles of the Fragile X Gene. Genome Res. 2013, 23, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höijer, I.; Tsai, Y.-C.; Clark, T.A.; Kotturi, P.; Dahl, N.; Stattin, E.-L.; Bondeson, M.-L.; Feuk, L.; Gyllensten, U.; Ameur, A. Detailed Analysis of HTT Repeat Elements in Human Blood Using Targeted Amplification-Free Long-Read Sequencing. Hum. Mutat. 2018, 39, 1262–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, S.; Matsumoto, N. Long-Read Sequencing for Rare Human Genetic Diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- E. Neville, C.; S. Mahadeva, M.; M. Barcelo, J.; Korneluk, R. High Resolution Genetic Analysis Suggests One Ancestral Predisposing Haplotype for the Origin of the Myotonic Dystrophy Mutation. Hum. Mol. Genet. 1994, 3, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Kretz, C.; Johnson, K.; Mandel, J.L. Origin of the Expansion Mutation in Myotonic Dystrophy. Nat. Genet. 1993, 4, 72–76. [Google Scholar] [CrossRef]
- Murillo-Melo, N.M.; Márquez-Quiróz, L.C.; Gómez, R.; Orozco, L.; Mendoza-Caamal, E.; Tapia-Guerrero, Y.S.; Camacho-Mejorado, R.; Cortés, H.; López-Reyes, A.; Santana, C.; et al. Origin of the Myotonic Dystrophy Type 1 Mutation in Mexican Population and Influence of Amerindian Ancestry on CTG Repeat Allelic Distribution. Neuromuscul. Disord. 2017, 27, 1106–1114. [Google Scholar] [CrossRef]
- Franck, S.; Barbé, L.; Ardui, S.; De Vlaeminck, Y.; Allemeersch, J.; Dziedzicka, D.; Spits, C.; Vanroye, F.; Hilven, P.; Duqué, G.; et al. MSH2 Knock-down Shows CTG Repeat Stability and Concomitant Upstream Demethylation at the DMPK Locus in Myotonic Dystrophy Type 1 Human Embryonic Stem Cells. Hum. Mol. Genet. 2020, ddaa250. [Google Scholar] [CrossRef]
- Hiatt, S.M.; Lawlor, J.M.J.; Handley, L.H.; Ramaker, R.C.; Rogers, B.B.; Partridge, E.C.; Boston, L.B.; Williams, M.; Plott, C.B.; Jenkins, J.; et al. Long-Read. Genome Sequencing for the Diagnosis of Neurodevelopmental Disorders. Hum. Genet. Genom. Adv. 2021, 2. [Google Scholar] [CrossRef]
- Lavedan, C.; Hofmann-Radvanyi, H.; Duros, C.; Savoy, D.; Dehaupas, I.; Luce, P.S.; Junien, C. Myotonic Dystrophy: Size- and Sex-Dependent Dynamics of CTG Meiotic Instability, and Somatic Mosaicism. Am. J. Hum. Genet. 1993, 52, 875–883. [Google Scholar] [PubMed]
- Morales, F.; Vásquez, M.; Cuenca, P.; Campos, D.; Santamaría, C.; del Valle, G.; Brian, R.; Sittenfeld, M.; Monckton, D.G. Parental Age Effects, but No Evidence for an Intrauterine Effect in the Transmission of Myotonic Dystrophy Type 1. Eur. J. Hum. Genet. 2015, 23, 646–653. [Google Scholar] [CrossRef]
- Joosten, I.B.T.; Hellebrekers, D.M.E.I.; de Greef, B.T.A.; Smeets, H.J.M.; de Die-Smulders, C.E.M.; Faber, C.G.; Gerrits, M.M. Parental Repeat Length Instability in Myotonic Dystrophy Type 1 Pre- and Protomutations. Eur. J. Hum. Genet. 2020, 28, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Johnson, K.; Boucher, C.A.; Baiget, M. Somatic Instability of the Myotonic Dystrophy (CTG)n Repeat during Human Fetal Development. Hum. Mol. Genet. 1997, 6, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Wöhrle, D.; Kennerknecht, I.; Wolf, M.; Enders, H.; Schwemmle, S.; Steinbach, P. Heterogeneity of DM Kinase Repeat Expansion in Different Fetal Tissues and Further Expansion during Cell Proliferation in Vitro: Evidence for a Casual Involvement of Methyl-Directed DNA Mismatch Repair in Triplet Repeat Stability. Hum. Mol. Genet. 1995, 4, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Hafford-Tear, N.J.; Tsai, Y.-C.; Sadan, A.N.; Sanchez-Pintado, B.; Zarouchlioti, C.; Maher, G.J.; Liskova, P.; Tuft, S.J.; Hardcastle, A.J.; Clark, T.A.; et al. CRISPR/Cas9-Targeted Enrichment and Long-Read Sequencing of the Fuchs Endothelial Corneal Dystrophy–Associated TCF4 Triplet Repeat. Genet. Med. 2019, 21, 2092–2102. [Google Scholar] [CrossRef] [PubMed]
- The Filnemus Myotonic Dystrophy Study Group; De Antonio, M.; Dogan, C.; Daidj, F.; Eymard, B.; Puymirat, J.; Mathieu, J.; Gagnon, C.; Katsahian, S.; Hamroun, D.; et al. The DM-Scope Registry: A Rare Disease Innovative Framework Bridging the Gap between Research and Medical Care. Orphanet J. Rare Dis. 2019, 14, 122. [Google Scholar] [CrossRef]
- Procedure & Checklist—Preparing SMRTbell Libraries Using PacBio Barcoded Universal Primers for Multiplexing Amplicons; PacBio: Menlo Park, CA, USA, 2020.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total CCS Reads | % CCS Reads on-Target | % on-Target Reads Full-Length | Full DM1 Reads | Total Reads Analyzed | Reads < 50 CTG | Reads ≥ 50 CTG | % Expanded Allele | Estimated Repeat Size (Mode) | CCG Interruptions |
|---|---|---|---|---|---|---|---|---|---|---|
| E1 | 94,411 | 99.37 | 84 | 79,224 | 10,000 | 724 | 9276 | 93 | 5, ~447 (1099 max rpt) | No obvious interruption |
| E2.1 | 110,578 | 99.54 | 87 | 95,756 | 9999 | 627 | 9372 | 94 | 13, ~383 (831 max rpt) | 2–3x CCG |
| E3 | 67,182 | 99.61 | 89 | 59,342 | 10,000 | 375 | 9625 | 96 | 31, ~173 and ~215 (478 max rpt) | 6x CCG |
| L2 | 48,310 | 99.10 | 79 | 37,727 | 10,000 | 1291 | 8709 | 87 | 5, ~957 (2138 max rpt) | No obvious interruption |
| L3 | 29,512 | 99.02 | 77 | 22,625 | 9999 | 1670 | 8329 | 83 | 5, ~1156 (2081 max rpt) | No obvious interruption |
| A4.1 | 50,300 | 99.62 | 89 | 44,745 | 10,000 | 721 | 9279 | 93 | 5, ~109 (245 max rpt) | 1x CAG |
| 1201 | 37,407 | 99.44 | 85 | 31,683 | 9999 | 1813 | 8186 | 82 | 5, ~118 (619 max rpt) | No obvious interruption |
| B2 | 57,255 | 99.3 | 84 | 47,745 | 9995 | 1075 | 8920 | 89 | 5, ~292 (802 max rpt) | 3x CCG |
| 5289 | 42,349 | 99.31 | 82 | 34,690 | 9994 | 1595 | 8399 | 84 | 5, ~185 (896 max rpt) | No obvious interruption |
| Polymorphism | RS2070736 | RS572634 | RS1799894 | RS4646995 (Alu Element) | RS16939 | RS527221 | RS915915 | CTG |
|---|---|---|---|---|---|---|---|---|
| Localization | Exon 1 | Intron 4 | Intron 5 | Intron 8 | Intron 9 | Exon 10 | Intron 11 | 3′UTR |
| G2.2 | T/T | T/T | T/T | Ins/ins | G/G | C/C | T/T | 5/37 |
| G3.1 | T/T | T/T | T/T | Ins/ins | G/G | C/C | T/T | 16/37 |
| DM1 haplotype A | T | T | T | Ins | G | C | T | >50 |
| Sample | Sex | Ethnic Origin | CTG Size | Transmission | Pattern | Localization |
|---|---|---|---|---|---|---|
| Mangin et al. | 2021 | |||||
| G2.1 | F | French | 37 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| G2.2 | M | French | 37 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| G2.3 | M | French | 37 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| G2.4 | M | French | 37 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| G3.1 | M | French | 37 | Paternal | (CTG)6(CCGCTG)13(CTG)5 | |
| G3.2 | F | French | 37 | Paternal | (CTG)6(CCGCTG)13(CTG)5 | |
| G4.1 | M | French | 37 | Maternal | (CTG)6(CCGCTG)13(CTG)5 | |
| E1 | M | French | 546 | N/A | (CTG)n | Pure |
| E2.1 | M | French | 415 | Paternal (de novo) | (CTG)n(CCGCTG)(CTG)7(CCGCTG)(CTG)11 | 3′ |
| E3 | M | French | 184/228 | Paternal | (CTG)n(CCGCTG)2(CTG)3(CCGCTG)2(CTG)5(CCGCTG)(CTG)7(CCGCTG)(CTG)11 | 3′ |
| Fontana et al. | 2020 | |||||
| I-2 | F | Italian | 41 | N/A | (CTG)6(CCGCTG)15(CTG)5 | |
| II-1 | M | Italian | 41 | Maternal | (CTG)6(CCGCTG)15(CTG)5 | |
| III-1 | F | Italian | 41 | Paternal | (CTG)6(CCGCTG)15(CTG)5 | |
| Pesovic et al. | 2018 | |||||
| DF1-1 | F | Serbian | 520 | N/A | (CTG)n(CCGCTG)3(CTG)4(CCGCTG)2CTG(CCGCTG)(CTG)16 | 3′ |
| DF1-2 | M | Serbian | 350 | Maternal | (CTG)n(CCGCTG)3(CTG)4(CCGCTG)2CTG(CCGCTG)(CTG)16 | 3′ |
| DF1-3 | M | Serbian | 450 | Maternal | (CTG)n(CCGCTG)3(CTG)4(CCGCTG)2CTG(CCGCTG)(CTG)16 | 3′ |
| DF2-1 | M | Serbian | 320 | N/A | (CTG)n(CCG)36(CTG)n(CCGCTG)(CTG)6(CCGCTG)(CTG)11 | 3′ |
| DF3-1 | F | Serbian | 240 | N/A | CTG)n(CCG)3(CTG)6(CCG)3(CTG)6(CTG)CCG(CTG)8CCG(CTG)8 | 3′ |
| DF3-2 | F | Serbian | ND | Maternal | CTG)n(CCG)3(CTG)6(CCG)3(CTG)6(CTG)CCG(CTG)8CCG(CTG)8 | 3′ |
| DF5-2 | F | Serbian | 250 | Paternal (de novo) | (CTG)nCTC(CTG)26 | 3′ |
| DF5-3 | F | Serbian | 300 | Paternal | (CTG)n (sister of DF5-2) | Pure |
| Cumming et al. | 2018 | |||||
| DMGV14 | F | Scotish | 381 | Paternal (de novo) | (CTG)180-240(CCGCTG)53-67(CTG)53-67 | 3′ |
| DMGV182 | M | Scotish | 293 | Paternal (de novo) | (CTG)200-300(CCG)(CTG)41-59 | 3′ |
| DMGV15 | F | Scotish | 327 | Paternal (de novo) | (CTG)260-320(CCGCTGCTG)10-14(CTG)15-23. | 3′ |
| De siena et al. | 2018 | |||||
| 2 patients | N/A | Italian | N/A | N/A | (CTG)nCTC-(CCGCTG)CTG-CCG(CTG)n | 5′ |
| 3 patients | N/A | Italian | N/A | N/A | (CTG)nCTC-(CCGCTG)CTG-CCG(CTG)n | 3′ |
| Tomé et al. | 2018 | |||||
| A1 | F | French | 170 | N/A | (CTG)n<32(CAG)1(CTG)n | 5′ |
| A2 | F | French | 150 | Maternal | (CTG)n<32(CAG)1(CTG)n | 5′ |
| A3 | F | French | 140 | Maternal | (CTG)n<32(CAG)1(CTG)n | 5′ |
| A4.1 | F | French | 125 | Maternal | (CTG)n<32(CAG)1(CTG)n | 5′ |
| A4.2 | F | French | 130 | Maternal | (CTG)n<32(CAG)1(CTG)n | 5′ |
| A4.3 | N/A | French | 125 | Maternal | (CTG)n<32(CAG)1(CTG)n | 5′ |
| B1 | F | French | 365 | N/A | (CTG)11(CCGCTG)(CTG)1(CCGCTG)(CTG)3(CCGCTG)(CTG)n | 5′ |
| B2 | F | French | 310 | Maternal | (CTG)11(CCGCTG)(CTG)1(CCGCTG)(CTG)3(CCGCTG)(CTG)n | 5′ |
| B3.1 | N/A | French | 300 | Maternal | (CTG)11(CCGCTG)(CTG)1(CCGCTG)(CTG)3(CCGCTG)(CTG)n | 5′ |
| B3.2 | N/A | French | 235 | Maternal | (CTG)11(CCGCTG)(CTG)1(CCGCTG)(CTG)3(CCGCTG)(CTG)n | 5′ |
| B3.3 | N/A | French | 250 | Maternal | (CTG)11(CCGCTG)(CTG)1(CCGCTG)(CTG)3(CCGCTG)(CTG)n | 5′ |
| Botta et al. | 2017 | |||||
| A1 | M | Italian | 1000–1400 | N/A | (CTG)880-1280(CTG)2(CCGCTG)(CTG)111(CCGCTG)(CTG)3 | 3′ |
| A2 | F | Italian | 475–640 | Paternal | (CTG)437-602(CTG)14(CCGCTG)(CTG)17(CCGCTG)(CTG)3 | 3′ |
| A3 | N/A | Italian | 500 | Maternal | (CTG)380(CTG)28(CCGCTG)(CTG)39(CTC)(CTG)36(CCGCTG)(CTG)7(CCGCTG)(CTG)3 | 3′ |
| B1 | F | Italian | 740–930 | N/A | (CTG)699-889(CCGCTG)2(CCG)2(CTG)3(CCGCTG)3(CTG)26 | 3′ |
| B2 | F | Italian | 450–550 | Maternal | (CTG)372-472(CTG)16(CCGCTG)(CTG)(CCGCTG)4(CTG)(CCGCTG)4(CCG)(CCGCTG)4(CCG)(CCGCTG)5(CTG)22 | 3′ |
| C1 | F | Italian | 140 | N/A | (CTG)30(CCG)2(CTG)2(CCGCTG)(CTG)104 | 5′ |
| C2 | F | Italian | 121 | Maternal | (CTG)28(CCG)2(CTG)2(CCGCTG)(CTG)87 | 5′ |
| C3 | N/A | Italian | 113 | Maternal | (CTG)31(CCG)2(CTG)2(CCGCTG)(CTG)12(CCGCTG)(CTG)62 | 5′ |
| D | F | Italian | 600–700 | N/A | (CTG)514-614(CTG)68(CCG)9(CTG)9 | 3′ |
| E | F | Italian | 500–660 | N/A | (CTG)404-564(CTG)33(CCGCTG)28(CTG)7 | 3′ |
| F | M | Italian | 250 | N/A | (CTG)208(CTG)5(CCGCTG)16(CTG)5 | 3′ |
| G | M | Italian | 400–580 | N/A | (CTG)330-510(CTG)8(CCGCTG)17(CTG)2(CCGCTG)(CTG)24 | 3′ |
| H | M | Italian | 175 | N/A | (CTG)133)(CTG)8(CCGCTG)(CTG)4(CCG)2(CTG)(CCG)4(CTG)2(CCG)4(CTG)(CCG)2(CTG)2(CCGCTG)(CTG)8 | 3′ |
| I | M | Italian | 260–722 | N/A | (CTG-188-650(CTG)(CCGCTG)(CTG)(CCGCTG)(CTG)6(CCGCTG)(CTG)27(CCGCTG)(CTG)6(CCGCTG)(CTG)21 | 3′ |
| Lian et al. | 2016 | |||||
| Individu 1 | N/A | ND | 520 | N/A | (CTG)n(CCG)50(CTG)9(CCGCTG)(CTG)6 | 3′ |
| Individu 2 | N/A | ND | 400–480 | N/A | (CTG)21(CCGCTG)(CTG)2(CCGCTG)(CTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)2(CCGCTG)2(CTG)2(CCGCTGCTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)n | 5′ |
| Santoro et al. | 2015 | |||||
| Pt58 | N/A | Italian | 118 | Maternal | (CTG)32(CCGCTG)(CTG)(CCGCTG)(CTG)5(CCG)2(CTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)24(CTG)31(CCG)2(CTG)2(CCGCTG)(CTG)21(CCGCTG)(CTG)33(CCG)2(CTG)2CCGCTC(CTG)4 | 3′ |
| Pt59 | N/A | Italian | 400–580 | Paternal | (CCGCTG)7TCGCTG(CCGCTG)7(CTG)20 | 3′ |
| Pt60 | N/A | Italian | 450–550 | Maternal | (CTG)16(CCGCTG)(CTG)1(CCGCTG)4CTG[(CCGCTG)4CCG]2(CCGCTG)5(CTG)22 | 3′ |
| Pt61 | N/A | Italian | 475–640 | Paternal | (CTG)2 G (CTG)9 G (CTG)20(CTG)23TTG(CTG)4 | 3′ |
| Pt62 | N/A | Italian | 550–700 | Paternal | (CTG)5(CCGCTGCTG)46 | 3′ |
| Pt63 | N/A | Italian | 600–700 | Paternal | (CTG)68(CCGCTG)(CTG)8 | 3′ |
| Pt64 | N/A | Italian | 600–830 | Paternal | (CTG)9(CCGCTGCTG)61 | 3′ |
| Pt65 | N/A | Italian | 740–930 | Paternal | (CCGCTG)2(CCG)2(CTG)3(CCGCTG)3(CTG)7(CCGCTG)18 | 3′ |
| Pt66 | N/A | Italian | 970 | Maternal | (CTG)12(CCGCTG)(CTG)5(CCGCTG)(CTG)4(CCGCTG)(CTG)4 | 3′ |
| Santoro et al. | 2013 | |||||
| pt1/pt62 | N/A | Italian | 550–700 | Paternal | (CTG)2(CCGCTGCTG)5(CCG)(CCGCTGCTG)46(CTG)5 | 3′ |
| pt2/pt64 | N/A | Italian | 600–830 | Paternal | (CTG)(CCGCTGCTG)4(CCG)(CCGCTGCTG)61(CTG)9 | 3′ |
| pt3 | N/A | Italian | 65 | N/A | (CTG)(CTG/CCG)(CTG)2(CTG/CCG)(CCGCTGCTG)5(CTG)3 | 3′ |
| pt4 | N/A | Italian | 900 | N/A | (CTG)16(CCGCTG)(CTG)3(CCGCTG)(CTG)7(CCGCTG)(CTG)4(CCGCTG)(CTG)(CCGCTG)(CTG)(CCGCTG)(CTG)7 | 3′ |
| pt5/pt66 | N/A | Italian | 970 | Maternal | (CTG)12(CCGCTG)(CTG)5(CCGCTG)(CTG)4(CCGCTG)(CTG)4 | 3′ |
| Addis et al. | 2012 | |||||
| AA | N/A | Sardinian | N/A | N/A | (CTG)3(CCGCTG)(CTG)2(CCG)CT en 3′ | 3′ |
| Radvansky et al. | 2011 | |||||
| Sample 7 | N/A | Czech | N/A | N/A | (CTG)n(CCG)38(CTG)22 | 3′ |
| Sample 8 | N/A | Czech | N/A | N/A | (CTG)nCTC(CTG)9(CCGCTG)2(CTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)12 | 3′ |
| Sample 9 | N/A | Czech | 43 | N/A | (CTG)6(CCGCTG)16(CTG)5 | |
| Sample 10 | N/A | Czech | 43 | N/A | (CTG)6(CCGCTG)16(CTG)5 | |
| Sample 11 | N/A | Czech | 39 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| Sample 12 | N/A | Czech | 41 | N/A | (CTG)6(CCGCTG)15(CTG)5 | |
| Musova et al. | 2009 | |||||
| A-1 | N/A | Czech | 230 | Maternal | (CTG)n(CTC)(CTG)9(CCGCTG)2(CTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)12 | 3′ |
| A-2 | F | Czech | 300 | Paternal | (CTG)n(CTC)(CTG)9(CCGCTG)2(CTG)2(CCGCTG)(CTG)4(CCGCTG)(CTG)12 | 3′ |
| A-3 | F | Czech | 400–500 | Paternal | (CTG)n(CTC)(CTG)7(CCGCTG)(CTG)4(CCGCTG)(CTG)4(CCGCTG)(CTG)12 | 3′ |
| A-4 | M | Czech | 600–800 | N/A | (CTG)n(CTC)(CTG)9(CCGCTG)(CTG)4(CCGCTG)(CTG)4(CCGCTG)(CTG)12 | 3′ |
| A-5 | F | Czech | 450–650 | N/A | (CTG)n(CTC)(CTG)9(CCGCTGCTG)6(CTG)8 | 3′ |
| A-6 | F | Czech | 650–750 | Maternal | (CTG)n(CTC)(CTG)9(CCGCTGCTG)5(CTG)8 | 3′ |
| A-7 | M | Czech | 270 | Maternal | (CTG)n(CTC)(CTG)9(CCGCTGCTG)6(CTG)8 | 3′ |
| B-1 | M | Czech | 450 | N/A | (CTG)n(CCGCTG)39(CCG)(CCGCTG)3(CTG)18 | 3′ |
| B-2 | M | Czech | 400 | Paternal | (CTG)n(CCGCTG)37(CCG)12(CTG)(CCGCTG)(CTG)10 | 3′ |
| C | F | Czech | 700 | N/A | (CTG)n(CCGCTG)2(CCG)8CTG(CCG)6CTG(CCG)6(CTG)(CCGCTG)(CCG)(CCGCTG)4(CCG)(CCGCTG)4(CTG)(CCGCTG)4(CTG)(CCGCTG)4(CCG)(CCGCTG)3(CTG)3(CCGCTG)2(CTG)10 | 3′ |
| D | M | Czech | 37 | N/A | (CTG)6(CCGCTG)13(CTG)5 | |
| E-1 | M | Czech | 43 | Paternal | (CTG)6(CCGCTG)16(CTG)5 | |
| E-2 | M | Czech | 43 | N/A | (CTG)6(CCGCTG)16(CTG)5 | |
| Leeflang et al. | 1995 | |||||
| 5048 | N/A | Caucasian | 37 | N/A | (CTG)4(CCGCTG)16(CTG)1 Alu Haplotype | |
| Braida et al. | 2010 | |||||
| III-9 | M | Dutch | 225/DM1-charcot | N/A | mutant allele: (CTG)n(GGC)3G(CCG)20(CCGCTG)14(CTG)35 and normal allele: (CTG)5(CCGCTG)14(CTG) | 3′ |
| IV-11 | F | Dutch | 38/Normal | Paternal | (CTG)5(CCGCTG)14(CTG) | |
| IV-12 | M | Dutch | 38/Normal | Paternal | (CTG)5(CCGCTG)14(CTG) | |
| DM1-Charcot family | N/A | Dutch | 500<CTG>200 | N/A | (CTG)n(GGC)3G(CCG)20(CCGCTG)14(CTG)35 | 3′ |
| DM1-UC1 | F | French | N/A | N/A | (CTG)n(CNG)(CTG)n(CNG)(CCGCTG)17(CTG)15 | 3′ |
| DM1-UC2 | M | French | N/A | Maternal | (CTG)222(CCG)5(CTG)5(CCG)5(CTG)5(CCG)5(CCGCTG)23(CTG)14 | 3′ |
| DM1-UC3 | N/A | French | N/A | N/A | (CTG)425(CCGCTG)4(CTG)2(CCGCTG)4(CTG)2(CCGCTG)4(CTG)4(CCGCTG)(CTG)4(CCGCTG)(CTG)4(CCGCTG)(CTG)14 | 3′ |
| DM1-UC4 | N/A | French | N/A | N/A | (CTG)318(CCGCTG)19(CTG)13 | 3′ |
| DM1-UC5 | N/A | French | N/A | N/A | (CTG)412(CCG)5(CTG)5(CCGCTG)(CCG)5(CTG)5(CCGCTG)3(CTG)4(CCGCTG)3(CTG)4(CCGCTG)3(CTG)4(CCGCTG)(CTG)5(CCGCTG)(CTG)5(CCGCTG)(CTG)5 | 3′ |
| DM1-UC6 | N/A | French | N/A | N/A | (CTG)516(CCG)3(CTG)(CCG)3(CTG)(CCG)2(CTG)5(CCGCTG)(CTG)5(CCGCTG)(CTG)5(CCG)3(CTG)6(CCG)3(CTG)6(CCG)3(CTG)6(CCG)3(CTG)6(CTG)26 | 3′ |
| DM1-UC7 | N/A | French | 41 | N/A | (CTG)6(CCGCTG)15(CTG)5 | |
| DM1-UC8 | N/A | French | N/A | N/A | (CTG)225(CCGCTG)(CTG)7(CCGCTG)(CTG)7(CCG)3(CTG)8(CCG)3(CTG)8(CCG)3(CTG)8(CCG)3(CTG)8(CTG)2(CCGCTG)(CTG)8 | 3′ |
| DM1-UC9 | N/A | French | 396 | N/A | (CTG)n pure et délétion 10bp en 3′ | N/A |
| DM1-UC10 | M | French | N/A | N/A | (CTG)166(CCGCTG)31(CTG)58 | 3′ |
| DM1-UC11 | N/A | French | N/A | Paternal | (CTG)105(CCGCTG)119(CTG)8 | 3′ |
| Leferink et al. | 2019 | |||||
| USN04034 | N/A | Dutch | >150 | N/A | (CTG)n(CCGCTG)~100(CTG)n | 3′ |
| USN08692 | N/A | Dutch | >150 | N/A | (CTG)n(CCGCTG)114(CTG)n | 3′ |
| USN00144 | N/A | Dutch | 174 | N/A | (CTG)24(CCGCTG)45(CTG)60 | 5′ |
| USN01084 | N/A | Dutch | >150 | N/A | (CTG)n(CCGCTG)167(CTG)n | 3′ |
| USN01299 | N/A | Dutch | >150 | N/A | (CTG)n(CCGCTG)37(CTG)n | 3′ |
| Ballester-Lopez et al. | 2020 | |||||
| Patient 1 | F | Spanish | 319 | N/A | (CTG)n(CCGCTG)(CTG)16(CCGCTG)(CTG)n | N/A |
| Patient 2 | F | Spanish | 241 | N/A | (CTG)n(CCGCTG)(CTG)8(CCG)(CCGCTG)(CTG)(CCGCTG)(CTG)3(CCGCTG)(CTG)n | N/A |
| Patient 3 | F | Spanish | 368 | N/A | (CTG)n(CCGCTG)3(CTG)3(CCGCTG)3(CTG)3(CCGCTG)2(CTG)n | N/A |
| Patient 4 | M | Spanish | 222 | Maternal | (CTG)n(CCGCTG)(CTG)8(CCG)(CCGCTG)(CTG)(CCGCTG)(CTG)3(CCGCTG)(CTG)n | N/A |
| Patient 5 | F | Spanish | 547 | Maternal | (CTG)n(CCGCTG)3(CTG)(CCG)2(CCGCTG)2(CTG)3(CCGCTG)2(CTG)n | N/A |
| Patients | Age (year) | Interruptions | PCR Product (bp) | 5′ Modification | Buffer Sequence | Barcode Name | Barcode | Primer ST-barcoded-300-F | Primer ST-barcoded-300-R |
|---|---|---|---|---|---|---|---|---|---|
| E1 | 56 | Yes (CCG) | 2551 | Phosphate | GGTAG | BC1018_Forward | TCACGTGCTCACTGTG | GGTAGTCACGTGCTCACTGTGGAACTGTCTTCGACTCCGGG | GGTAGTCACGTGCTCACTGTGGCACTTTGCGAACCAACGAT |
| E2.1 | 29 | Yes (CCG) | 1864 | Phosphate | GGTAG | BC1019_Forward | ACACACTCTATCAGAT | GGTAGACACACTCTATCAGATGAACTGTCTTCGACTCCGGG | GGTAGACACACTCTATCAGATGCACTTTGCGAACCAACGAT |
| E3 | 0 | Yes (CCG) | 1150 | Phosphate | GGTAG | BC1020_Forward | CACGACACGACGATGT | GGTAGCACGACACGACGATGTGAACTGTCTTCGACTCCGGG | GGTAGCACGACACGACGATGTGCACTTTGCGAACCAACGAT |
| L2 | 31 | No | >5000 | Phosphate | GGTAG | BC1012_Forward | ACACTAGATCGCGTGT | GGTAGACACTAGATCGCGTGTGAACTGTCTTCGACTCCGGG | GGTAGACACTAGATCGCGTGTGCACTTTGCGAACCAACGAT |
| L3 | 26 | No | >5000 | Phosphate | GGTAG | BC1013_Forward | CTCTCGCATACGCGAG | GGTAGCTCTCGCATACGCGAGGAACTGTCTTCGACTCCGGG | GGTAGCTCTCGCATACGCGAGGCACTTTGCGAACCAACGAT |
| A4.1 | 32 | Yes (CAG) | 760 | Phosphate | GGTAG | BC1016_Forward | CATAGAGAGATAGTAT | GGTAGCATAGAGAGATAGTATGAACTGTCTTCGACTCCGGG | GGTAGCATAGAGAGATAGTATGCACTTTGCGAACCAACGAT |
| 1201 | 32 | No | 796 | Phosphate | GGTAG | BC1017_Forward | CACACGCGCGCTATAT | GGTAGCACACGCGCGCTATATGAACTGTCTTCGACTCCGGG | GGTAGCACACGCGCGCTATATGCACTTTGCGAACCAACGAT |
| B2 | 33 | Yes (CCG) | 1291 | Phosphate | GGTAG | BC1014_Forward | CTCACTACGCGCGCGT | GGTAGCTCACTACGCGCGCGTGAACTGTCTTCGACTCCGGG | GGTAGCTCACTACGCGCGCGTGCACTTTGCGAACCAACGAT |
| 5289 | 37 | No | 1141 | Phosphate | GGTAG | BC1015_Forward | CGCATGACACGTGTGT | GGTAGCGCATGACACGTGTGTGAACTGTCTTCGACTCCGGG | GGTAGCGCATGACACGTGTGTGCACTTTGCGAACCAACGAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangin, A.; de Pontual, L.; Tsai, Y.-C.; Monteil, L.; Nizon, M.; Boisseau, P.; Mercier, S.; Ziegle, J.; Harting, J.; Heiner, C.; et al. Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052616
Mangin A, de Pontual L, Tsai Y-C, Monteil L, Nizon M, Boisseau P, Mercier S, Ziegle J, Harting J, Heiner C, et al. Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1. International Journal of Molecular Sciences. 2021; 22(5):2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052616
Chicago/Turabian StyleMangin, Antoine, Laure de Pontual, Yu-Chih Tsai, Laetitia Monteil, Mathilde Nizon, Pierre Boisseau, Sandra Mercier, Janet Ziegle, John Harting, Cheryl Heiner, and et al. 2021. "Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1" International Journal of Molecular Sciences 22, no. 5: 2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052616