
The Role of Immunogenetics in COVID-19

 ,
,  ,
, 
 and
and
Abstract
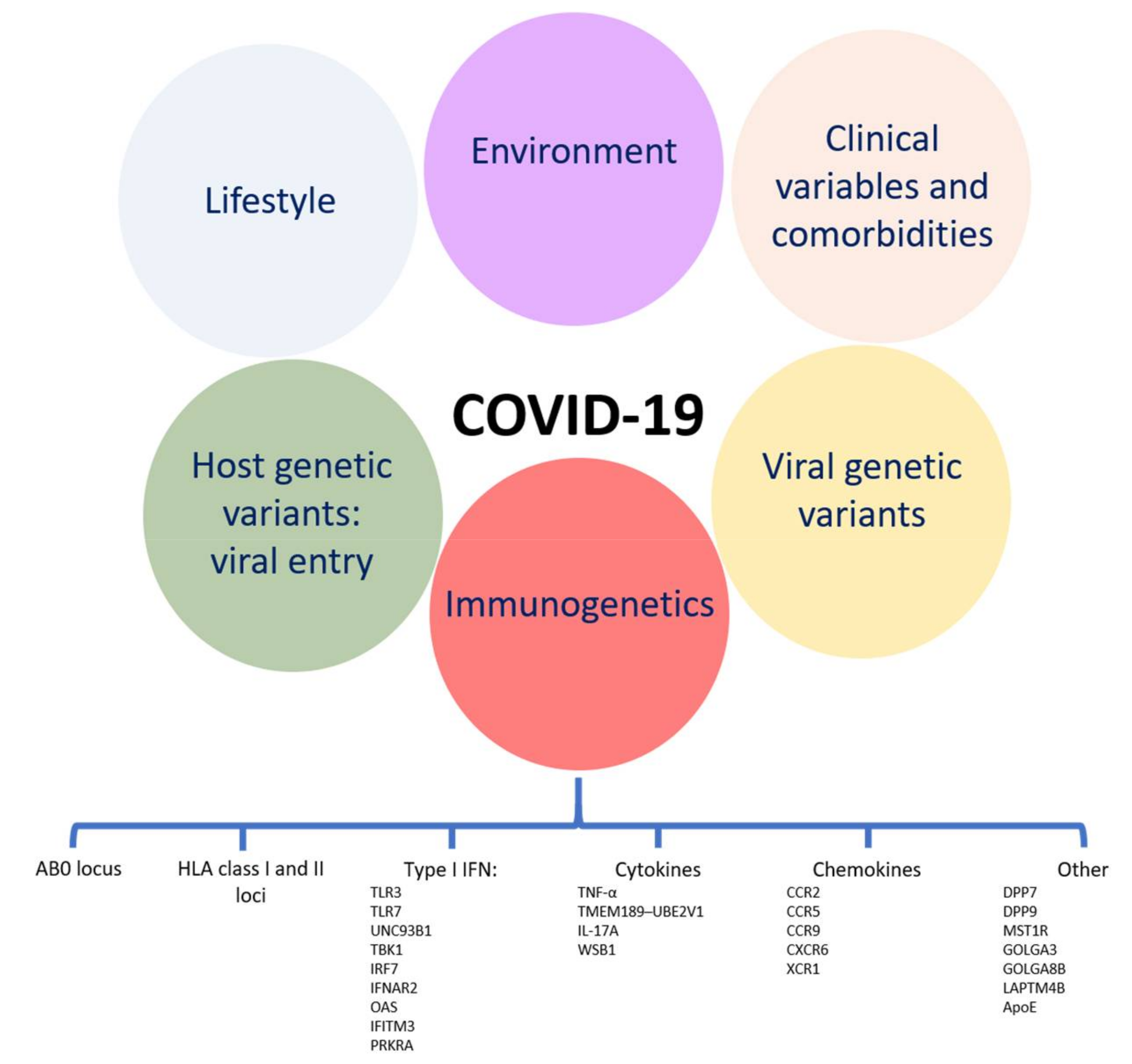
:1. Introduction
2. AB0 Groups
3. HLA
3.1. Incidence and Susceptibility
3.2. Disease Severity
3.3. Mortality
3.4. Transplanted Patients
3.5. Functional and Mechanistic Considerations
4. Other Immune Response Genes
4.1. Type I Interferons and Players of Their Molecular Pathways
4.2. Other Cytokines, Chemokines and Their Signaling Pathways
4.3. Other Genetic Determinants of Establishment/Maintenance/Resolution of the Immune Response and Antigen Presentation
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| AD | Autosomal-dominant |
| AH | Ancestral haplotype |
| AIDS | Acquired immunodeficiency syndrome |
| APACHE | Acute Physiology and Chronic Health Evaluation |
| ApoE | Apoliprotein E |
| AR | Autosomal-recessive |
| ARDS | Acute respiratory distress syndrome |
| CD40L | CD40 ligand |
| CDC | Centers for Disease Control and Prevention |
| CCR2 | C-C Motif Chemokine Receptor 2 |
| CCR3 | C-C Motif Chemokine Receptor 3 |
| CCR5 | C-C Motif Chemokine Receptor 5 |
| CCR9 | C-C Motif Chemokine Receptor 9 |
| COVID-19 | Coronavirus disease 19 |
| CRP | C-reactive protein |
| CXCR3 | C-X-C Motif Chemokine Receptor 3 |
| CXCR6 | C-X-C Motif Chemokine Receptor 6 |
| DPP | Dipeptidyl peptidase |
| EBV | Epstein-Barr Virus |
| ECMO | Extracorporeal membrane oxygenation |
| eQTL | Expression quantitative locus |
| FOXP3 | Forkhead box p3 |
| GM | Immunoglobulin G heavy chain |
| GOLGA3 | Golgin subfamily A 3 |
| GOLGA8B | Golgin A8 family member B |
| GWAS | Genome wide association study |
| HIV | Human immunodeficiency virus |
| HLA | Human Leukocyte Antigens |
| ICU | Intensive care unit |
| IFITM3 | Interferon-induced transmembrane protein 3 |
| IFNB1 | Interferon β 1 |
| IFN-α | Interferon-α |
| IFN-β | Interferon-β |
| IFN-γ | Interferon-γ |
| IFNAR1 | IFN-α receptor 1 |
| IFNAR2 | IFN-α receptor 2 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| IL-10 | Interleukin-10 |
| IL-17A | Interleukin-17A |
| IL-17F | Interleukin-17F |
| IL-21 | Interleukin-21 |
| IP-10 | Interferon γ-induced protein-10 |
| IRF | IFN regulatory factors |
| ISG15 | Ubiquitin-like modifier ISG15 |
| KIR | Killer Immunoglobulin Receptors |
| KIR2DL2 | Killer Cell Immunoglobulin-Like Receptor 2DL2 |
| KIR2DL3 | Killer Cell Immunoglobulin-Like Receptor 2DL3 |
| LAPTM4B | Lysosomal Protein Transmembrane 4 β |
| MIP-1α | Macrophage inflammatory protein 1α |
| MIP-1β | Macrophage inflammatory protein 1β |
| MMR | Measles, mumps, and rubella |
| MST1R | Macrophage stimulating 1 receptor |
| NEMO/IKBKG | NF-κB essential modulator |
| NK | Natural Killer |
| OAS | 2’-5’-Oligoadenylate synthetase |
| PHA | Phytohemagglutinin |
| PKR | IFN-induced, double-stranded RNA-activated protein kinase |
| pLOF | Predicted to be loss-of-function |
| PRKRA | Protein Activator of The Interferon-Induced Protein Kinase |
| RBC | Red blood cells |
| SNP | Single nucleotide polymorphism |
| SOFA | Sepsis-related Organ Failure Assessment |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT2 | Signal transducer and activator of transcription 2 |
| TBK1 | TANK binding kinase 1 |
| TICAM1/TRIF | TIR-domain containing adaptor inducing IFN-β |
| TLR | Toll like receptor |
| TMEM189 | Transmembrane protein 189 |
| TMPRSS2 | Transmembrane protease serine protease 2 |
| TNF-α | Tumor necrosis factor-α |
| TRAF3 | TNF Receptor Associated Factor 3 |
| UBE2V1 | Ubiquitin-conjugating enzyme E2 variant 1 |
| UNC93B1 | Unc-93 homolog B1 |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
| VWF | von Willebrand factor |
| WSB1 | WD Repeat And SOCS Box Containing 1 |
| XCL1 | X-C Motif Chemokine Ligand 1 |
| XCR1 | X-C Motif Chemokine Receptor 1 |
References
- Guo, G.; Ye, L.; Pan, K.; Chen, Y.; Xing, D.; Yan, K.; Chen, Z.; Ding, N.; Li, W.; Huang, H.; et al. New Insights of Emerging SARS-CoV-2: Epidemiology, Etiology, Clinical Features, Clinical Treatment, and Prevention. Front. Cell Dev. Biol. 2020, 410. PMCID: PMC7256189. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020, 1–14, PMCID: PMC7537588. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2020. PMCID: PMC7361342. [Google Scholar] [CrossRef] [PubMed]
- Tizaoui, K.; Zidi, I.; Lee, K.H.; Ghayda, R.A.; Hong, S.H.; Li, H.; Smith, L.; Koyanagi, A.; Jacob, L.; Kronbichler, A.; et al. Update of the current knowledge on genetics, evolution, immunopathogenesis, and transmission for coronavirus disease 19 (COVID-19). Int. J. Biol. Sci. 2020, 16, 2906–2923, PMCID: PMC7545713. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Hilgenfeld, R.; Zabel, P. Molecular mechanisms of severe acute respiratory syndrome (SARS). Respir. Res. 2005, 6, 8, PMCID: PMC548145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groneberg, D.A.; Poutanen, S.M.; Low, D.E.; Lode, H.; Welte, T.; Zabel, P. Treatment and vaccines for severe acute respiratory syndrome. Lancet Infect. Dis. 2005, 5, 147–155, PMCID:PMC7106466. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Tabarsi, P.; Varahram, M.; Folkerts, G.; Adcock, I.M. The Immune Response and Immunopathology of COVID-19. Front. Immunol. 2020, 11, 2037, PMCID: PMC7479965. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, S. Understand variability of COVID-19 through population and tissue variations in expression of SARS-CoV-2 host genes. Inf. Med. Unlocked 2020, 21, 100443, PMCID: PMC7550072. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Bandopadhyay, A.; Das, D.; Pandey, R.K.; Singh, V.; Khanam, N.; Srivastava, N.; Singh, P.P.; Dubey, P.K.; Pathak, A.; et al. Genetic Association of ACE2 rs2285666 Polymorphism With COVID-19 Spatial Distribution in India. Front. Genet. 2020, 11, 564741, PMCID: PMC7545580. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef]
- Barash, A.; Machluf, Y.; Ariel, I.; Dekel, Y. The Pursuit of COVID-19 Biomarkers: Putting the Spotlight on ACE2 and TMPRSS2 Regulatory Sequences. Front. Med. 2020, 7, 582793, PMCID: PMC7661736. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Sreenivasulu, K.; Mitra, P.; Misra, S.; Sharma, P. Role of Genetic Variants and Gene Expression in the Susceptibility and Severity of COVID-19. Ann. Lab. Med. 2021, 41, 129–138, PMCID: PMC7591285. [Google Scholar] [CrossRef] [PubMed]
- Chiappelli, F. CoViD-19 Susceptibility. Bioinformation 2020, 16, 501–504, PMCID: PMC7505245. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 40, PMCID: PMC7578581. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469, PMCID: PMC7477538. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Carrington, M. Immunogenetics of viral infections. Curr. Opin. Immunol. 2005, 17, 510–516. [Google Scholar] [CrossRef] [PubMed]
- McCoy, K.; Peterson, A.; Tian, Y.; Sang, Y. Immunogenetic Association Underlying Severe COVID-19. Vaccines 2020, 8, 700. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Caruso, C.; Vasto, S. Possible role of ABO system in age-related diseases and longevity: A narrative review. Immun. Ageing 2014, 11, 16, PMCID: PMC4265994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasto, S.; Caruso, C.; Castiglia, L.; Duro, G.; Monastero, R.; Rizzo, C. Blood group does not appear to affect longevity a pilot study in centenarians from Western Sicily. Biogerontology 2011, 12, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. Clin. Infect. Dis. 2020, ciaa1150, PMCID: PMC7454371. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A. Genetic Risk of Severe Covid-19. N. Engl. J. Med. 2020, 383, 1590–1591, PMCID: PMC7583681. [Google Scholar] [CrossRef] [PubMed]
- Zietz, M.; Zucker, J.; Tatonetti, N.P. Associations between blood type and COVID-19 infection, intubation, and death. Nat. Commun. 2020, 11, 5761, PMCID: PMC7666188. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, W.; Li, B.; Li, D.J.; Zhang, J.; Zhao, F. Association between ABO Blood Group System and COVID-19 Susceptibility in Wuhan. Front. Cell Infect. Microbiol. 2020, 10, 404, PMCID: PMC7385064. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-Diaz, E.; Llopis, J.; Parra, R.; Roig, I.; Ferrer, G.; Grifols, J.; Millán, A.; Ene, G.; Ramiro, L.; Maglio, L.; et al. Relationship between the ABO blood group and COVID-19 susceptibility, severity and mortality in two cohorts of patients. Blood Transfus. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.B.; Gu, D.Z.; Yu, J.N.; Yang, J.; Shen, W.Q. Association between ABO blood groups and COVID-19 infection, severity and demise: A systematic review and meta-analysis. Infect. Genet. Evol. 2020, 84, 104485, PMCID: PMC7391292. [Google Scholar] [CrossRef] [PubMed]
- Barnkob, M.B.; Pottegård, A.; Støvring, H.; Haunstrup, T.M.; Homburg, K.; Larsen, R.; Hansen, M.B.; Titlestad, K.; Aagaard, B.; Møller, B.K.; et al. Reduced prevalence of SARS-CoV-2 infection in ABO blood group O. Blood Adv. 2020, 4, 4990–4993, PMCID: PMC7594382. [Google Scholar] [CrossRef] [PubMed]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534, PMCID: PMC7315890. [Google Scholar] [CrossRef]
- Amoroso, A.; Magistroni, P.; Vespasiano, F.; Bella, A.; Bellino, S.; Puoti, F.; Alizzi, S.; Vaisitti, T.; Boros, S.; Grossi, P.A.; et al. HLA and AB0 Polymorphisms May Influence SARS-CoV-2 Infection and COVID-19 Severity. Transplantation 2020. [Google Scholar] [CrossRef] [PubMed]
- Latz, C.A.; DeCarlo, C.; Boitano, L.; Png, C.Y.M.; Patell, R.; Conrad, M.F.; Eagleton, M.; Dua, A. Blood type and outcomes in patients with COVID-19. Ann. Hematol. 2020, 99, 2113–2118, PMCID: PMC7354354. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Albaiceta, G.M.; Clemente, M.G.; Gómez, J. Lack of association between SNPsrs8176719(O blood group) and COVID-19: Data from Spanish age matched patients and controls. Transfusion 2020. [Google Scholar] [CrossRef] [PubMed]
- Licastro, F.; Candore, G.; Lio, D.; Porcellini, E.; Colonna-Romano, G.; Franceschi, C.; Caruso, C. Innate immunity and inflammation in ageing: A key for understanding age-related diseases. Immun. Ageing 2005, 2, 8, PMCID: PMC1166571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lio, D.; Scola, L.; Giarratana, R.M.; Candore, G.; Colonna-Romano, G.; Caruso, C.; Balistreri, C.R. SARS CoV2 infection _The longevity study perspectives. Ageing Res. Rev. 2021, 67, 101299, PMCID: PMC7885677. [Google Scholar] [CrossRef] [PubMed]
- Cooling, L. Blood Groups in Infection and Host Susceptibility. Clin. Microbiol. Rev. 2015, 28, 801–870, PMCID: PMC4475644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liumbruno, G.M.; Franchini, M. Beyond immunohaematology: The role of the ABO blood group in human diseases. Blood Transfus. 2013, 11, 491–499, PMCID: PMC3827391. [Google Scholar] [CrossRef] [PubMed]
- Handunnetthi, L.; Ramagopalan, S.V.; Ebers, G.C.; Knight, J.C. Regulation of major histocompatibility complex class II gene expression, genetic variation and disease. Genes Immun. 2010, 11, 99–112, PMCID: PMC2987717. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Yang, G.-y.; Song, Y.; Zhao, X.; So, C.; Liao, J.; Wang, L.-D.; Yang, C.S. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis 2001, 22, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Banerjee, S. Downregulation of HLA-ABC expression through promoter hypermethylation and downmodulation of MIC-A/B surface expression in LMP2A-positive epithelial carcinoma cell lines. Sci. Rep. 2020, 10, 5415. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Mutti, L.; Pentimalli, F.; Baglio, G.; Saladino, R.E.; Sileri, P.; Giordano, A. HLA-B*44 and C*01 Prevalence Correlates with Covid19 Spreading across Italy. Int. J. Mol. Sci. 2020, 21, 5205, PMCID: PMC7432860. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Franco, A.; Barrios, Y.; Cáceres, J.J.; Solé-Violán, J.; Perez, A.; Marcos, Y.; Ramos, J.A.; Ramos-Gómez, L.; et al. HLA genetic polymorphisms and prognosis of patients with COVID-19. Med. Intensiva. 2021, 45, 96–103, PMCID: PMC7474921. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Andreani, M.; Biancolella, M.; Liberatoscioli, L.; Passarelli, C.; Colona, V.L.; Rogliani, P.; Leonardis, F.; Campana, A.; Carsetti, R.; et al. HLA allele frequencies and susceptibility to COVID-19 in a group of 99 Italian patients. HLA 2020, 96, 610–614, PMCID: PMC7461491. [Google Scholar] [CrossRef] [PubMed]
- Kachuri, L.; Francis, S.S.; Morrison, M.; Wendt, G.A.; Bossé, Y.; Cavazos, T.B.; Rashkin, S.R.; Ziv, E.; Witte, J.S. The landscape of host genetic factors involved in infection to common viruses and SARS-CoV-2. medRxiv 2020, 12, 93, Update in Genome Med. 2020, 12, 93. PMCID: PMC7273301. [Google Scholar] [CrossRef]
- Benlyamani, I.; Venet, F.; Coudereau, R.; Gossez, M.; Monneret, G. Monocyte HLA-DR Measurement by Flow Cytometry in COVID-19 Patients: An Interim Review. Cytom. A 2020. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Pittet, J.F. Human Leukocyte Antigen-DR Deficiency and Immunosuppression-Related End-Organ Failure in SARS-CoV2 Infection. Anesth. Analg. 2020, 131, 989–992, PMCID: PMC7386673. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Zhang, J.; He, J.; Zhu, F. Distribution of HLA allele frequencies in 82 Chinese individuals with coronavirus disease-2019 (COVID-19). HLA 2020, 96, 194–196, PMCID: PMC7276866. [Google Scholar] [CrossRef]
- Yung, Y.L.; Cheng, C.K.; Chan, H.Y.; Xia, J.T.; Lau, K.M.; Wong, R.S.M.; Wu, A.K.L.; Chu, R.W.; Wong, A.C.C.; Chow, E.Y.D.; et al. Association of HLA-B22 serotype with SARS-CoV-2 susceptibility in Hong Kong Chinese patients. HLA 2020. [Google Scholar] [CrossRef] [PubMed]
- Poulton, K.; Wright, P.; Hughes, P.; Savic, S.; Welberry Smith, M.; Guiver, M.; Morton, M.; van Dellen, D.; Tholouli, E.; Wynn, R.; et al. A role for human leucocyte antigens in the susceptibility to SARS-Cov-2 infection observed in transplant patients. Int. J. Immunogenet. 2020, 47, 324–328, PMCID: PMC7361549. [Google Scholar] [CrossRef]
- Rosenbaum, J.T.; Hamilton, H.; Weisman, M.H.; Reveille, J.D.; Winthrop, K.L.; Choi, D. The Effect of HLA-B27 on Susceptibility and Severity of COVID-19. J. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KIR2DL2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KIR2DL3&keywords=KIR2DL3 (accessed on 26 February 2021).
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, e00510-20, PMCID: PMC7307149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olwenyi, O.A.; Dyavar, S.R.; Acharya, A.; Podany, A.T.; Fletcher, C.V.; Ng, C.L.; Reid, S.P.; Byrareddy, S.N. Immuno-epidemiology and pathophysiology of coronavirus disease 2019 (COVID-19). J. Mol. Med. 2020, 98, 1369–1383, PMCID: PMC7431311. [Google Scholar] [CrossRef]
- Tomita, Y.; Ikeda, T.; Sato, R.; Sakagami, T. Association between HLA gene polymorphisms and mortality of COVID-19: An in silico analysis. Immun. Inflamm. Dis. 2020, 8, 684–694, PMCID: PMC7654404. [Google Scholar] [CrossRef]
- Iturrieta-Zuazo, I.; Rita, C.G.; García-Soidán, A.; de Malet Pintos-Fonseca, A.; Alonso-Alarcón, N.; Pariente-Rodríguez, R.; Tejeda-Velarde, A.; Serrano-Villar, S.; Castañer-Alabau, J.L.; Nieto-Gañán, I. Possible role of HLA class-I genotype in SARS-CoV-2 infection and progression: A pilot study in a cohort of Covid-19 Spanish patients. Clin. Immunol. 2020, 219, 108572, PMCID: PMC7428760. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, J.P.; Carnalla-Cortés, M.; Pacheco-Olvera, D.L.; Ocampo-Godínez, J.M.; Oliva-Ramírez, J.; Moreno-Manjón, J.; Bernal-Alferes, B.; López-Olmedo, N.; García-Latorre, E.; Domínguez-López, M.L.; et al. A bioinformatic prediction of antigen presentation from SARS-CoV-2 spike protein revealed a theoretical correlation of HLA-DRB1*01 with COVID-19 fatality in Mexican population: An ecological approach. J. Med. Virol. 2020. PMCID: PMC7537233. [Google Scholar] [CrossRef]
- Pisanti, S.; Deelen, J.; Gallina, A.M.; Caputo, M.; Citro, M.; Abate, M.; Sacchi, N.; Vecchione, C.; Martinelli, R. Correlation of the two most frequent HLA haplotypes in the Italian population to the differential regional incidence of Covid-19. J. Transl. Med. 2020, 18, 352, PMCID: PMC7491019. [Google Scholar] [CrossRef] [PubMed]
- Gambino, C.M.; Aiello, A.; Accardi, G.; Caruso, C.; Candore, G. Autoimmune diseases and 8.1 ancestral haplotype: An update. HLA 2018, 92, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Habel, J.R.; Nguyen, T.H.O.; van de Sandt, C.E.; Juno, J.A.; Chaurasia, P.; Wragg, K.; Koutsakos, M.; Hensen, L.; Jia, X.; Chua, B.; et al. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A*02:01 phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 24384–24391, PMCID: PMC7533701. [Google Scholar] [CrossRef]
- Caruso, C.; Candore, G.; Romano, G.C.; Lio, D.; Bonafè, M.; Valensin, S.; Franceschi, C. Immunogenetics of longevity. Is major histocompatibility complex polymorphism relevant to the control of human longevity? A review of literature data. Mech. Ageing Dev. 2001, 122, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Player, M.A.; Barracchini, K.C.; Simonis, T.B.; Rivoltini, L.; Arienti, F.; Castelli, C.; Mazzocchi, A.; Belli, F.; Parmiani, G.; Marincola, F.M. Differences in frequency distribution of HLA-A2 subtypes between North American and Italian white melanoma patients: Relevance for epitope specific vaccination protocols. J. Immunother. Emphas. Tumor Immunol. 1996, 19, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Accardi, G.; Caruso, C. Genetic variation in human leukocyte antigen and susceptibility to acute myeloid leukemia. Acta Haematol. 2015, 133, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Joosten, S.A.; Sullivan, L.C.; Ottenhoff, T.H. Characteristics of HLA-E Restricted T-Cell Responses and Their Role in Infectious Diseases. J. Immunol. Res. 2016, 2016, 2695396, PMCID: PMC5028793. [Google Scholar] [CrossRef] [Green Version]
- Kanevskiy, L.; Erokhina, S.; Kobyzeva, P.; Streltsova, M.; Sapozhnikov, A.; Kovalenko, E. Dimorphism of HLA-E and its Disease Association. Int. J. Mol. Sci. 2019, 20, 5496, PMCID: PMC6862560. [Google Scholar] [CrossRef] [Green Version]
- Lopez, L.; Sang, P.C.; Tian, Y.; Sang, Y. Dysregulated Interferon Response Underlying Severe COVID-19. Viruses 2020, 12, 1433. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G. The Role of Type I Interferons in the Pathogenesis and Treatment of COVID-19. Front. Immunol. 2020, 11, 595739, PMCID: PMC7561359. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2020, 65, 101205, PMCID: PMC7604159. [Google Scholar] [CrossRef] [PubMed]
- Angioni, R.; Sánchez-Rodríguez, R.; Munari, F.; Bertoldi, N.; Arcidiacono, D.; Cavinato, S.; Marturano, D.; Zaramella, A.; Realdon, S.; Cattelan, A.; et al. Age- severity matched cytokine profiling reveals specific signatures in Covid-19 patients. Cell Death Dis. 2020, 11, 957, PMCID: PMC7646225. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. Immunesenescence: A Predisposing Risk Factor for the Development of COVID-19? Front. Immunol. 2020, 11, 573662, PMCID: PMC7573102. [Google Scholar] [CrossRef]
- Pietrobon, A.J.; Teixeira, F.M.E.; Sato, M.N. Immunosenescence and Inflammaging: Risk Factors of Severe COVID-19 in Older People. Front. Immunol. 2020, 11, 579220, PMCID: PMC7656138. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch. Immunol. Ther. Exp. 2016, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247, PMCID: PMC6773825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouadma, L.; Wiedemann, A.; Patrier, J.; Surénaud, M.; Wicky, P.H.; Foucat, E.; Diehl, J.L.; Hejblum, B.P.; Sinnah, F.; de Montmollin, E.; et al. Immune Alterations in a Patient with SARS-CoV-2-Related Acute Respiratory Distress Syndrome. J. Clin. Immunol. 2020, 40, 1082–1092, PMCID: PMC7443154. [Google Scholar] [CrossRef] [PubMed]
- Jurado, A.; Martín, M.C.; Abad-Molina, C.; Orduña, A.; Martínez, A.; Ocaña, E.; Yarce, O.; Navas, A.M.; Trujillo, A.; Fernández, L.; et al. COVID-19: Age, Interleukin-6, C-reactive protein, and lymphocytes as key clues from a multicentre retrospective study. Immun Ageing. 2020, 17, 22, PMCID: PMC7426672. [Google Scholar] [CrossRef]
- Files, J.K.; Boppana, S.; Perez, M.D.; Sarkar, S.; Lowman, K.E.; Qin, K.; Sterrett, S.; Carlin, E.; Bansal, A.; Sabbaj, S.; et al. Sustained cellular immune dysregulation in individuals recovering from SARS-CoV-2 infection. J. Clin. Investig. 2020, 140491. [Google Scholar] [CrossRef] [PubMed]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020, 5, eabd6832, PMCID: PMC7665314. [Google Scholar] [CrossRef]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Life Sci. 2020, 258, 118167, PMCID: PMC7387997. [Google Scholar] [CrossRef] [PubMed]
- Cacciapuoti, S.; De Rosa, A.; Gelzo, M.; Megna, M.; Raia, M.; Pinchera, B.; Pontarelli, A.; Scotto, R.; Scala, E.; Scarano, F.; et al. Immunocytometric analysis of COVID patients: A contribution to personalized therapy? Life Sci. 2020, 261, 118355, PMCID: PMC7456265. [Google Scholar] [CrossRef]
- Cantenys-Molina, S.; Fernández-Cruz, E.; Francos, P.; Lopez Bernaldo de Quirós, J.C.; Muñoz, P.; Gil-Herrera, J. Lymphocyte subsets early predict mortality in a large series of hospitalized COVID-19 patients in Spain. Clin. Exp. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, W.; Dong, Y.; Wang, X.; Dai, D.; Liu, X.; Wu, Y.; Li, M.; Zhang, W.; Zhou, H.; et al. Elevated Exhaustion Levels of NK and CD8+ T Cells as Indicators for Progression and Prognosis of COVID-19 Disease. Front. Immunol. 2020, 11, 580237, PMCID: PMC7591707. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Luo, M.; Liu, J.; Wu, L.; Lin, X.; Li, R.; Wang, Z.; Zhong, H.; Zheng, W.; et al. Lymphopenia predicted illness severity and recovery in patients with COVID-19: A single-center, retrospective study. PLoS ONE 2020, 15, e0241659, PMCID: PMC7673513. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wang, Z.; Yin, Y.; Zhao, Y.; Tao, P.; Zhong, P. Association of Peripheral Lymphocyte and the Subset Levels With the Progression and Mortality of COVID-19: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7, 558545, PMCID: PMC7546210. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.; Faridi, R.M.; Sligl, W.; Shabani-Rad, M.T.; Dharmani-Khan, P.; Parker, A.; Kalra, A.; Tripathi, M.B.; Storek, J.; Cohen Tervaert, J.W.; et al. Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv. 2020, 4, 5035–5039, PMCID: PMC7594380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, L.; Liu, J.; Chen, L.; Zhou, F.; Jin, T.; Jiang, L.; Li, X.; Yang, M.; Wang, H. The characteristics and predictive role of lymphocyte subsets in COVID-19 patients. Int. J. Infect. Dis. 2020, 99, 92–99, PMCID: PMC7398035. [Google Scholar] [CrossRef]
- Zingaropoli, M.A.; Perri, V.; Pasculli, P.; Dezza, F.C.; Nijhawan, P.; Savelloni, G.; La Torre, G.; D’Agostino, C.; Mengoni, F.; Lichtner, M.; et al. Major reduction of NKT cells in patients with severe COVID-19 pneumonia. Clin. Immunol. 2020, 222, 108630, PMCID: PMC7661928. [Google Scholar] [CrossRef]
- Naumova, E.; Ivanova, M.; Pawelec, G. Immunogenetics of ageing. Int. J. Immunogenet. 2011, 38, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.D.; Conneely, K.N. The role of DNA methylation and hydroxymethylation in immunosenescence. Ageing Res. Rev. 2019, 51, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Allan, R.S. Epigenomic drivers of immune dysfunction in aging. Aging Cell 2019, 18, e12878, PMCID: PMC6351880. [Google Scholar] [CrossRef] [Green Version]
- Ostan, R.; Bucci, L.; Capri, M.; Salvioli, S.; Scurti, M.; Pini, E.; Monti, D.; Franceschi, C. Immunosenescence and immunogenetics of human longevity. Neuroimmunomodulation 2008, 15, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Atlante, S.; Mongelli, A.; Barbi, V.; Martelli, F.; Farsetti, A.; Gaetano, C. The epigenetic implication in coronavirus infection and therapy. Clin. Epigenet. 2020, 12, 156, PMCID: PMC7576975. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Kinoshita, N.; Ide, S.; Nomoto, H.; Nakamoto, T.; Saito, S.; Ishikane, M.; Kutsuna, S.; Hayakawa, K.; Hashimoto, M.; et al. Serum CCL17 level becomes a predictive marker to distinguish between mild/moderate and severe/critical disease in patients with COVID-19. Gene 2021, 766, 145145, PMCID: PMC7489253. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362, Erratum in 2020, 20, 448, doi:10.1038/s41577-020-0353-y. PMCID: PMC7201395. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Moosavi, M. Cytokine-targeted therapy in severely ill COVID-19 patients: Options and cautions. EJMO 2020, 4, 179–181. [Google Scholar] [CrossRef]
- Neumann, J.; Prezzemolo, T.; Vanderbeke, L.; Roca, C.P.; Gerbaux, M.; Janssens, S.; Willemsen, M.; Burton, O.; Van Mol, P.; Van Herck, Y.; et al. Increased IL-10-producing regulatory T cells are characteristic of severe cases of COVID-19. Clin. Transl. Immunol. 2020, 9, e1204, PMCID: PMC7662088. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb Pathog. 2021, 18, 104799, PMCID: PMC7889464. [Google Scholar] [CrossRef]
- Pontali, E.; Volpi, S.; Signori, A.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Montale, A.; Bustaffa, M.; Angelelli, A.; Caorsi, R.; et al. Efficacy of early anti-inflammatory treatment with high doses IV Anakinra with or without glucocorticoids in patients with severe COVID-19 pneumonia. J. Allergy Clin. Immunol. 2021. PMCID: PMC7865089. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Savale, L.; Dossier, A.; Ghosn, J.; Taillé, C.; Visseaux, B.; Jebreen, K.; Diallo, A.; Tesmoingt, C.; Morer, L.; et al. Glucocorticoids with low-dose anti-IL1 anakinra rescue in severe non-ICU COVID-19 infection: A cohort study. PLoS ONE 2020, 15, e0243961, PMCID: PMC7743937. [Google Scholar] [CrossRef]
- Bozzi, G.; Mangioni, D.; Minoia, F.; Aliberti, S.; Grasselli, G.; Barbetta, L.; Castelli, V.; Palomba, E.; Alagna, L.; Lombardi, A.; et al. Anakinra combined with methylprednisolone in patients with severe COVID-19 pneumonia and hyperinflammation: An observational cohort study. J. Allergy Clin. Immunol. 2021, 147, 561–566.e4, PMCID: PMC7674131. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.A.; Stewart, I.; Fabbri, L.; Moss, S.; Robinson, K.; Smyth, A.R.; Jenkins, G. Systematic review and meta-analysis of anakinra, sarilumab, siltuximab and tocilizumab for COVID-19. Thorax 2021. [Google Scholar] [CrossRef]
- The REMAP-CAP Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19—Preliminary report. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.01.07.21249390v1 (accessed on 26 February 2021). [CrossRef]
- Lauder, S.N.; Jones, E.; Smart, K.; Bloom, A.; Williams, A.S.; Hindley, J.P.; Ondondo, B.; Taylor, P.R.; Clement, M.; Fielding, C.; et al. Interleukin-6 limits influenza-induced inflammation and protects against fatal lung pathology. Eur. J. Immunol. 2013, 43, 2613–2625, PMCID: PMC3886386. [Google Scholar] [CrossRef] [Green Version]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456, PMCID: PMC7719805. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2020, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, M.; Manet, C.; Montagutelli, X. Host genetic susceptibility to viral infections: The role of type I interferon induction. Genes Immun. 2020, 1–15, PMCID: PMC7677911. [Google Scholar] [CrossRef]
- Lee, B.L.; Barton, G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014, 24, 360–369, PMCID: PMC4037363. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFNAR1&keywords=IFNAR1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IFNAR2&keywords=IFNAR2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=STAT1&keywords=stat1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=STAT2&keywords=stat2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TICAM1&keywords=TICAM1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TBK1&keywords=TBK1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TRAF3&keywords=traf3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=UNC93B1&keywords=UNC93B1 (accessed on 26 February 2021).
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IKBKG&keywords=nemo (accessed on 26 February 2021).
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020, 289, 198163, PMCID: PMC7480444. [Google Scholar] [CrossRef] [PubMed]
- Di Maria, E.; Latini, A.; Borgiani, P.; Novelli, G. Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): Rapid systematic review and field synopsis. Hum. Genom. 2020, 14, 30, PMCID: PMC7484929. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in Covid-19. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qin, L.; Zhao, Y.; Zhang, P.; Xu, B.; Li, K.; Liang, L.; Zhang, C.; Dai, Y.; Feng, Y.; et al. Interferon-Induced Transmembrane Protein 3 Genetic Variant rs12252-C Associated With Disease Severity in Coronavirus Disease 2019. J. Infect. Dis. 2020, 222, 34–37, PMCID: PMC7197559. [Google Scholar] [CrossRef]
- Gómez, J.; Albaiceta, G.M.; Cuesta-Llavona, E.; García-Clemente, M.; López-Larrea, C.; Amado-Rodríguez, L.; López-Alonso, I.; Melón, S.; Alvarez-Argüelles, M.E.; Gil-Peña, H.; et al. The Interferon-induced transmembrane protein 3 gene (IFITM3) rs12252 C variant is associated with COVID-19. Cytokine 2020, 137, 155354. [Google Scholar] [CrossRef] [PubMed]
- Benetti, E.; Giliberti, A.; Emiliozzi, A.; Valentino, F.; Bergantini, L.; Fallerini, C.; Anedda, F.; Amitrano, S.; Conticini, E.; Tita, R.; et al. Clinical and molecular characterization of COVID-19 hospitalized patients. PLoS ONE 2020, 15, e0242534, PMCID: PMC7673557. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Sultan, A.; Elashry, M.A.; Farag, A.; Mortada, M.I.; Ghannam, M.A.; Saed, A.M.; Ghoneem, S. Association of TNF-α G-308 a Promoter Polymorphism with the Course and Outcome of COVID-19 Patients. Immunol. Investig. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232, PMCID: PMC7314139. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. ApoE e4e4 Genotype and Mortality With COVID-19 in UK Biobank. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1801–1803, PMCID: PMC7337688. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.L.; Conn, G.L. RNA regulation of the antiviral protein 2′-5′-oligoadenylate synthetase. Wiley Interdiscip. Rev. RNA 2019, 10, e1534, PMCID: PMC6585406. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.omim.org/entry/605579?search=ifitm3&highlight=ifitm3 (accessed on 26 February 2021).
- Wellington, D.; Laurenson-Schafer, H.; Abdel-Haq, A.; Dong, T. IFITM3: How genetics influence influenza infection demographically. Biomed. J. 2019, 42, 19–26, PMCID: PMC6468115. [Google Scholar] [CrossRef]
- Lee, J.; Robinson, M.E.; Ma, N.; Artadji, D.; Ahmed, M.A.; Xiao, G.; Sadras, T.; Deb, G.; Winchester, J.; Cosgun, K.N.; et al. IFITM3 functions as a PIP3 scaffold to amplify PI3K signalling in B cells. Nature 2020, 588, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.G.; O’Keeffe, M.; Reading, P.C.; Wakim, L.M. Rapid interferon independent expression of IFITM3 following T cell activation protects cells from influenza virus infection. PLoS ONE 2019, 14, e0210132, PMCID: PMC6334895. [Google Scholar] [CrossRef]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455, PMCID: PMC7095036. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/603424?search=PRKRA&highlight=prkra (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=PRKRA&keywords=PRKRA (accessed on 26 February 2021).
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390, PMCID: PMC1170771. [Google Scholar] [CrossRef]
- Al-Meghaiseeb, E.S.; Al-Robayan, A.A.; Al-Otaibi, M.M.; Arfin, M.; Al-Asmari, A.K. Association of tumor necrosis factor-α and -β gene polymorphisms in inflammatory bowel disease. J. Inflamm. Res. 2016, 9, 133–140, PMCID: PMC4918894. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cui, X.; Ning, L.; Wei, D. The effects of tumor necrosis factor-α (TNF-α) rs1800629 and rs361525 polymorphisms on sepsis risk. Oncotarget 2017, 8, 111456–111469, PMCID: PMC5762335. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Zhao, H. Association between Tumor Necrosis Factor-308 G/A Polymorphism and Chronic Obstructive Pulmonary Disease Risk in Chinese Population: Evidence from a Meta-Analysis. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.K.; Han, Y.R.; Hong, D.; Chon, J.; Chung, J.H.; Hong, S.J.; Park, M.S.; Ban, J.Y. Promoter Polymorphism (-308G/A) of Tumor Necrosis Factor-Alpha (TNF-α) Gene and Asthma Risk: An Updated Meta-Analysis. Genet. Test. Mol. Biomark. 2019, 23, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.C.D.; Gomes, T.N.; Góis, I.A.F.; Oliveira, J.S.; Coelho, L.F.L.; Ferreira, G.P.; Silva, F.R.P.D.; Pereira, A.C.T.D.C. Association of single nucleotide polymorphisms in TNF-α (-308G/A and -238G/A) to dengue: Case-control and meta-analysis study. Cytokine 2020, 134, 155183. [Google Scholar] [CrossRef] [PubMed]
- Ferdosian, F.; Dastgheib, S.A.; Hosseini-Jangjou, S.H.; Nafei, Z.; Lookzadeh, M.H.; Noorishadkam, M.; Mirjalili, S.R.; Neamatzadeh, H. Association of TNF-α rs1800629, CASP3 rs72689236 and FCGR2A rs1801274 Polymorphisms with Susceptibility to Kawasaki Disease: A Comprehensive Meta-Analysis. Fetal Pediatr. Pathol. 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.Z.; Ezzat, D.A. Association of-308G/A Polymorphism and Serum Level of TNF-α with Bronchial asthma in Children. Egypt. J. Immunol. 2018, 25, 117–124. [Google Scholar] [PubMed]
- Thriveni, K.; Raju, A.; Ramaswamy, G.; Krishnamurthy, S. Impact of gene polymorphism of TNF-α rs 1800629 and TNF-β rs 909253 on plasma levels of South Indian breast cancer patients. Indian J. Cancer. 2018, 55, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Hurtado, I.A.; Puebla-Pérez, A.M.; Delgado-Saucedo, J.I.; Figuera, L.E.; Zúñiga-González, G.M.; Gomez-Mariscal, K.; Ronquillo-Carreón, C.A.; Gallegos-Arreola, M.P. Association between TNF-α-308G>A and -238G>A gene polymorphisms and TNF-α serum levels in Mexican colorectal cancer patients. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Culjak, M.; Perkovic, M.N.; Uzun, S.; Strac, D.S.; Erjavec, G.N.; Leko, M.B.; Simic, G.; Tudor, L.; Konjevod, M.; Kozumplik, O.; et al. The Association between TNF-alpha, IL-1 alpha and IL-10 with Alzheimer’s Disease. Curr. Alzheimer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Sharif, D.; Jaf, M.; Amin, D.M. Effect of TNF-α -308G/A (rs1800629) Promoter Polymorphism on the Serum Level of TNF-α Among Iraqi Patients with Generalized Vitiligo. Clin. Cosmet. Investig. Dermatol. 2020, 13, 825–835, PMCID: PMC7671505. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR9&keywords=ccr9 (accessed on 26 February 2021).
- Pathak, M.; Lal, G. The Regulatory Function of CCR9+ Dendritic Cells in Inflammation and Autoimmunity. Front. Immunol. 2020, 11, 536326, PMCID: PMC7566413. [Google Scholar] [CrossRef]
- Biswas, S.; Bryant, R.V.; Travis, S. Interfering with leukocyte trafficking in Crohn’s disease. Best Pract. Res. Clin. Gastroenterol. 2019, 38–39, 1016. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CXCR6&keywords=cxcr6 (accessed on 26 February 2021).
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 2019, 216, 2748–2762, PMCID: PMC6888981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=XCR1&keywords=XCR1 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=XCL1&keywords=xcl1 (accessed on 26 February 2021).
- Lei, Y.; Takahama, Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012, 14, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR2&keywords=ccr2 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR5&keywords=ccr5 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CCR3&keywords=ccr3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=PEDS1-UBE2V1 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/610994?search=tmem189&highlight=tmem189 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/602995?search=ube2v1&highlight=ube2v1 (accessed on 26 February 2021).
- Mackelprang, R.D.; Bamshad, M.J.; Chong, J.X.; Hou, X.; Buckingham, K.J.; Shively, K.; deBruyn, G.; Mugo, N.R.; Mullins, J.I.; McElrath, M.J.; et al. Whole genome sequencing of extreme phenotypes identifies variants in CD101 and UBE2V1 associated with increased risk of sexually acquired HIV-1. PLoS Pathog. 2017, 13, e1006703, Erratum in 2019, 15, e1007588. PMCID: PMC5690691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL10&keywords=il10 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL17A&keywords=il17a (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL17F&keywords=il17f (accessed on 26 February 2021).
- Karcıoğlu Batur, L.; Hekim, N. Correlations of IL-6, IL-6R, IL-10 and IL-17 gene polymorphisms with the prevalence of COVID-2019 infection and its mortality rate. medRxiv 2020. Available online: https://www.researchsquare.com/article/rs-82662/v1 (accessed on 26 February 2021). [CrossRef]
- Gonçalves de Albuquerque, S.D.C.; da Costa Oliveira, C.N.; Vaitkevicius-Antão, V.; Silva, A.C.; Luna, C.F.; de Lorena, V.M.B.; de Paiva-Cavalcanti, M. Study of association of the rs2275913 IL-17A single nucleotide polymorphism and susceptibility to cutaneous leishmaniasis caused by Leishmania braziliensis. Cytokine 2019, 123, 154784. [Google Scholar] [CrossRef] [PubMed]
- Rolandelli, A.; Hernández Del Pino, R.E.; Pellegrini, J.M.; Tateosian, N.L.; Amiano, N.O.; de la Barrera, S.; Casco, N.; Gutiérrez, M.; Palmero, D.J.; García, V.E. The IL-17A rs2275913 single nucleotide polymorphism is associated with protection to tuberculosis but related to higher disease severity in Argentina. Sci. Rep. 2017, 7, 40666, PMCID: PMC5241634. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.G.; Wang, B.Z.; Li, J.; Ding, Z.L.; Wang, K. Association between interleukin-17 genetic polymorphisms and tuberculosis susceptibility: An updated meta-analysis. Int. J. Tuberc. Lung Dis. 2017, 21, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Cheng, B.; Ding, Y.; Wang, C.; Chen, J. Correlations of IL-17 and NF-κB gene polymorphisms with susceptibility and prognosis in acute respiratory distress syndrome in a chinese population. Biosci. Rep. 2019, 39, BSR20181987, PMCID: PMC6367126. [Google Scholar] [CrossRef] [Green Version]
- Zhai, C.; Li, S.; Feng, W.; Shi, W.; Wang, J.; Wang, Q.; Chai, L.; Zhang, Q.; Yan, X.; Li, M. Association of interleukin-17a rs2275913 gene polymorphism and asthma risk: A meta-analysis. Arch. Med. Sci. 2018, 14, 1204–1211, PMCID: PMC6209699. [Google Scholar] [CrossRef]
- Holster, A.; Teräsjärvi, J.; Lauhkonen, E.; Törmänen, S.; Helminen, M.; Koponen, P.; Korppi, M.; Peltola, V.; He, Q.; Nuolivirta, K. IL-17A gene polymorphism rs2275913 is associated with the development of asthma after bronchiolitis in infancy. Allergol. Int. 2018, 67, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Han, J.C.; Zhang, Y.J.; Qi, G.B.; Li, H.B.; Zhang, Y.J.; Cai, S. Single-Nucleotide Polymorphisms of IL-17 Gene Are Associated with Asthma Susceptibility in an Asian Population. Med. Sci. Monit. 2016, 22, 780–787, PMCID: PMC4793684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=WSB1&keywords=wsb1v (accessed on 26 February 2021).
- Hu, J.; Li, C.; Wang, S.; Li, T.; Zhang, H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK Biobank data. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.11.05.20226761v1 (accessed on 26 February 2021). [CrossRef]
- Haque, M.; Kendal, J.K.; MacIsaac, R.M.; Demetrick, D.J. WSB1: From homeostasis to hypoxia. J. Biomed. Sci. 2016, 23, 61, PMCID: PMC4992216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL21R&keywords=il-21 (accessed on 26 February 2021).
- Nara, H.; Onoda, T.; Rahman, M.; Araki, A.; Juliana, F.M.; Tanaka, N.; Asao, H. WSB-1, a novel IL-21 receptor binding molecule, enhances the maturation of IL-21 receptor. Cell Immunol. 2011, 269, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Ma, C.S. Regulation of the germinal center and humoral immunity by interleukin-21. J. Exp. Med. 2020, 217, e20191638, PMCID: PMC7037251. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.M.; Lukacher, A.E. IL-21 in Homeostasis of Resident Memory and Exhausted CD8 T Cells during Persistent Infection. Int. J. Mol. Sci. 2020, 21, 6966, PMCID: PMC7554897. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/608258 (accessed on 26 February 2021).
- Waumans, Y.; Baerts, L.; Kehoe, K.; Lambeir, A.M.; De Meester, I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front. Immunol. 2015, 6, 387, PMCID: PMC4528296. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=DPP9&keywords=dpp9 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=DPP7&keywords=dpp7 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/610537?search=dpp7&highlight=dpp7 (accessed on 26 February 2021).
- Mele, D.A.; Bista, P.; Baez, D.V.; Huber, B.T. Dipeptidyl peptidase 2 is an essential survival factor in the regulation of cell quiescence. Cell Cycle 2009, 8, 2425–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HIPC-CHI Signatures Project Team; HIPC-I Consortium. Multicohort analysis reveals baseline transcriptional predictors of influenza vaccination responses. Sci Immunol. 2017, 2, eaal4656, PMCID: PMC5800877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org/entry/602581?search=golga3&highlight=golga3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GOLGA3&keywords=golga3 (accessed on 26 February 2021).
- Ovsyannikova, I.G.; Kennedy, R.B.; O’Byrne, M.; Jacobson, R.M.; Pankratz, V.S.; Poland, G.A. Genome-wide association study of antibody response to smallpox vaccine. Vaccine 2012, 30, 4182–4189, PMCID: PMC3367131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/609619?search=golga8b&highlight=golga8b (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GOLGA8B&keywords=golga8b (accessed on 26 February 2021).
- Available online: https://www.iaf.urmc.rochester.edu/results?search=rs200975425; https://www.iaf.urmc.rochester.edu/about.html (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/600168?search=mst1r&highlight=mst1r (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MST1R&keywords=MST1R (accessed on 26 February 2021).
- Chaudhuri, A. Regulation of Macrophage Polarization by RON Receptor Tyrosine Kinase Signaling. Front. Immunol. 2014, 5, 546, PMCID: PMC4215628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.B.; Ray, M.; Lutz, M.; Sharda, D.; Xu, J.; Hankey, P.A. The RON receptor tyrosine kinase regulates IFN-gamma production and responses in innate immunity. J. Immunol. 2008, 181, 2303–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, A.C.; Wilson, C.B.; Ray, M.; Correll, P.H. Macrophage-stimulating protein, the ligand for the stem cell-derived tyrosine kinase/RON receptor tyrosine kinase, inhibits IL-12 production by primary peritoneal macrophages stimulated with IFN-gamma and lipopolysaccharide. J. Immunol. 2004, 172, 1825–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.H.; Zhou, Y.Q.; Chen, Y.Q. Macrophage-stimulating protein and RON receptor tyrosine kinase: Potential regulators of macrophage inflammatory activities. Scand. J. Immunol. 2002, 56, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/613296?search=laptm4b&highlight=laptm4b (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=LAPTM4B&keywords=LAPTM4B (accessed on 26 February 2021).
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. LAPTMs regulate lisosomal function and interact with mucolipin 1: New clues for understanding mucolipidosis type IV. J. Cell Sci. 2011, 124, 459–468, PMCID: PMC3022000. [Google Scholar] [CrossRef] [Green Version]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250, PMCID: PMC4455107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/107741?search=apoe&highlight=apoe (accessed on 26 February 2021).
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188, PMCID: PMC2674716. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Janmohamed, K.; Nyhan, K.; Martins, S.S.; Cerda, M.; Hasin, D.; Scott, J.; Pates, R.; Ghandour, L.; Wazaify, M.; et al. Substance use and substance use disorder, in relation to COVID-19: Protocol for a scoping review. Syst. Rev. 2021, 10, 48, PMCID: PMC7857102. [Google Scholar] [CrossRef] [PubMed]
- Brickhill-Atkinson, M.; Hauck, F.R. Impact of COVID-19 on Resettled Refugees. Prim. Care 2021, 48, 57–66, PMCID: PMC7538065. [Google Scholar] [CrossRef]
- Fronteira, I.; Sidat, M.; Magalhães, J.P.; de Barros, F.P.C.; Delgado, A.P.; Correia, T.; Daniel-Ribeiro, C.T.; Ferrinho, P. The SARS-CoV-2 pandemic: A syndemic perspective. One Health 2021, 12, 100228, PMCID: PMC7887445. [Google Scholar] [CrossRef]
- Emeny, R.T.; Carpenter, D.O.; Lawrence, D.A. Health disparities: Intracellular consequences of social determinants of health. Toxicol. Appl. Pharmacol. 2021, 115444. [Google Scholar] [CrossRef] [PubMed]
- Bourdrel, T.; Annesi-Maesano, I.; Alahmad, B.; Maesano, C.N.; Bind, M.A. The impact of outdoor air pollution on COVID-19: A review of evidence from in vitro, animal, and human studies. Eur. Respir. Rev. 2021, 30, 200242, PMCID: PMC7879496. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, E.D.; Engin, A. The effect of environmental pollution on immune evasion checkpoints of SARS-CoV-2. Environ. Toxicol. Pharmacol. 2021, 81, 103520, PMCID: PMC7580701. [Google Scholar] [CrossRef]
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat. Immunol. 2020, 21, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Quinete, N.; Hauser-Davis, R.A. Drinking water pollutants may affect the immune system: Concerns regarding COVID-19 health effects. Environ. Sci. Pollut. Res. Int. 2021, 28, 1235–1246, PMCID: PMC7644792. [Google Scholar] [CrossRef] [PubMed]
- Zahra, A.; Sisu, C.; Silva, E.; De Aguiar Greca, S.C.; Randeva, H.S.; Chatha, K.; Kyrou, I.; Karteris, E. Is There a Link between Bisphenol A (BPA), a Key Endocrine Disruptor, and the Risk for SARS-CoV-2 Infection and Severe COVID-19? J. Clin. Med. 2020, 9, 3296, PMCID: PMC7602132. [Google Scholar] [CrossRef]
- Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; Graham, B.S.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. BioRxiv 2021. PMCID: PMC7852271. [Google Scholar] [CrossRef]
- Ortuso, F.; Mercatelli, D.; Guzzi, P.H.; Giorgi, F.M. Structural genetics of circulating variants affecting the SARS-CoV-2 spike/human ACE2 complex. J. Biomol. Struct. Dyn. 2021, 1–11, PMCID: PMC7885719. [Google Scholar] [CrossRef] [PubMed]
- Brookman, S.; Cook, J.; Zucherman, M.; Broughton, S.; Harman, K.; Gupta, A. Effect of the new SARS-CoV-2 variant B.1.1.7 on children and young people. Lancet Child. Adolesc. Health 2021. [Google Scholar] [CrossRef]
- Available online: https://covid19.who.int/ (accessed on 2 February 2021).
- Abduljalil, J.M.; Abduljalil, B.M. Epidemiology, genome, and clinical features of the pandemic SARS-CoV-2: A recent view. New Microbes New Infect. 2020, 35, 100672, PMCID: PMC7171182. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Lawrence, D.A. Susceptibility to COVID-19 in populations with health disparities: Posited involvement of mitochondrial disorder, socioeconomic stress, and pollutants. J. Biochem. Mol. Toxicol. 2020, e22626. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Harrison, E.M.; Ho, A.; Docherty, A.B.; Knight, S.R.; van Smeden, M.; Abubakar, I.; Lipman, M.; Quartagno, M.; Pius, R.; et al. Development and validation of the ISARIC 4C Deterioration model for adults hospitalised with COVID-19: A prospective cohort study. Lancet Respir. Med. 2021. PMCID: PMC7832571. [Google Scholar] [CrossRef] [PubMed]
- Márquez, E.J.; Chung, C.-H.; Marches, R.; Rossi, R.J.; Nehar-Belaid, D.; Eroglu, A.; Mellert, D.J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. Sexual-dimorphism in human immune system aging. Nat. Commun. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez, E.J.; Trowbridge, J.; Kuchel, G.A.; Banchereau, J.; Ucar, D. The lethal sex gap: COVID-19. Immun. Ageing 2020, 17, 13, PMCID: PMC7240166. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Gadi, N.; Wu, S.C.; Spihlman, A.P.; Moulton, V.R. What’s Sex Got to Do With COVID-19? Gender-Based Differences in the Host Immune Response to Coronaviruses. Front. Immunol. 2020, 11, 2147, PMCID: PMC7485092. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/300386 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/300292?search=foxp3&highlight=foxp3 (accessed on 26 February 2021).
- Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CXCR3&keywords=cxcr3 (accessed on 26 February 2021).
- Dattilo, M. The role of host defences in Covid 19 and treatments thereof. Mol. Med. 2020, 26, 90, PMCID: PMC7522454. [Google Scholar] [CrossRef]
- Van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA 2020, 324, 1–11, PMCID: PMC7382021. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/147640?search=IFNB1&highlight=ifnb1 (accessed on 26 February 2021).
- Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/entry/147571?search=isg15&highlight=isg15 (accessed on 26 February 2021).
- Aiello, A.; Accardi, G.; Candore, G.; Caruso, C.; Colomba, C.; Di Bona, D.; Duro, G.; Gambino, C.M.; Ligotti, M.E.; Pandey, J.P. Role of Immunogenetics in the Outcome of HCMV Infection: Implications for Ageing. Int. J. Mol. Sci. 2019, 20, 685, PMCID: PMC6386818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, A.; Candore, G.; Accardi, G.; Caruso, C.; Colomba, C.; Duro, G.; Gambino, C.M.; Ligotti, M.E.; Di Bona, D. Translation of Basic Research into Clinics: Killer Immunoglobulin-like Receptors Genes in Autoimmune and Infectious Diseases. Curr. Pharm. Des. 2018, 24, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C.; Pandey, J.P.; Puca, A.A. Genetics of exceptional longevity: Possible role of GM allotypes. Immun. Ageing 2018, 15, 25, PMCID: PMC6219196. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Netea, M.G. Trained Innate Immunity, Epigenetics, and Covid-19. N. Engl. J. Med. 2020, 383, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Papp, N.; Chou, J.T.; Hana, D.; Mackiewicz, A.; Kaczmarek, M. SARS-CoV-2: Pathogenesis, and Advancements in Diagnostics and Treatment. Front. Immunol. 2020, 11, 570927, PMCID: PMC7573101. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Luxami, V.; Tandon, R.; Paul, K. Recent Approaches of Repositioning and Traditional Drugs for the Treatment of COVID-19 Pandemic Outbreak. Mini Rev. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Jethava, Y.; Vikas, P. Repurposing Anti-Cancer Drugs for COVID-19 Treatment. Drug Des. Dev. Ther. 2020, 14, 5045–5058, PMCID: PMC7680713. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharm. Rep. 2020, 1–30, PMCID: PMC7474498. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Cordoba, V. Drug Repositioning for COVID-19. Colomb. Med. 2020, 51, e4279, PMCID: PMC7518729. [Google Scholar] [CrossRef]


{kind=link}
| Group/Locus | Variant | Outcome | p | OR (95% CI) | RR (95%CI) | Refs. |
|---|---|---|---|---|---|---|
| A | ||||||
| Susceptibility | 0.027 | 1.21 (1.02–1.43) | [21] | |||
| Susceptibility | * 0.04 | * 1.33 (1.02–1.73) | [24] | |||
| Susceptibility | 0.0024 | 1.23 (1.08–1.41) | [25] | |||
| Susceptibility | <0.001 | 1.09 (1.02–1.13) | [27] | |||
| Susceptibility (1) | <0.001 | 1.249 (1.114–1.440) | [26] | |||
| Susceptibility (2) | 0.03 | 1.3 (1.02–1.66) | [29] | |||
| Respiratory failure | * 1.48×10−4 | * 1.45 (1.20–1.75) | [28] | |||
| Mortality | 0.008 | 1.482 (1.113–1.972) | [21] | |||
| AB | ||||||
| Susceptibility | * 0.035 | * 1.37 (1.02–1.83) | [30] | |||
| B | ||||||
| Susceptibility | * 0.004 | * 1.28 (1.08–1.52) | [30] | |||
| 0 | ||||||
| Susceptibility | <0.001 | 0.67 (0.60–0.75) | [21] | |||
| Susceptibility | 0.0006 | 0.787 (0.69–0.90) | [25] | |||
| Susceptibility | <0.001 | 0.87 (0.82–0.91) | [27] | |||
| Susceptibility | * 0.007 | 0.84 (0.75–0.95) | [30] | |||
| Susceptibility (1) | <0.001 | 0.699 (0.635–0.770) | [26] | |||
| Respiratory failure | * 1.06×10−5 | * 0.65 (0.53–0.79) | [28] | |||
| Mortality | 0.014 | 0.660 (0.479–0.911) | [21] | |||
| Locus AB0 | ||||||
| rs657152 (A) | Respiratory failure | * 5.35×10−7 | * 1.39 (1.22–1.59) | [28] |
| Variable | Allele/Serotype | p | OR (95%CI) | Regression Coefficient | Growth Rate (95%CI) | Refs. |
|---|---|---|---|---|---|---|
| Incidence | B*44 | 0.05 | 0.1484 | 1.16 (1–1.35%) | [39] | |
| C*01 | 0.042 | 0.1747 | 1.19 (1.01–1.41%) | [39] | ||
| Susceptibility | B*15:27 | 0.030 | 3.59 (1.72–7.50) § | [45] | ||
| B46 | n.s. | [46] | ||||
| B22 | 0.032 | 1.71 (1.23–2.38) § | [46] | |||
| C*07:29 | 0.025 | 130.20 (5.28–3211) § | [45] | |||
| DQB1*06 | 0.0468 # | 1.96 (1.19–3.22) #§ | [47] | |||
| DRB1*08 | 0.010 # | 1.814 (1.151–2.860) # | [29] | |||
| Severity | A | n.s. | [28] | |||
| A*11:01 | 0.008 | 2.33 | [10] | |||
| B22 | n.s. | [46] | ||||
| B27 | n.s. | [46,48] | ||||
| B*27:07 | * 0.004 | [41] | ||||
| B46 | n.s. | [46] | ||||
| B*51:01 | 0.007 | 3.38 | [10] | |||
| C | n.s. | [28] | ||||
| C*14:02 | 0.003 | 4.75 | [10] | |||
| DPB1*03:01 | 0.037 | 0.09 | [10] | |||
| DQA1*01:01 | 0.039 | 6.05 | [10] | |||
| DQB1 | n.s. | [28] | ||||
| DQB1*06:02 | 0.016 | [41] | ||||
| DRB1 | n.s. | [28] | ||||
| DRB1*01:01 | 0.02 | 13.7 | [10] | |||
| DRB1*12:01 | 0.045 | 0.18 | [10] | |||
| DRB1*14:04 | 0.01 | 15.1 | [10] | |||
| DRB1*15:01 | 0.048 | [37] | ||||
| A*11 | 0.04 (1) | 7.693 (1.063–55.65) (1) | [40] | |||
| 0.02 (2) | 11.858 (1.524–92.273) (2) | [40] | ||||
| Mortality | C*01 | 0.04 (1) | 11.182 (1.053–118.7) (1) | [40] | ||
| 0.02 (2) | 17.604 (1.629–190.211) (2) | [40] | ||||
| DQB1*04 | 0.03 (1) | 9.963 (1.235–80.358) (1) | [40] | |||
| DRB1*08 | 0.01 # | 8.6 (1.7–43.9) # | [29] |
| Gene/Locus | Variant/ Position | Reference/ Other Allele | Altered/ Risk Allele | Protein Variant | Type | p | OR (95%CI) | Outcome | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| TLR3 | § 187003852 | AT | A | p.Ser339fs | pLOF | 0.01 | 8.28 (1.04–65.64) | severity | [113] |
| TLR3 | § 187005146 | G | A | p.Trp769 * | pLOF | ||||
| UNC93B1 | § 67770598 | C | A | p.Glu96 * | pLOF | ||||
| TBK1 | § 64875731 | C | T | p.Arg308 * | pLOF | ||||
| IRF7 | § 615095 | A | C | p.Arg7fs | pLOF | ||||
| IRF7 | § 614300 | G | A | p.Gln185 * | pLOF | ||||
| IRF7 | § 613966 | CGGGCTGGGGCCCG | C | p.Pro246fs | pLOF | ||||
| IRF7 | § 613353 | G | GC | p.Pro364fs | pLOF | ||||
| IFNAR2 | § 34621038 | AGATTGTTGGTTTT | A | p.Glu140fs | pLOF | ||||
| IFNAR2 | rs2236757 | G | A | 4.99 × 10−8 | 1.28 | severity | [117] | ||
| OAS3 | rs10735079 | G | A | 1.65 × 10−8 | 1.29 | severity | [117] | ||
| IFITM3 | rs12252 | T | C | # 0.0093 | # 6.37 | severity | [118] | ||
| 0.025 | 1.93 (1.09–3.46) | severity | [119] | ||||||
| PRKRA | I226N | 0.02 | severity | [120] | |||||
| TNF-α | rs1800629 | G | A | ° <0.001 | age > 60 | [121] | |||
| ° <0.001 | lymphopenia | [121] | |||||||
| ° 0.009 | high CRP | [121] | |||||||
| ° <0.001 | high ferritin | [121] | |||||||
| ° <0.001 | severity | [121] | |||||||
| † 0.045 | severity | [121] | |||||||
| 3p21.31 | rs11385942 | G | GA | 1.15 × 10−10 | 1.77 (1.48–2.11) | respiratory failure | [28] | ||
| 0.003 | 1.56 (1.17–2.01) | mechanical ventilation | [28] | ||||||
| TMEM189UBE2V1 | rs6020298 | G | A | 4.1 × 10–6 | 1.2 | severity | [10] | ||
| DPP9 | rs2109069 | G | A | 3.98 × 10−12 | 1.36 | severity | [117] | ||
| GOLGA8B | rs200975425 | C | T | 9.4 × 10–10 | 5.4 | susceptibility | [10] | ||
| LAPTM4B | P219L | 0.029 | [120] | ||||||
| P220L | |||||||||
| I109F | |||||||||
| P50T | |||||||||
| ApoE | e3 | e4 | 1.19 × 10–6 | 2.31 (1.65–3.24) | severity | [122] | |||
| ^ 0.009 | ^ 1.20 (1.05–1.37) | severity | [123] | ||||||
| 3.24 × 10−9 | 2.24 (1.72–2.93) | severity | [123] | ||||||
| 1.22 × 10–6 | 4.29 (2.38–7.72) | mortality | [123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pojero, F.; Candore, G.; Caruso, C.; Di Bona, D.; Groneberg, D.A.; Ligotti, M.E.; Accardi, G.; Aiello, A. The Role of Immunogenetics in COVID-19. Int. J. Mol. Sci. 2021, 22, 2636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052636
Pojero F, Candore G, Caruso C, Di Bona D, Groneberg DA, Ligotti ME, Accardi G, Aiello A. The Role of Immunogenetics in COVID-19. International Journal of Molecular Sciences. 2021; 22(5):2636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052636
Chicago/Turabian StylePojero, Fanny, Giuseppina Candore, Calogero Caruso, Danilo Di Bona, David A. Groneberg, Mattia E. Ligotti, Giulia Accardi, and Anna Aiello. 2021. "The Role of Immunogenetics in COVID-19" International Journal of Molecular Sciences 22, no. 5: 2636. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052636