
Neuroinflammation and Hypothalamo-Pituitary Dysfunction: Focus of Traumatic Brain Injury

 , , , , and
, , , , and
Abstract
:1. Introduction
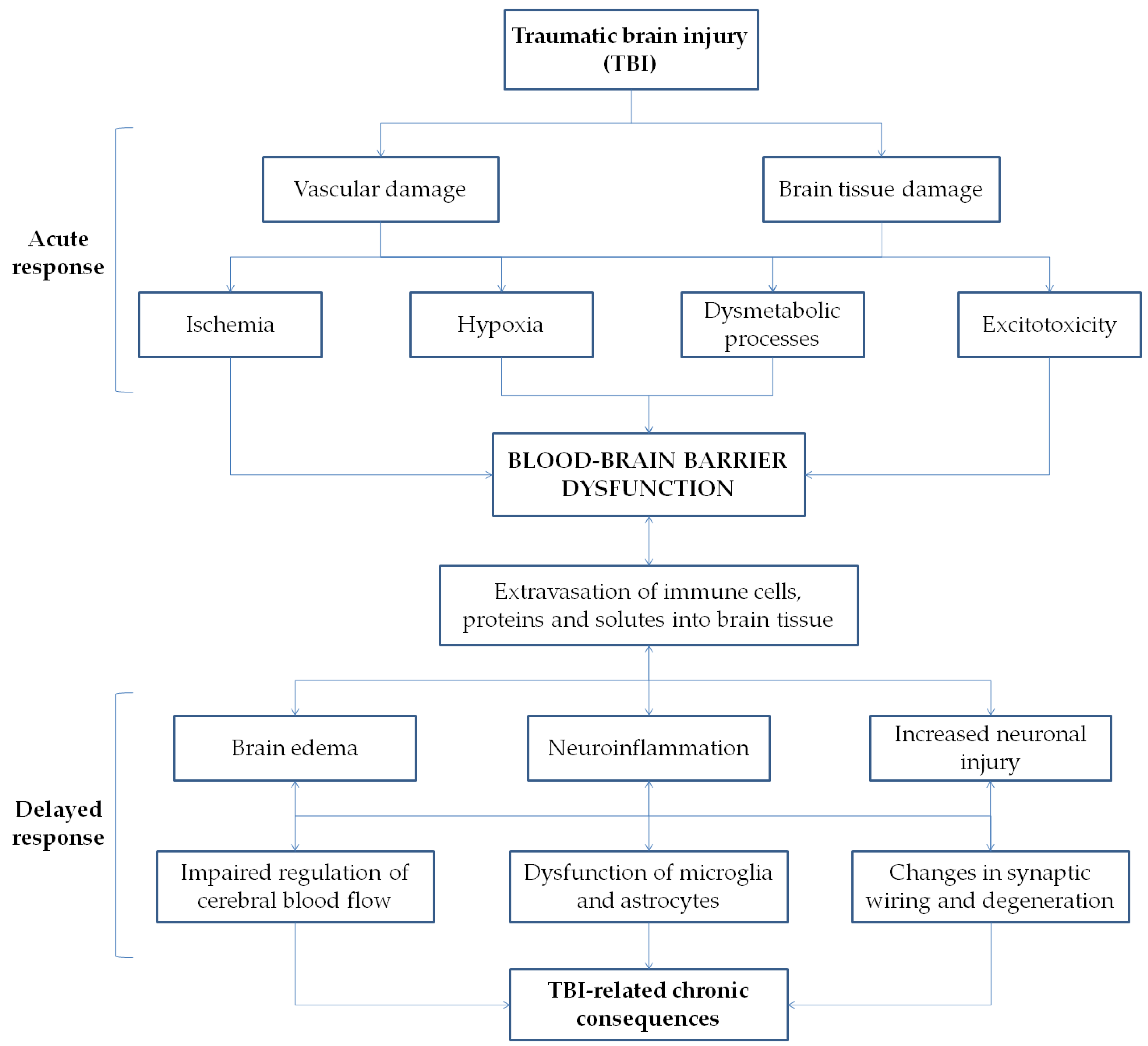
2. CNS Immunometabolism, Neuroinflammation and TBI
Post-TBI BBB Dysfunction in the Hypothalamic Area
3. Molecular Patterns: Inflammasome and Inflammaging
3.1. Inflammasome
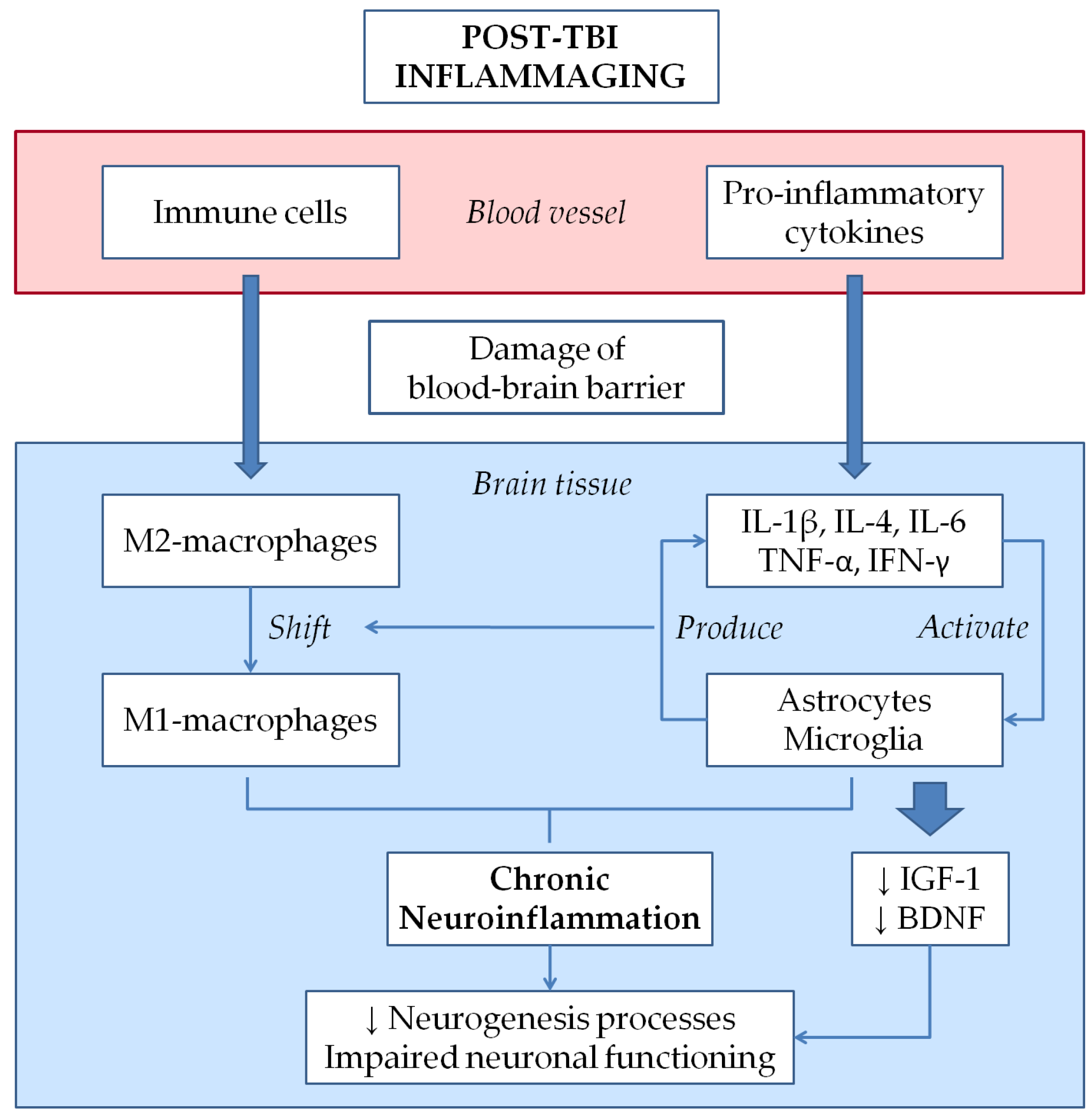
3.2. Inflammaging
4. The Clinical Involvement of TBI on Pituitary Functions
5. Dynamics of Post-TBI Pituitary Damage and Neuroinflammation
6. The Interplay between Neural Post-TBI Damage, Residual Pituitary Activity and Rehabilitation Outcomes
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| AD | Alzheimer’s disease |
| AHA | Autoantibodies against the hypothalamus |
| APA | Autoantibodies against pituitary |
| APOE | Apolipoprotein-E |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CTE | Chronic traumatic encephalopathy |
| DAMPs | Damage-associated molecular patterns |
| FIM | Functional independence measure |
| GCS | Glasgow Coma Scale |
| GFAP | Glial fibrillary acidic protein |
| GHD | Growth hormone deficiency |
| HMGB1 | High mobility group box 1 |
| HPA | Hypothalamus-pituitary-adrenal |
| IGF-1 | Insulin-like growth factor-1 |
| IL | Interleukin |
| MAC | Membrane attack complex |
| MMSE | Mini-mental state examination |
| NOD | Nucleotide oligomerization domain |
| P-gp | P-glycopreotein |
| PAMPs | Pathogen-associated molecular patterns |
| PRRs | Pattern recognition receptors |
| PTSD | Post-traumatic stress disorder |
| QoL | Quality of life |
| rhGH | Recombinant human GH |
| ROS | Reactive oxygen species |
| SIRS | Systemic inflammatory response syndrome |
| TBI | Traumatic brain injury |
| TNF- α | Tumour necrosis factor-α |
References
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain-Immune Interactions and Neuroinflammation after Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef]
- Hinzman, J.M.; Wilson, J.A.; Mazzeo, A.T.; Bullock, M.R.; Hartings, J.A. Excitotoxicity and Metabolic Crisis Are Associated with Spreading Depolarizations in Severe Traumatic Brain Injury Patients. J. Neurotrauma 2016, 33, 1775–1783. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [Green Version]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Chandran, R.; Kim, T.; Mehta, S.L.; Udho, E.; Chanana, V.; Cengiz, P.; Kim, H.; Kim, C.; Vemuganti, R. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1818–1827. [Google Scholar] [CrossRef]
- Shi, K.; Zhang, J.; Dong, J.F.; Shi, F.D. Dissemination of brain inflammation in traumatic brain injury. Cell Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailer, N.P. Immunosuppression after traumatic or ischemic CNS damage: It is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 2008, 84, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Plesnila, N. The immune system in traumatic brain injury. Curr. Opin. Pharmacol. 2016, 26, 110–117. [Google Scholar] [CrossRef]
- Needham, E.J.; Helmy, A.; Zanier, E.R.; Jones, J.L.; Coles, A.J.; Menon, D.K. The immunological response to traumatic brain injury. J. Neuroimmunol. 2019, 332, 112–125. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop, V.; Badaut, J. A neurovascular perspective for long-term changes after brain trauma. Transl. Stroke Res. 2011, 2, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.H.; Johnson, V.E.; Stewart, W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 2013, 9, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, R.; Fiest, K.M.; McChesney, J.; Kwon, C.S.; Jette, N.; Frolkis, A.D.; Atta, C.; Mah, S.; Dhaliwal, H.; Reid, A.; et al. The International Incidence of Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Can. J. Neurol. Sci. 2016, 43, 774–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. What are the leading causes of TBI? Available online: http://www.cdc.gov/traumaticbraininjury/causes.html (accessed on 6 January 2021).
- Gennarelli, T.A.; Champion, H.R.; Copes, W.S.; Sacco, W.J. Comparison of mortality, morbidity, and severity of 59,713 head injured patients with 114,447 patients with extracranial injuries. J. Trauma 1994, 37, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Entezami, P.; Shah, K.; Medhkour, A. Predictors of Outcomes in Traumatic Brain Injury. World Neurosurg. 2016, 90, 525–529. [Google Scholar] [CrossRef]
- Jain, S.; Iverson, L.M. Glasgow Coma Scale; StatPearls: Treasure Island, FL, USA, 2021. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK513298/ (accessed on 14 January 2021).
- Pavlovic, D.; Pekic, S.; Stojanovic, M.; Popovic, V. Traumatic brain injury: Neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 2019, 22, 270–282. [Google Scholar] [CrossRef]
- Melmed, S.; Jameson, J.L. Disorders of the anterior pituitary and hypothalamus. In Harrison’s Principles of Internal Medicine, 16th ed.; McGraw-Hill: New York, NY, USA, 2005; pp. 2076–2097. [Google Scholar]
- Kelly, D.F.; Gonzalo, I.T.; Cohan, P.; Berman, N.; Swerdloff, R.; Wang, C. Hypopituitarism following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A preliminary report. J. Neurosurg. 2000, 93, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, S.A.; Oberoi, A.L.; Gilkison, C.R.; Masel, B.E.; Urban, R.J. Prevalence of neuroendocrine dysfunction in patients recovering from traumatic brain injury. J. Clin. Endocrinol. Metab. 2001, 86, 2752–2756. [Google Scholar] [CrossRef]
- Bondanelli, M.; De Marinis, L.; Ambrosio, M.R.; Monesi, M.; Valle, D.; Zatelli, M.C.; Fusco, A.; Bianchi, A.; Farneti, M.; degli Uberti, E.C. Occurrence of pituitary dysfunction following traumatic brain injury. J. Neurotrauma 2004, 21, 685–696. [Google Scholar] [CrossRef]
- Aimaretti, G.; Ambrosio, M.R.; Di Somma, C.; Fusco, A.; Cannavò, S.; Gasperi, M.; Scaroni, C.; De Marinis, L.; Benvenga, S.; degli Uberti, E.C.; et al. Traumatic brain injury and subarachnoid haemorrhage are conditions at high risk for hypopituitarism: Screening study at 3 months after the brain injury. Clin. Endocrinol. 2004, 61, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Sherlock, M.; O’Kelly, P.; Tormey, W.; Phillips, J.; Thompson, C.J. Anterior pituitary dysfunction in survivors of traumatic brain injury. J. Clin. Endocrinol. Metab. 2004, 89, 4929–4936. [Google Scholar] [CrossRef] [Green Version]
- Popovic, V.; Pekic, S.; Pavlovic, D.; Maric, N.; Jasovic-Gasic, M.; Djurovic, B.; Medic Stojanoska, M.; Zivkovic, V.; Stojanovic, M.; Doknic, M.; et al. Hypopituitarism as a consequence of traumatic brain injury (TBI) and its possible relation with cognitive disabilities and mental distress. J. Endocrinol. Invest. 2004, 27, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Aimaretti, G.; Ghigo, E. Traumatic brain injury and hypopituitarism. Sci. World J. 2005, 5, 777–781. [Google Scholar] [CrossRef]
- Tanriverdi, F.; Senyurek, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J. Clin. Endocrinol. Metab. 2006, 91, 2105–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurismal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar] [CrossRef]
- Schneider, H.J.; Schneider, M.; Saller, B.; Petersenn, S.; Uhr, M.; Husemann, B.; von Rosen, F.; Stalla, G.K. Prevalence of anterior pituitary insufficiency 3 and 12 months after traumatic brain injury. Eur. J. Endocrinol. 2006, 154, 259–265. [Google Scholar] [CrossRef]
- Park, K.D.; Kim, D.Y.; Lee, J.K.; Nam, H.S.; Park, Y.G. Anterior pituitary dysfunction in moderate-to-severe chronic traumatic brain injury patients and the influence on functional outcome. Brain Inj. 2010, 24, 1330–1335. [Google Scholar] [CrossRef]
- Caputo, M.; Mele, C.; Prodam, F.; Marzullo, P.; Aimaretti, G. Clinical picture and the treatment of TBI-induced hypopituitarism. Pituitary 2019, 22, 261–269. [Google Scholar] [CrossRef]
- Kokshoorn, N.E.; Wassenaar, M.J.; Biermasz, N.R.; Roelfsema, F.; Smit, J.W.; Romijn, J.A.; Pereira, A.M. Hypopituitarism following traumatic brain injury: Prevalence is affected by the use of different dynamic tests and different normal values. Eur. J. Endocrinol. 2010, 162, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; He, B.; Tong, W. Dynamic pituitary hormones change after traumatic brain injury. Neurol. India 2014, 62, 280–284. [Google Scholar] [CrossRef]
- Tan, C.L.; Alavi, S.A.; Baldeweg, S.E.; Belli, A.; Carson, A.; Feeney, C.; Goldstone, A.P.; Greenwood, R.; Menon, D.K.; Simpson, H.L.; et al. The screening and management of pituitary dysfunction following traumatic brain injury in adults: British Neurotrauma Group guidance. J. Neurol. Neurosurg. Psychiatry 2017, 88, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Popovic, V.; Aimaretti, G.; Casanueva, F.F.; Ghigo, E. Hypopituitarism following traumatic brain injury. Front. Horm. Res. 2005, 33, 33–44. [Google Scholar] [PubMed]
- Schneider, M.; Schneider, H.J.; Stalla, G.K. Anterior pituitary hormone abnormalities following traumatic brain injury. J. Neurotrauma 2005, 22, 937–946. [Google Scholar] [CrossRef]
- Heather, N.; Cutfield, W. Traumatic brain injury: Is the pituitary out of harm’s way? J. Pediatr. 2011, 159, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; De Bellis, A.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Pane, E.; Bellastella, A.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; et al. Antipituitary antibodies after traumatic brain injury: Is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur. J. Endocrinol. 2008, 159, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Tanriverdi, F.; Taheri, S.; Ulutabanca, H.; Caglayan, A.O.; Ozkul, Y.; Dundar, M.; Selcuklu, A.; Unluhizarci, K.; Casanueva, F.F.; Kelestimur, F. Apolipoprotein E3/E3 genotype decreases the risk of pituitary dysfunction after traumatic brain injury due to various causes: Preliminary data. J. Neurotrauma 2008, 25, 1071–1077. [Google Scholar] [CrossRef]
- Karaca, Z.; Tanrıverdi, F.; Ünlühızarcı, K.; Kelestimur, F. GH and Pituitary Hormone Alterations After Traumatic Brain Injury. Prog. Mol. Biol. Transl. Sci. 2016, 138, 167–191. [Google Scholar] [PubMed]
- Tanriverdi, F.; Unluhizarci, K.; Kelestrimur, F. Persistent neuroinflammation may be involved in the pathogenesis of traumatic brain injury (TBI)-induced hypopituitarism: Potential genetic and autoimmune factors. J. Neurotrauma 2010, 27, 301–302. [Google Scholar] [CrossRef]
- Tanriverdi, F.; De Bellis, A.; Ulutabanca, H.; Bizzarro, A.; Sinisi, A.A.; Bellastella, G.; Amoresano Paglionico, V.; Dalla Mora, L.; Selcuklu, A.; Unluhizarci, K.; et al. A five year prospective investigation of anterior pituitary function after traumatic brain injury: Is hypopituitarism long-term after head trauma associated with autoimmunity? J. Neurotrauma 2013, 30, 1426–1433. [Google Scholar] [CrossRef]
- Gilis-Januszewska, A.; Kluczyński, Ł.; Hubalewska-Dydejczyk, A. Traumatic brain injuries induced pituitary dysfunction: A call for algorithms. Endocr. Connect. 2020, 9, R112–R123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Puntambekar, S.S.; Saber, M.; Lamb, B.T.; Kokiko-Cochran, O.N. Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain. Behav. Immun. 2018, 71, 9–17. [Google Scholar] [CrossRef] [Green Version]
- van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Başkaya, M.K.; Rao, A.M.; Doğan, A.; Donaldson, D.; Dempsey, R.J. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 1997, 226, 33–36. [Google Scholar] [CrossRef]
- McKee, C.A.; Lukens, J.R. Emerging Roles for the Immune System in Traumatic Brain Injury. Front. Immunol. 2016, 7, 556. [Google Scholar] [CrossRef] [Green Version]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neurology 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Shetty, A.K.; Mishra, V.; Kodali, M.; Hattiangady, B. Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell Neurosci. 2014, 8, 232. [Google Scholar] [PubMed] [Green Version]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflammation 2016, 13, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michinaga, S.; Kimura, A.; Hatanaka, S.; Minami, S.; Asano, A.; Ikushima, Y.; Matsui, S.; Toriyama, Y.; Fujii, M.; Koyama, Y. Delayed Administration of BQ788, an ETB Antagonist, after Experimental Traumatic Brain Injury Promotes Recovery of Blood-Brain Barrier Function and a Reduction of Cerebral Edema in Mice. J. Neurotrauma 2018, 35, 1481–1494. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, A.M.; Nagai, A.; Ryu, J.K.; McLarnon, J.G.; Kim, S.U.; Masuda, J. Lysophosphatidylcholine induces glial cell activation: Role of rho kinase. Glia 2009, 57, 898–907. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammosome: A molecular platform triggering activation of inflammatory caspases and processing proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Invest. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of immune response by head injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhang, J.; Hu, X.; Zhang, L.; Mao, L.; Jiang, X.; Liou, A.K.; Leak, R.K.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Bell-Temin, H.; Culver-Cochran, A.E.; Chaput, D.; Carlson, C.M.; Kuehl, M.; Burkhardt, B.R.; Bickford, P.C.; Liu, B.; Stevens, S.M., Jr. Novel Molecular Insights into Classical and Alternative Activation States of Microglia as Revealed by Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-based Proteomics. Mol. Cell Proteomics 2015, 14, 3173–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Febinger, H.Y.; Thomasy, H.E.; Pavlova, M.N.; Ringgold, K.M.; Barf, P.R.; George, A.M.; Grillo, J.N.; Bachstetter, A.D.; Garcia, J.A.; Cardona, A.E.; et al. Time-dependent effects of CX3CR1 in a mouse model of mild traumatic brain injury. J. Neuroinflammation 2015, 12, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanier, E.R.; Marchesi, F.; Ortolano, F.; Perego, C.; Arabian, M.; Zoerle, T.; Sammali, E.; Pischiutta, F.; De Simoni, M.G. Fractalkine Receptor Deficiency Is Associated with Early Protection but Late Worsening of Outcome following Brain Trauma in Mice. J. Neurotrauma 2016, 33, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsi, S.; Piaggio, T.; Croci, N.; Noble, F.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: A twelve-week follow-up study. J. Neurotrauma 2010, 27, 911–921. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.-V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef] [Green Version]
- Bonneh-Barkay, D.; Zagadailov, P.; Zou, H.; Niyonkuru, C.; Figley, M.; Starkey, A.; Wang, G.; Bissel, S.J.; Wiley, C.A.; Wagner, A.K. YKL-40 expression in traumatic brain injury: An initial analysis. J. Neurotrauma 2010, 27, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S232–S240. [Google Scholar] [CrossRef] [Green Version]
- Thal, S.C.; Neuhaus, W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Hans, V.H.; Lenzlinger, P.M.; Dubs, R.; Ludwig, E.; Trentz, O.; Kossmann, T. TGF-beta is elevated in the CSF of patients with severe traumatic brain injuries and parallels blood-brain barrier function. J. Neurotrauma 1999, 16, 617–628. [Google Scholar] [CrossRef]
- Clausen, F.; Hånell, A.; Björk, M.; Hillered, L.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2009, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Clausen, F.; Hånell, A.; Israelsson, C.; Hedin, J.; Ebendal, T.; Mir, A.K.; Gram, H.; Marklund, N. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 2011, 34, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Tehranian, R.; Andell-Jonsson, S.; Beni, S.M.; Yatsiv, I.; Shohami, E.; Bartfai, T.; Lundkvist, J.; Iverfeldt, K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. Neurotrauma 2002, 19, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Lin, J.W.; Chang, M.W.; Wang, C.C.; Kuo, J.R.; Yang, C.Z.; Chang, C.P. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J. Neurochem. 2010, 115, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.; Schiding, J.K.; Kaczorowski, S.L.; Marion, D.W.; Kochanek, P.M. Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight drop and controlled cortical impact models. J. Neurotrauma 1994, 11, 499–506. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Holmin, S.; Mathiesen, T. Biphasic edema development after experimental brain contusion in rat. Neurosci. Lett. 1995, 194, 97–100. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Yoles, E.; Hauben, E.; Palgi, O.; Agranov, E.; Gothilf, A.; Cohen, A.; Kuchroo, V.; Cohen, I.R.; Weiner, H.; Schwartz, M. Protective autoimmunity is a physiological response to CNS trauma. J. Neurosci. 2001, 21, 3740–3748. [Google Scholar] [CrossRef] [PubMed]
- Hauben, E.; Nevo, U.; Yoles, E.; Moalem, G.; Agranov, E.; Mor, F.; Akselrod, S.; Neeman, M.; Cohen, I.R.; Schwartz, M. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet 2000, 355, 286–287. [Google Scholar] [CrossRef]
- Walsh, J.T.; Hendrix, S.; Boato, F.; Smirnov, I.; Zheng, J.; Lukens, J.R.; Gadani, S.; Hechler, D.; Gölz, G.; Rosenberger, K.; et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J. Clin. Invest. 2015, 125, 2547. [Google Scholar] [CrossRef] [PubMed]
- Hammarberg, H.; Lidman, O.; Lundberg, C.; Eltayeb, S.Y.; Gielen, A.W.; Muhallab, S.; Svenningsson, A.; Lindå, H.; van Der Meide, P.H.; Cullheim, S.; et al. Neuroprotection by encephalomyelitis: Rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J. Neurosci. 2000, 20, 5283–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.A.; Steiner, B.; Akpinarli, A.; Kammertoens, T.; Nassenstein, C.; Braun, A.; Blankenstein, T.; Kempermann, G. CD4-positive T lymphocytes provide a neuroimmunological link in the control of adult hippocampal neurogenesis. J. Immunol. 2009, 182, 3979–3984. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; Mcgeachy, M.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. Neuroinflammation in the evolution of secondary injury, repair, and chronic neurodegeneration after traumatic brain injury. Nat. Rev. Neurol. 2018, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Weckbach, S.; Neher, M.; Losacco, J.T.; Bolden, A.L.; Kulik, L.; Flierl, M.A.; Bell, S.E.; Holers, V.M.; Stahel, P.F. Challenging the role of adaptive immunity in neurotrauma: Rag1(-/-) mice lacking mature B and T cells do not show neuroprotection after closed head injury. J. Neurotrauma 2012, 29, 1233–1242. [Google Scholar] [CrossRef] [Green Version]
- Ertürk, A.; Mentz, S.; Stout, E.E.; Hedehus, M.; Dominguez, S.L.; Neumaier, L.; Krammer, F.; Llovera, G.; Srinivasan, K.; Hansen, D.V.; et al. Interfering with the Chronic Immune Response Rescues Chronic Degeneration After Traumatic Brain Injury. J. Neurosci. 2016, 36, 9962–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramlackhansingh, A.F.; Brooks, D.J.; Greenwood, R.J.; Bose, S.K.; Turkheimer, F.E.; Kinnunen, K.M.; Gentleman, S.; Heckemann, R.A.; Gunanayagam, K.; Gelosa, G.; et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; McNeal, D.W.; Morecraft, R.J.; Combs, C.K. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 2007, 24, 1719–1742. [Google Scholar] [CrossRef]
- Giunta, B.; Obregon, D.; Velisetty, R.; Sanberg, P.R.; Borlongan, C.V.; Tan, J. The immunology of traumatic brain injury: A prime target for Alzheimer’s disease prevention. J. Neuroinflammation 2012, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.E.; Byers, A.L.; Gardner, R.C.; Seal, K.H.; Boscardin, W.J.; Yaffe, K. Association of Mild Traumatic Brain Injury With and Without Loss of Consciousness With Dementia in US Military Veterans. JAMA Neurol. 2018, 75, 1055–1061. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Shitaka, Y.; Tran, H.T.; Bennett, R.E.; Sanchez, L.; Levy, M.A.; Dikranian, K.; Brody, D.L. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol. 2011, 70, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mohapatra, S.; Mohapatra, S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J. Neuroinflammation 2012, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Goh, S.J.; Tng, P.Y.; Deng, Y.Y.; Ling, E.A.; Moochhala, S. Systemic inflammatory response following acute traumatic brain injury. Front. Biosci. 2009, 14, 3795–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keel, M.; Trentz, O. Pathophysiology of polytrauma. Injury 2005, 36, 691–709. [Google Scholar] [CrossRef]
- Jacome, T.; Tatum, D. Systemic Inflammatory Response Syndrome (SIRS) Score Independently Predicts Poor Outcome in Isolated Traumatic Brain Injury. Neurocrit. Care 2018, 28, 110–116. [Google Scholar] [CrossRef]
- Barrientos, R.M.; Kitt, M.M.; Watkins, L.R.; Maier, S.F. Neuroinflammation in the normal aging hippocampus. Neuroscience 2015, 309, 84–99. [Google Scholar] [CrossRef] [Green Version]
- Hänsel, A.; Hong, S.; Cámara, R.J.; von Känel, R. Inflammation as a psychophysiological biomarker in chronic psychosocial stress. Neurosci. Biobehav. Rev. 2010, 35, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Yang, M.; Enikolopov, G.; Iacovitti, L. Circumventricular organs: A novel site of neural stem cells in the adult brain. Mol. Cell Neurosci. 2009, 41, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, E.M.; Blázquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Akmayev, I.G.; Popov, A.P. Morphological aspects of the hypothalamic-hypophyseal system. VII. The tanycytes: Their relation to the hypophyseal adrenocorticotrophic function. An ultrastructural study. Cell Tissue Res. 1977, 180, 263–282. [Google Scholar] [CrossRef]
- Vallet, P.G.; Charnay, Y.; Boura, C.; Kiss, J.Z. Colocalization of delta sleep inducing peptide and luteinizing hormone releasing hormone in neurosecretory vesicles in rat median eminence. Neuroendocrinology 1991, 53, 103–106. [Google Scholar] [CrossRef]
- Osterstock, G.; El Yandouzi, T.; Romanò, N.; Carmignac, D.; Langlet, F.; Coutry, N.; Guillou, A.; Schaeffer, M.; Chauvet, N.; Vanacker, C.; et al. Sustained alterations of hypothalamic tanycytes during posttraumatic hypopituitarism in male mice. Endocrinology 2014, 155, 1887–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevot, V.; Bellefontaine, N.; Baroncini, M.; Sharif, A.; Hanchate, N.K.; Parkash, J.; Campagne, C.; de Seranno, S. Gonadotrophin-releasing hormone nerve terminals, tanycytes and neurohaemal junction remodelling in the adult median eminence: Functional consequences for reproduction and dynamic role of vascular endothelial cells. J. Neuroendocrinol. 2010, 22, 639–649. [Google Scholar] [PubMed] [Green Version]
- Devanney, N.A.; Stewart, A.N.; Gensel, J.C. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp. Neurol. 2020, 329, 113310. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflammation 2020, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; Russo, M.; Pallio, G.; Bitto, A.; Mannino, F.; Minutoli, L.; Altavilla, D.; Squadrito, F. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 6204. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Marcillo, A.; Nonner, D.; Dietrich, W.D.; Keane, R.W. Neuroprotective effects of bone morphogenetic protein 7 (BMP7) treatment after spinal cord injury. Neurosci. Lett. 2009, 465, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Khanlarkhani, N.; Beyer, C.; Zendedel, A. Inflammasome: Its role in traumatic brain and spinal cord injury. J. Cell Physiol. 2018, 233, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.P.; Xiong, G.P.; Lin, Q.; Chen, X.W.; Zhang, L.Q.; Shi, J.X.; Ke, Q.F.; Lin, J.H. Heme oxygenase-1 promotes neuron survival through down-regulation of neuronal NLRP1 expression after spinal cord injury. J. Neuroinflammation 2016, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.D.; Li, W.; Chen, Z.R.; Hu, Y.C.; Zhang, D.D.; Shen, W.; Zhou, M.L.; Zhu, L.; Hang, C.H. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem. Res. 2013, 38, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Bao, Z.; Hou, L.; Liu, Y.; Wang, X.; You, Y.; et al. Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp. Neurol. 2017, 290, 115–122. [Google Scholar] [CrossRef]
- Wallisch, J.S.; Simon, D.W.; Bayır, H.; Bell, M.J.; Kochanek, P.M.; Clark, R.S.B. Cerebrospinal Fluid NLRP3 is Increased After Severe Traumatic Brain Injury in Infants and Children. Neurocrit. Care 2017, 27, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Pop, V.; Sorensen, D.W.; Kamper, J.E.; Ajao, D.O.; Murphy, M.P.; Head, E.; Hartman, R.E.; Badaut, J. Early brain injury alters the blood-brain barrier phenotype in parallel with β-amyloid and cognitive changes in adulthood. J. Cereb. Blood Flow Metab. 2013, 33, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Fann, J.R.; Ribe, A.R.; Pedersen, H.S.; Fenger-Grøn, M.; Christensen, J.; Benros, M.E.; Vestergaard, M. Long-term risk of dementia among people with traumatic brain injury in Denmark: A population-based observational cohort study. Lancet. Psychiatry 2018, 5, 424–431. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Pablo-Bernal, R.S.; Cañizares, J.; Rosado, I.; Galvá, M.I.; Alvarez-Ríos, A.I.; Carrillo-Vico, A.; Ferrando-Martínez, S.; Muñoz-Fernández, M.Á.; Rafii-El-Idrissi Benhnia, M.; Pacheco, Y.M.; et al. Monocyte Phenotype and Polyfunctionality Are Associated With Elevated Soluble Inflammatory Markers, Cytomegalovirus Infection, and Functional and Cognitive Decline in Elderly Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
- Schnydrig, S.; Korner, L.; Landweer, S.; Ernst, B.; Walker, G.; Otten, U.; Kunz, D. Peripheral lipopolysaccharide administration transiently affects expression of brain-derived neurotrophic factor, corticotropin and proopiomelanocortin in mouse brain. Neurosci. Lett. 2007, 429, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Fang, J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav. Immun. 2006, 20, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Tumati, S.; Burger, H.; Martens, S.; van der Schouw, Y.T.; Aleman, A. Association between Cognition and Serum Insulin-Like Growth Factor-1 in Middle-Aged & Older Men: An 8 Year Follow-Up Study. PLoS ONE 2016, 11, e0154450. [Google Scholar]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Murray, L.; Landon, M.; Graham, D.I. Beta amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Ikonomovic, M.D.; Uryu, K.; Abrahamson, E.E.; Ciallella, J.R.; Trojanowski, J.Q.; Lee, V.M.; Clark, R.S.; Marion, D.W.; Wisniewski, S.R.; DeKosky, S.T. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 2004, 190, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Ubukata, S.; Oishi, N.; Higashi, T.; Kagawa, S.; Yamauchi, H.; Okuyama, C.; Watanabe, H.; Ono, M.; Saji, H.; Aso, T.; et al. Spatial Patterns of Amyloid Deposition in Patients with Chronic Focal or Diffuse Traumatic Brain Injury Using 18F-FPYBF-2 PET. Neuropsychiatr. Dis. Treat. 2020, 16, 2719–2732. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar]
- Jullienne, A.; Roberts, J.M.; Pop, V.; Paul Murphy, M.; Head, E.; Bix, G.J.; Badaut, J. Juvenile traumatic brain injury induces long-term perivascular matrix changes alongside amyloid-beta accumulation. J. Cereb. Blood Flow Metab. 2014, 34, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Graham, D.I. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991, 338, 1422–1423. [Google Scholar] [CrossRef]
- Blasko, I.; Beer, R.; Bigl, M.; Apelt, J.; Franz, G.; Rudzki, D.; Ransmayr, G.; Kampfl, A.; Schliebs, R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J. Neural. Transm. 2004, 111, 523–536. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef]
- Chen, X.H.; Siman, R.; Iwata, A.; Meaney, D.F.; Trojanowski, J.Q.; Smith, D.H. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004, 165, 357–371. [Google Scholar] [CrossRef]
- Lepelletier, F.X.; Mann, D.M.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Crehan, H.; Holton, P.; Wray, S.; Pocock, J.; Guerreiro, R.; Hardy, J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 2012, 217, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Lashkari, K.; Teague, G.; Chen, H.; Lin, Y.Q.; Kumar, S.; McLaughlin, M.M.; López, F.J. A monoclonal antibody targeting amyloid β (Aβ) restores complement factor I bioactivity: Potential implications in age-related macular degeneration and Alzheimer’s disease. PLoS ONE 2018, 13, e0195751. [Google Scholar] [CrossRef]
- Bradt, B.M.; Kolb, W.P.; Cooper, N.R. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J. Exp. Med. 1998, 188, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neurology 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Alawieh, A.; Langley, E.F.; Weber, S.; Adkins, D.; Tomlinson, S. Identifying the Role of Complement in Triggering Neuroinflammation after Traumatic Brain Injury. J. Neurosci. 2018, 38, 2519–2532. [Google Scholar] [CrossRef]
- Cryan, E. Pituitary demage due to skull base fracture. Dtsch. Med. Wochenschr. 1918, 44, 1261–1270. [Google Scholar]
- Prodam, F.; Caputo, M.; Mele, C.; Marzullo, P.; Aimaretti, G. Insights into non-classic and emerging causes of hypopituitarism. Nat. Rev. Endocrinol. 2020. Online ahead of print. [Google Scholar]
- Tanriverdi, F.; Agha, A.; Aimaretti, G.; Casanueva, F.F.; Kelestimur, F.; Klose, M.; Masel, B.E.; Pereira, A.M.; Popovic, V.; Schneider, H.J. Manifesto for the current understanding and management of traumatic brain injury-induced hypopituitarism. J. Endocrinol. Invest. 2011, 34, 541–543. [Google Scholar]
- Hannon, M.J.; Crowley, R.K.; Behan, L.A.; O’Sullivan, E.P.; O’Brien, M.M.; Sherlock, M.; Rawluk, D.; O’Dwyer, R.; Tormey, W.; Thompson, C.J. Acute glucocorticoid deficiency and diabetes insipidus are common after acute traumatic brain injury and predict mortality. J. Clin. Endocrinol. Metab. 2013, 98, 3229–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.J.; Aimaretti, G.; Kreitschmann-Andermahr, I.; Stalla, G.K.; Ghigo, E. Hypopituitarism. Lancet 2007, 369, 1461–1470. [Google Scholar] [CrossRef]
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary dysfunction after traumatic brain injury: A clinical and pathophysiological approach. Endocr. Rev. 2015, 36, 305–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, A.; Phillips, J.; O’Kelly, P.; Tormey, W.; Thompson, C.J. The natural history of post-traumatic hypopituitarism: Implications for assessment and treatment. Am. J. Med. 2005, 118, 1416. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, B.L.; Rehder, J.; Kahlke, S.; Wiedemayer, H.; Doerfler, A.; Ischebeck, W.; Laumer, R.; Forsting, M.; Stolke, D.; Mann, K. Hypopituitarism following severe traumatic brain injury. Exp. Clin. Endocrinol. Diabetes 2006, 114, 316–321. [Google Scholar] [CrossRef]
- Klose, M.; Juul, A.; Poulsgaard, L.; Kosteljanetz, M.; Brennum, J.; Feldt-Rasmussen, U. Prevalence and predictive factors of post-traumatic hypopituitarism. Clin. Endocrinol. 2007, 67, 193–201. [Google Scholar] [CrossRef]
- Glynn, N.; Agha, A. The frequency and the diagnosis of pituitary dysfunction after traumatic brain injury. Pituitary 2019, 22, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Thornton, E.; O’Kelly, P.; Tormey, W.; Phillips, J.; Thompson, C.J. Posterior pituitary dysfunction after traumatic brain injury. J. Clin. Endocrinol. Metab. 2004, 89, 5987–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tritos, N.A.; Yuen, K.C.; Kellym, D.F.; AACE Neuroendocrine and Pituitary Scientific Committee. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: A neuroendocrine approach to patients with traumatic brain injury. Endocr. Pract. 2015, 21, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenske, W.; Allolio, B. Clinical review: Current state and future perspectives in the diagnosis of diabetes insipidus: A clinical review. J. Clin. Endocrinol. Metab. 2012, 97, 3426–3437. [Google Scholar] [CrossRef] [Green Version]
- Aimaretti, G.; Ambrosio, M.R.; Di Somma, C.; Gasperi, M.; Cannavò, S.; Scaroni, C.; Fusco, A.; Del Monte, P.; De Menis, E.; Faustini-Fustini, M.; et al. Residual pituitary function after brain injury-induced hypopituitarism: A prospective 12-month study. J. Clin. Endocrinol. Metab. 2005, 90, 6085–6092. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.; Agha, A. Post-Traumatic Hypopituitarism-Who Should Be Screened, When, and How? Front. Endocrinol. 2018, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Krewer, C.; Schneider, M.; Schneider, H.J.; Kreitschmann-Andermahr, I.; Buchfelder, M.; Faust, M.; Berg, C.; Wallaschofski, H.; Renner, C.; Uhl, E.; et al. Neuroendocrine Disturbances One to Five or More Years after Traumatic Brain Injury and Aneurysmal Subarachnoid Hemorrhage: Data from the German Database on Hypopituitarism. J. Neurotrauma 2016, 33, 1544–1553. [Google Scholar] [CrossRef]
- Jonasdottir, A.D.; Sigurjonsson, P.; Olafsson, I.H.; Karason, S.; Sigthorsson, G.; Sigurjonsdottir, H.A. Hypopituitarism 3 and 12 months after traumatic brain injury and subarachnoid haemorrhage. Brain Inj. 2018, 32, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fleseriu, M.; Hashim, I.A.; Karavitaki, N.; Melmed, S.; Murad, M.H.; Salvatori, R.; Samuels, M.H. Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 3888–3921. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Watt, T.; Brennum, J.; Feldt-Rasmussen, U. Posttraumatic hypopituitarism s associated with an unfavorable body composition and lipid profile, and decreased quality of life 12 months after injury. J. Clin. Endocrinol. Metab. 2007, 92, 3861–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodam, F.; Caputo, M.; Belcastro, S.; Garbaccio, V.; Zavattaro, M.; Samà, M.T.; Bellone, S.; Pagano, L.; Bona, G.; Aimaretti, G. Quality of life, mood disturbances and psychological parameters in adult patients with GH deficiency. Panminerva. Med. 2012, 54, 323–331. [Google Scholar] [PubMed]
- León-Carrión, J.; Leal-Cerro, A.; Cabezas, F.M.; Atutxa, A.M.; Gomez, S.G.; Cordero, J.M.; Moreno, A.S.; Ferrari, M.D.; Domínguez-Morales, M.R. Cognitive deterioration due to GH deficiency in patients with traumatic brain injury: A preliminary report. Brain Inj. 2007, 21, 871–875. [Google Scholar] [CrossRef]
- Kelly, D.F.; McArthur, D.L.; Levin, H.; Swimmer, S.; Dusick, J.R.; Cohan, P.; Wang, C.; Swerdloff, R. Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J. Neurotrauma 2006, 23, 928–942. [Google Scholar] [CrossRef]
- Ozdemir, D.; Baykara, B.; Aksu, I.; Kiray, M.; Sisman, A.R.; Cetin, F.; Dayi, A.; Gurpinar, T.; Uysal, N.; Arda, M.N. Relationship between circulating IGF-1 levels and traumatic brain injury-induced hippocampal damage and cognitive dysfunction in immature rats. Neurosci. Lett. 2012, 507, 84–89. [Google Scholar] [CrossRef]
- Dusick, J.R.; Wang, C.; Cohan, P.; Swerdloff, R.; Kelly, D.F. Pathophysiology of hypopituitarism in the setting of brain injury. Pituitary 2012, 15, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef]
- Ceballos, R. Pituitary changes in head trauma (analysis of 102 consecutive cases of head injury). Ala. J. Med. Sci. 1966, 3, 185–198. [Google Scholar]
- Daniel, P.M.; Prichard, M.M.; Treip, C.S. Traumatic infarction of the anterior lobe of the pituitary gland. Lancet 1959, 2, 927–931. [Google Scholar] [CrossRef]
- Kornblum, R.N.; Fisher, R.S. Pituitary lesions in craniocerebral injuries. Arch. Pathol. 1969, 88, 242–248. [Google Scholar]
- Dubourg, J.; Messerer, M. Sports-related chronic repetitive head trauma as a cause of pituitary dysfunction. Neurosurg. Focus 2011, 31, E2. [Google Scholar] [CrossRef]
- Richmond, E.; Rogol, A.D. Traumatic brain injury: Endocrine consequences in children and adults. Endocrine 2014, 45, 3–8. [Google Scholar] [CrossRef]
- Salehi, F.; Kovacs, K.; Scheithauer, B.W.; Pfeifer, E.A.; Cusimano, M. Histologic study of the human pituitary gland in acute traumatic brain injury. Brain Inj. 2007, 21, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, H.L. The recognition of chronic hypopituitarism resulting from postpartum pituitary necrosis. Am. J. Obstet. Gynecol. 1971, 111, 852–854. [Google Scholar] [CrossRef]
- Kasturi, B.S.; Stein, D.G. Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. J. Neurotrauma 2009, 26, 1315–1324. [Google Scholar] [CrossRef] [Green Version]
- Laskowitz, D.T.; Goel, S.; Bennett, E.R.; Matthew, W.D. Apolipoprotein E suppresses glial cell secretion of TNF alpha. J. Neuroimmunol. 1997, 76, 70–74. [Google Scholar] [CrossRef]
- Nishida, Y.; Yoshioka, M.; St-Amand, J. The top 10 most abundant transcripts are sufficient to characterize the organs functional specificity: Evidences from the cortex, hypothalamus and pituitary gland. Gene 2005, 344, 133–141. [Google Scholar] [CrossRef]
- Lynch, J.R.; Wang, H.; Mace, B.; Leinenweber, S.; Warner, D.S.; Bennett, E.R.; Vitek, M.P.; McKenna, S.; Laskowitz, D.T. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp. Neurol. 2005, 192, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Laskowitz, D.T.; Thekdi, A.D.; Thekdi, S.D.; Han, S.K.; Myers, J.K.; Pizzo, S.V.; Bennett, E.R. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 2001, 167, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Rudehill, S.; Muhallab, S.; Wennersten, A.; von Gertten, C.; Al Nimer, F.; Sandberg-Nordqvist, A.C.; Holmin, S.; Mathiesen, T. Autoreactive antibodies against neurons and basal lamina found in serum following experimental brain contusion in rats. Acta Neurochir. 2006, 148, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Müller-Fielitz, H.; Stahr, M.; Bernau, M.; Richter, M.; Abele, S.; Krajka, V.; Benzin, A.; Wenzel, J.; Kalies, K.; Mittag, J.; et al. Tanycytes control the hormonal output of the hypothalamic-pituitary-thyroid axis. Nat. Commun. 2017, 8, 484. [Google Scholar] [CrossRef]
- Prevot, V.; Hanchate, N.K.; Bellefontaine, N.; Sharif, A.; Parkash, J.; Estrella, C.; Allet, C.; de Seranno, S.; Campagne, C.; de Tassigny, X.; et al. Function-related structural plasticity of the GnRH system: A role for neuronal-glial-endothelial interactions. Front. Neuroendocrinol. 2010, 31, 241–258. [Google Scholar] [CrossRef]
- Vennekens, A.; Vankelecom, H. Traumatic brain injury and resultant pituitary dysfunction: Insights from experimental animal models. Pituitary 2019, 22, 212–219. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury--an inflammatory disease? Brain Res. Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef]
- Riggio, S.; Wong, M. Neurobehavioral sequelae of traumatic brain injury. Mt. Sinai. J. Med. 2009, 76, 163–172. [Google Scholar] [CrossRef]
- Prince, C.; Bruhns, M.E. Evaluation and Treatment of Mild Traumatic Brain Injury: The Role of Neuropsychology. Brain Sci. 2017, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, A.R.; Levin, H.S. Cognitive sequelae of traumatic brain injury. Psychiatr. Clin. N. Am. 2014, 37, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Li, W.B. Cognitive impairment after traumatic brain injury: The role of MRI and possible pathological basis. J. Neurol. Sci. 2016, 370, 244–250. [Google Scholar] [CrossRef]
- Deijen, J.B.; de Boer, H.; Blok, G.J.; van der Veen, E.A. Cognitive impairments and mood disturbances in growth hormone deficient men. Psychoneuroendocrinology 1996, 21, 313–322. [Google Scholar] [CrossRef]
- Bülow, B.; Hagmar, L.; Ørbaek, P.; Osterberg, K.; Erfurth, E.M. High incidence of mental disorders, reduced mental well-being and cognitive function in hypopituitary women with GH deficiency treated for pituitary disease. Clin. Endocrinol. 2002, 56, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Falleti, M.G.; Maruff, P.; Burman, P.; Harris, A. The effects of growth hormone (GH) deficiency and GH replacement on cognitive performance in adults: A meta-analysis of the current literature. Psychoneuroendocrinology 2006, 31, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Burman, P.; Broman, J.E.; Hetta, J.; Wiklund, I.; Erfurth, E.M.; Hagg, E.; Karlsson, F.A. Quality of life in adults with growth hormone (GH) deficiency: Response to treatment with recombinant human GH in a placebo-controlled 21-month trial. J. Clin. Endocrinol. Metab. 1995, 80, 3585–3590. [Google Scholar] [CrossRef]
- Burman, P.; Hetta, J.; Wide, L.; Månsson, J.E.; Ekman, R.; Karlsson, F.A. Growth hormone treatment affects brain neurotransmitters and thyroxine [see comment]. Clin. Endocrinol. 1996, 44, 319–324. [Google Scholar] [CrossRef]
- High, W.M., Jr.; Briones-Galang, M.; Clark, J.A.; Gilkison, C.; Mossberg, K.A.; Zgaljardic, D.J.; Masel, B.E.; Urban, R.J. Effect of growth hormone replacement therapy on cognition after traumatic brain injury. J. Neurotrauma 2010, 27, 1565–1575. [Google Scholar] [CrossRef]
- Devesa, J.; Díaz-Getino, G.; Rey, P.; García-Cancela, J.; Loures, I.; Nogueiras, S.; Hurtado de Mendoza, A.; Salgado, L.; González, M.; Pablos, T.; et al. Brain Recovery after a Plane Crash: Treatment with Growth Hormone (GH) and Neurorehabilitation: A Case Report. Int. J. Mol. Sci. 2015, 16, 30470–30482. [Google Scholar] [CrossRef] [Green Version]
- Devesa, J.; Reimunde, P.; Devesa, P.; Barberá, M.; Arce, V. Growth hormone (GH) and brain trauma. Horm. Behav. 2013, 63, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Moreau, O.K.; Cortet-Rudelli, C.; Yollin, E.; Merlen, E.; Daveluy, W.; Rousseaux, M. Growth hormone replacement therapy in patients with traumatic brain injury. J. Neurotrauma 2013, 30, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Mossberg, K.A.; Durham, W.J.; Zgaljardic, D.J.; Gilkison, C.R.; Danesi, C.P.; Sheffield-Moore, M.; Masel, B.E.; Urban, R.J. Functional Changes after Recombinant Human Growth Hormone Replacement in Patients with Chronic Traumatic Brain Injury and Abnormal Growth Hormone Secretion. J. Neurotrauma 2017, 34, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubiel, R.; Callender, L.; Dunklin, C.; Harper, C.; Bennett, M.; Kreber, L.; Auchus, R.; Diaz-Arrastia, R. Phase 2 Randomized, Placebo-Controlled Clinical Trial of Recombinant Human Growth Hormone (rhGH) During Rehabilitation From Traumatic Brain Injury. Front. Endocrinol. 2018, 9, 520. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Hormones Deficiency | Clinical Features | Finding | Diagnosis |
|---|---|---|---|
| ACTH | Acute phase: Life-threatening adrenal crises:
|
| Acute phase: Serum cortisol ≤10 μg/dL Chronic phase: - Serum cortisol ≤3 μg/dL is diagnostic - Serum cortisol ≥18 μg/dL exclude diagnosis - Serum cortisol 3–18 μg/dL consider stimulation test (corticotrophin) |
| TSH | Chronic phase:
|
| fT4 below the reference range with low or inappropriately normal TSH |
| FSH/LH | Chronic phase: Men:
| Men:
| Men: Low or inappropriately normal gonadotropins with low serum testosterone Women:
|
| GH | Chronic phase:
|
|
|
| ADH | Acute and chronic phase:
|
| 24 h output of 3.5 L or more of hypotonic urine and serum sodium above the reference range |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mele, C.; Pingue, V.; Caputo, M.; Zavattaro, M.; Pagano, L.; Prodam, F.; Nardone, A.; Aimaretti, G.; Marzullo, P. Neuroinflammation and Hypothalamo-Pituitary Dysfunction: Focus of Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 2686. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052686
Mele C, Pingue V, Caputo M, Zavattaro M, Pagano L, Prodam F, Nardone A, Aimaretti G, Marzullo P. Neuroinflammation and Hypothalamo-Pituitary Dysfunction: Focus of Traumatic Brain Injury. International Journal of Molecular Sciences. 2021; 22(5):2686. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052686
Chicago/Turabian StyleMele, Chiara, Valeria Pingue, Marina Caputo, Marco Zavattaro, Loredana Pagano, Flavia Prodam, Antonio Nardone, Gianluca Aimaretti, and Paolo Marzullo. 2021. "Neuroinflammation and Hypothalamo-Pituitary Dysfunction: Focus of Traumatic Brain Injury" International Journal of Molecular Sciences 22, no. 5: 2686. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052686