
ATP13A2 Regulates Cellular α-Synuclein Multimerization, Membrane Association, and Externalization

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
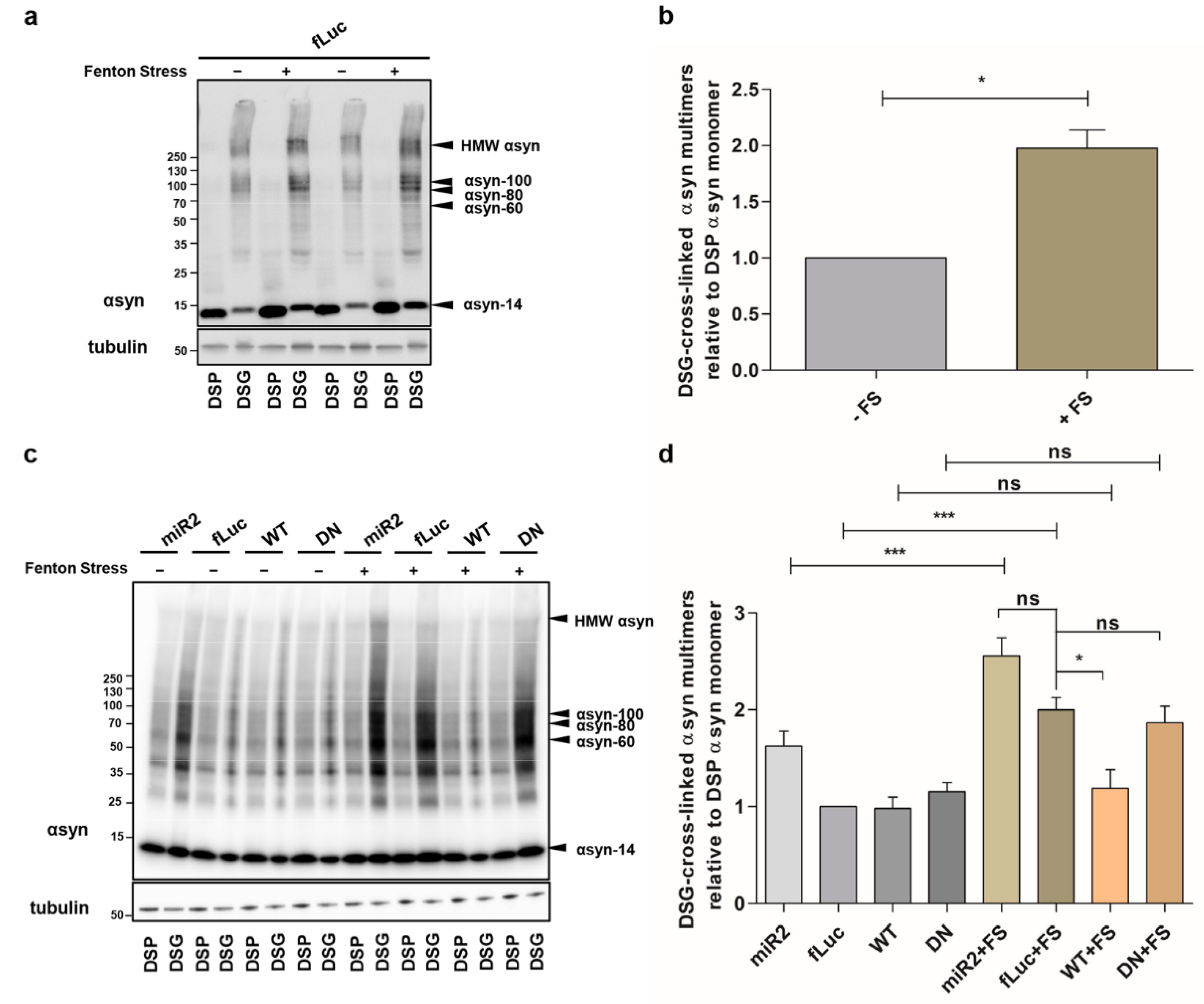
2.1. ATP13A2 KD Results in Upregulation of αsyn Multimers
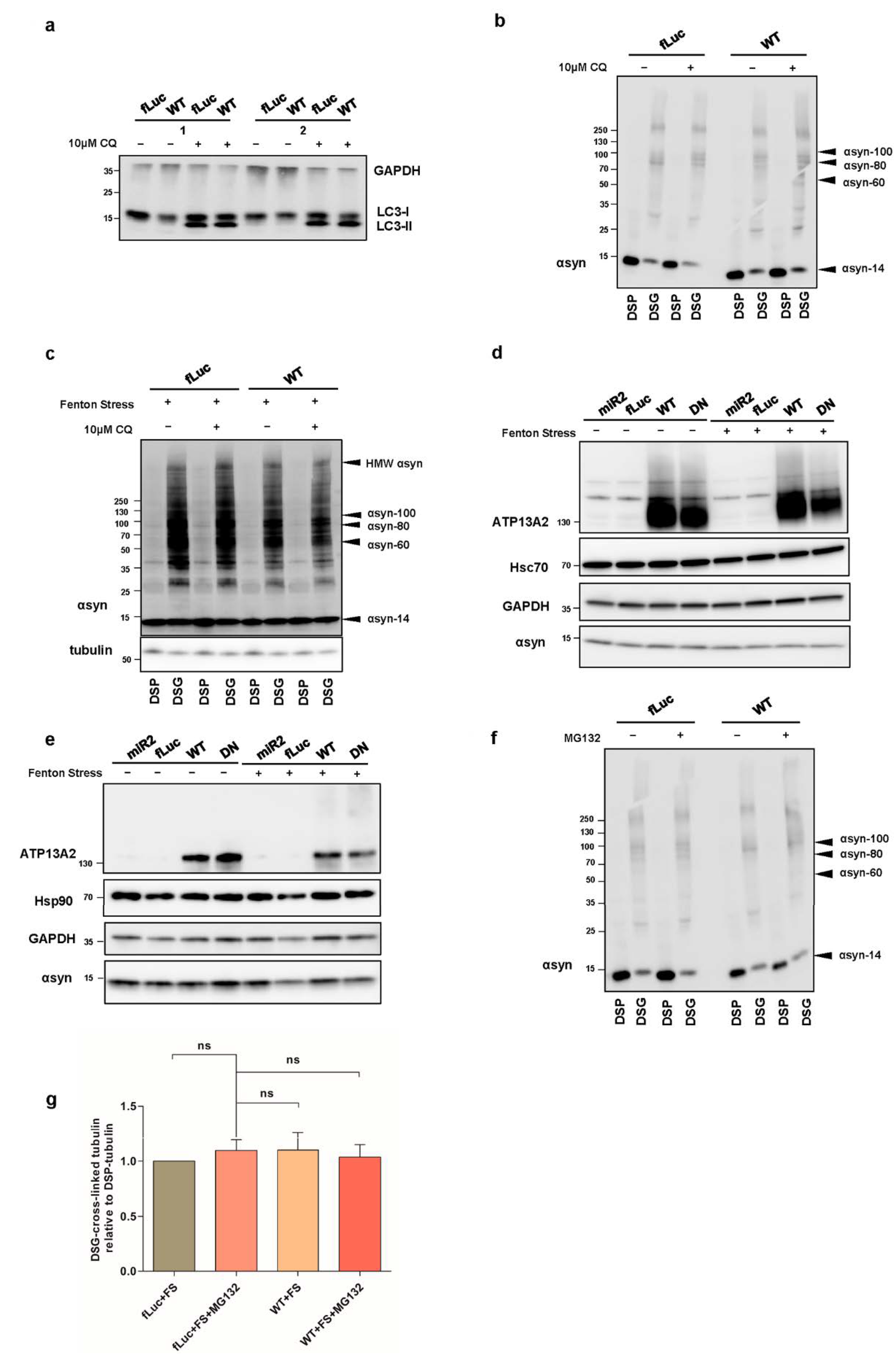
2.2. ATP13A2 Regulates Oxidative Stress-Induced αsyn Multimerization
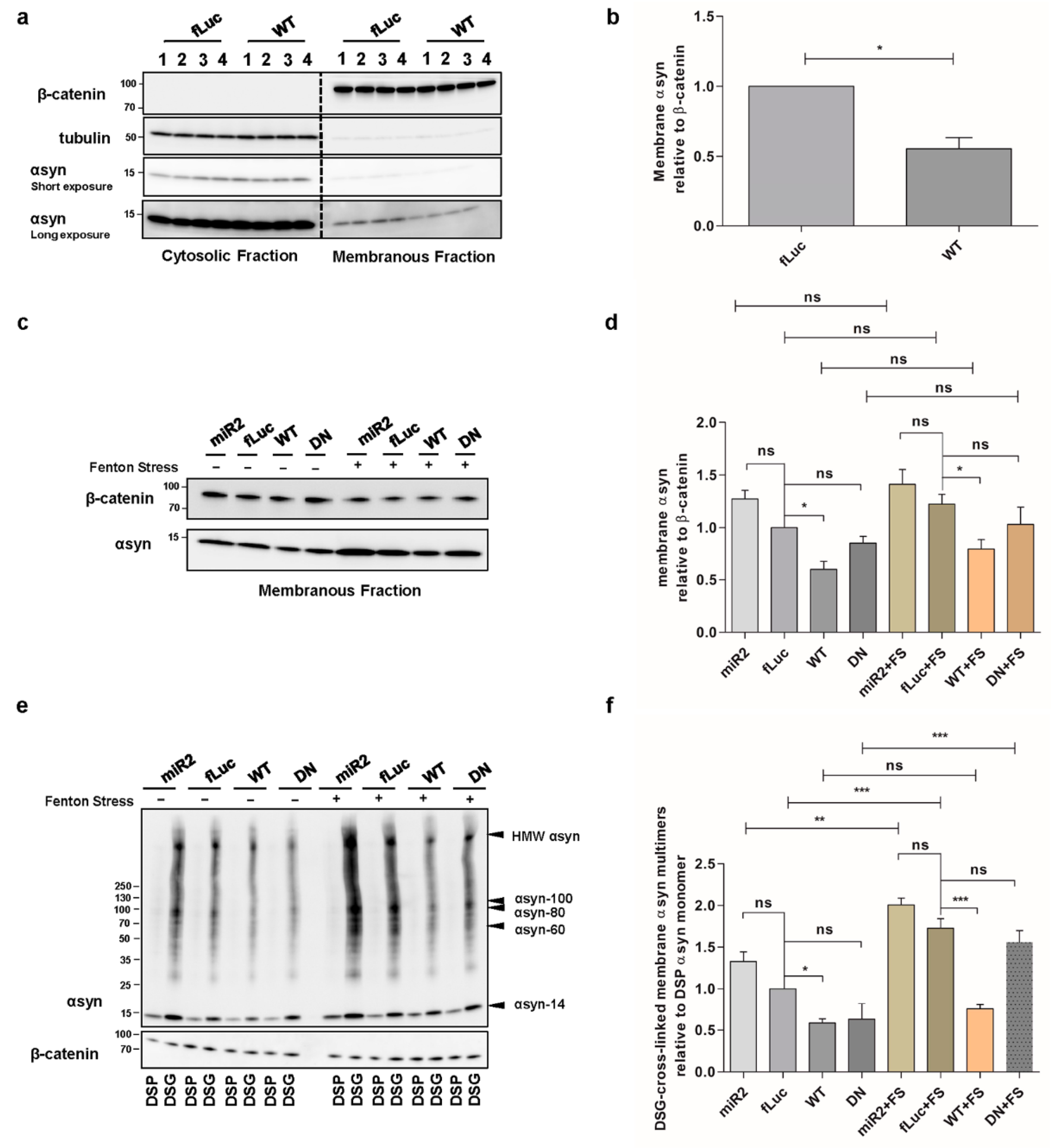
2.3. ATP13A2 Regulates αsyn Membrane Association and Multimerization
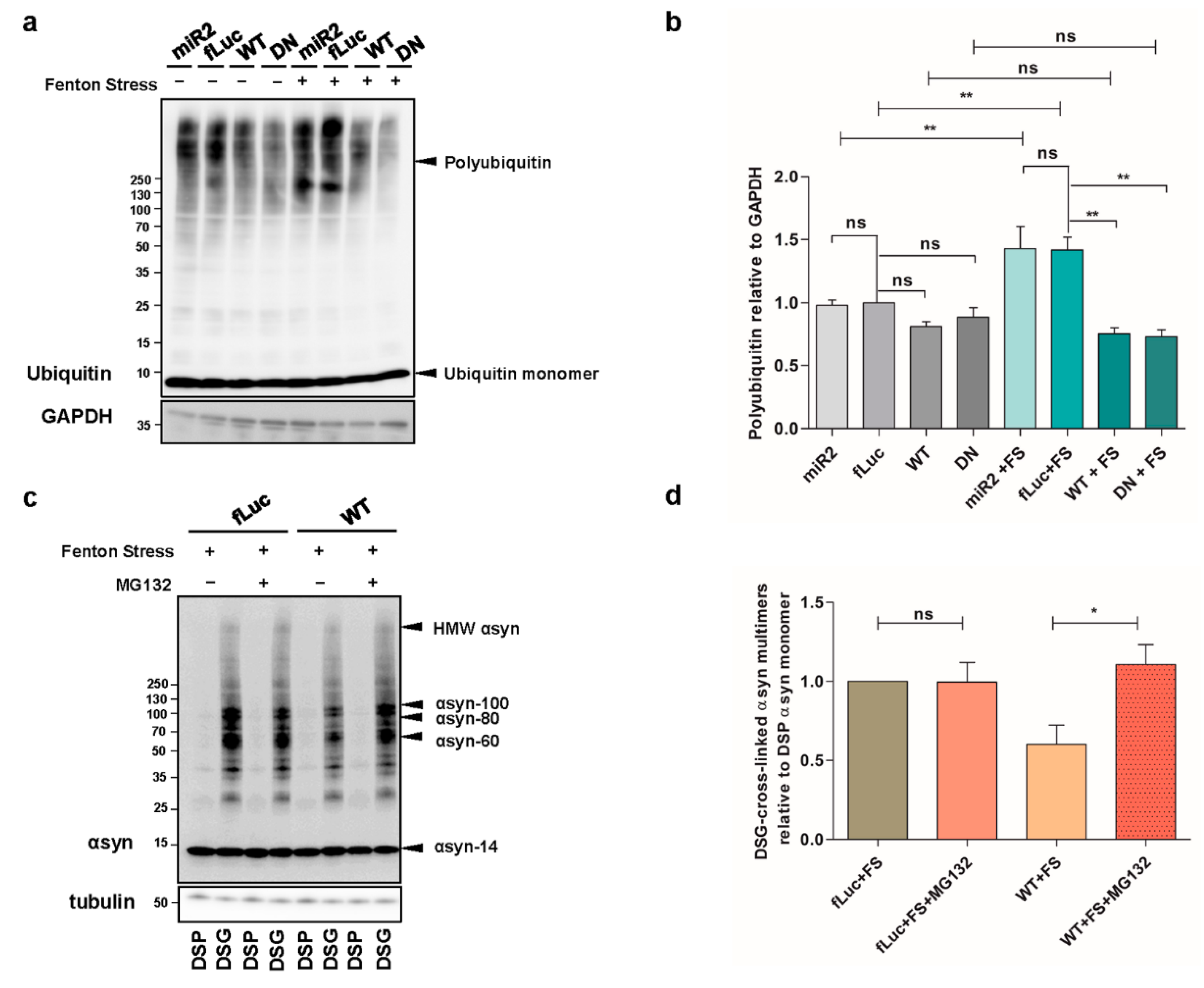
2.4. ATP13A2 Inhibits αsyn Multimerization via Regulating the UPS
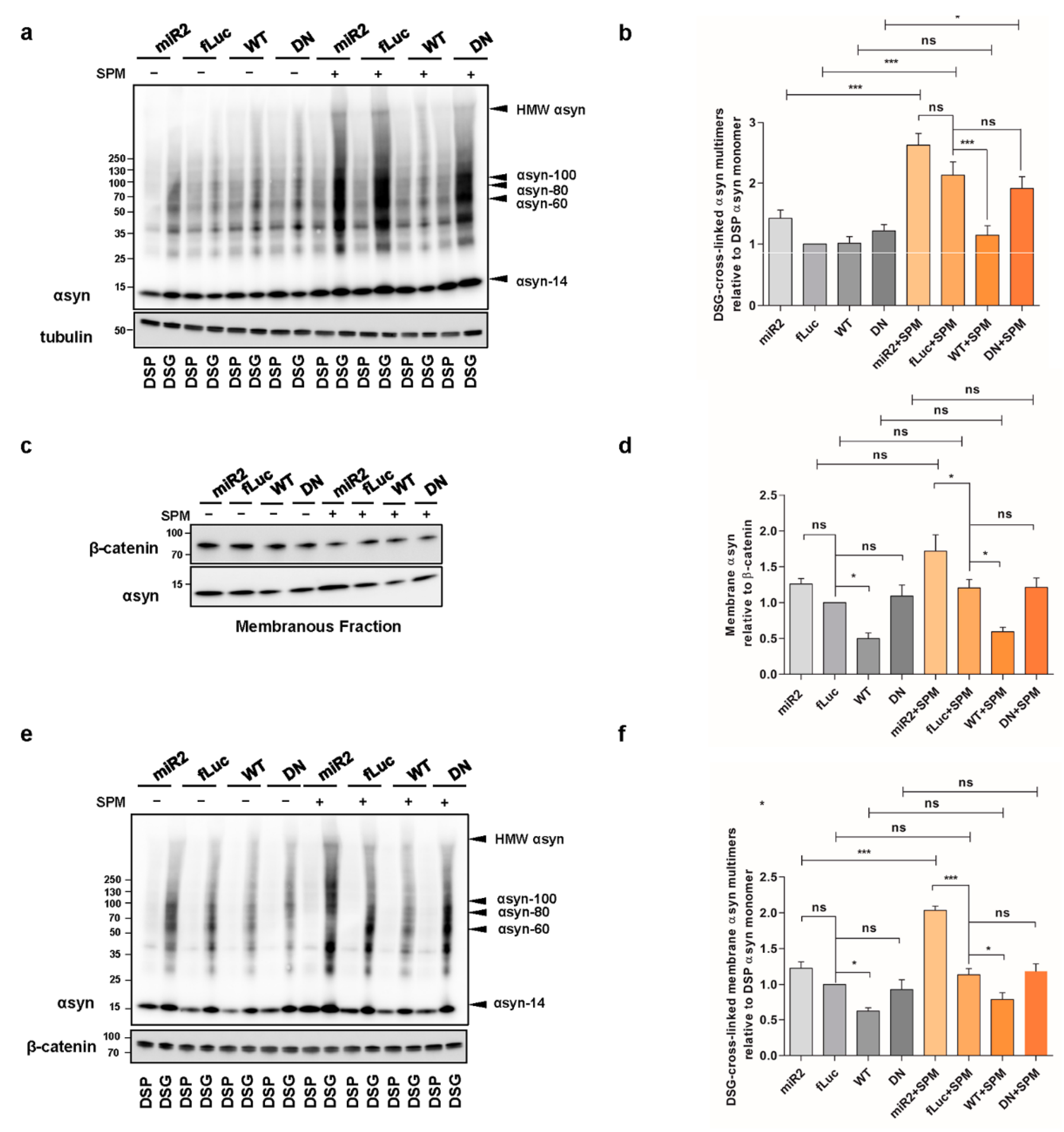
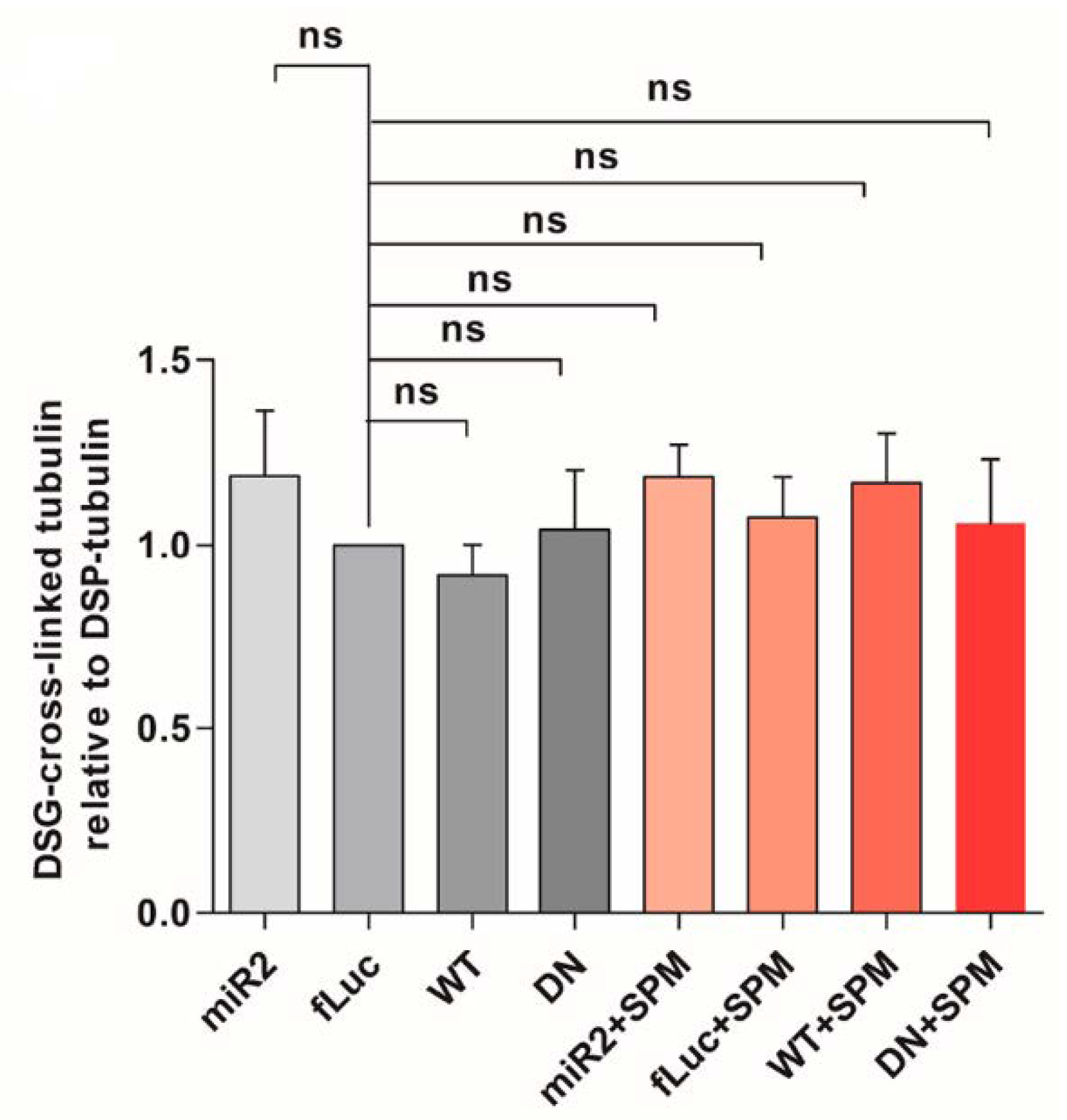
2.5. ATP13A2 Regulates SPM-Induced αsyn Multimerization
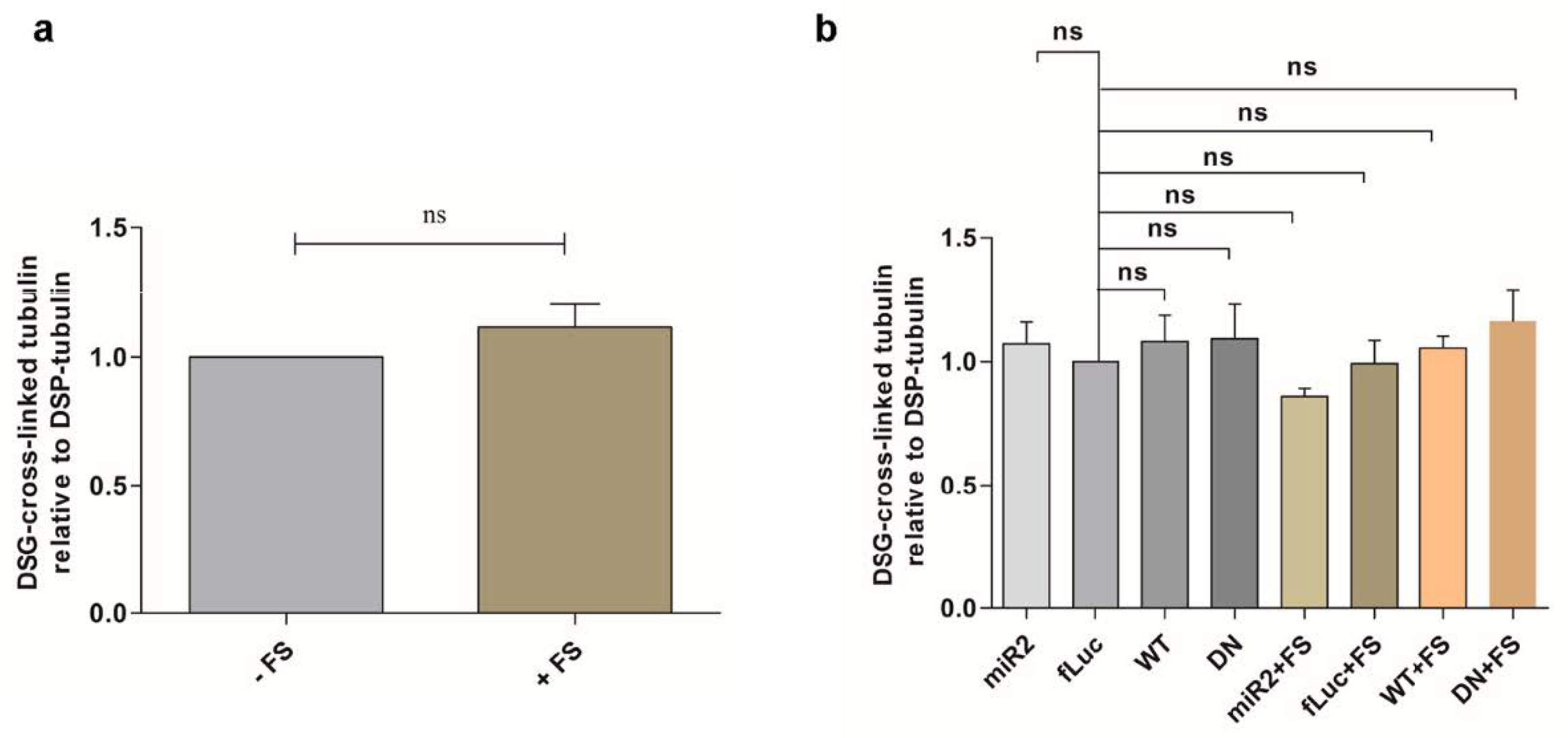
2.6. Impaired Lysosomal Membrane Integrity Is Linked to αsyn Multimerization
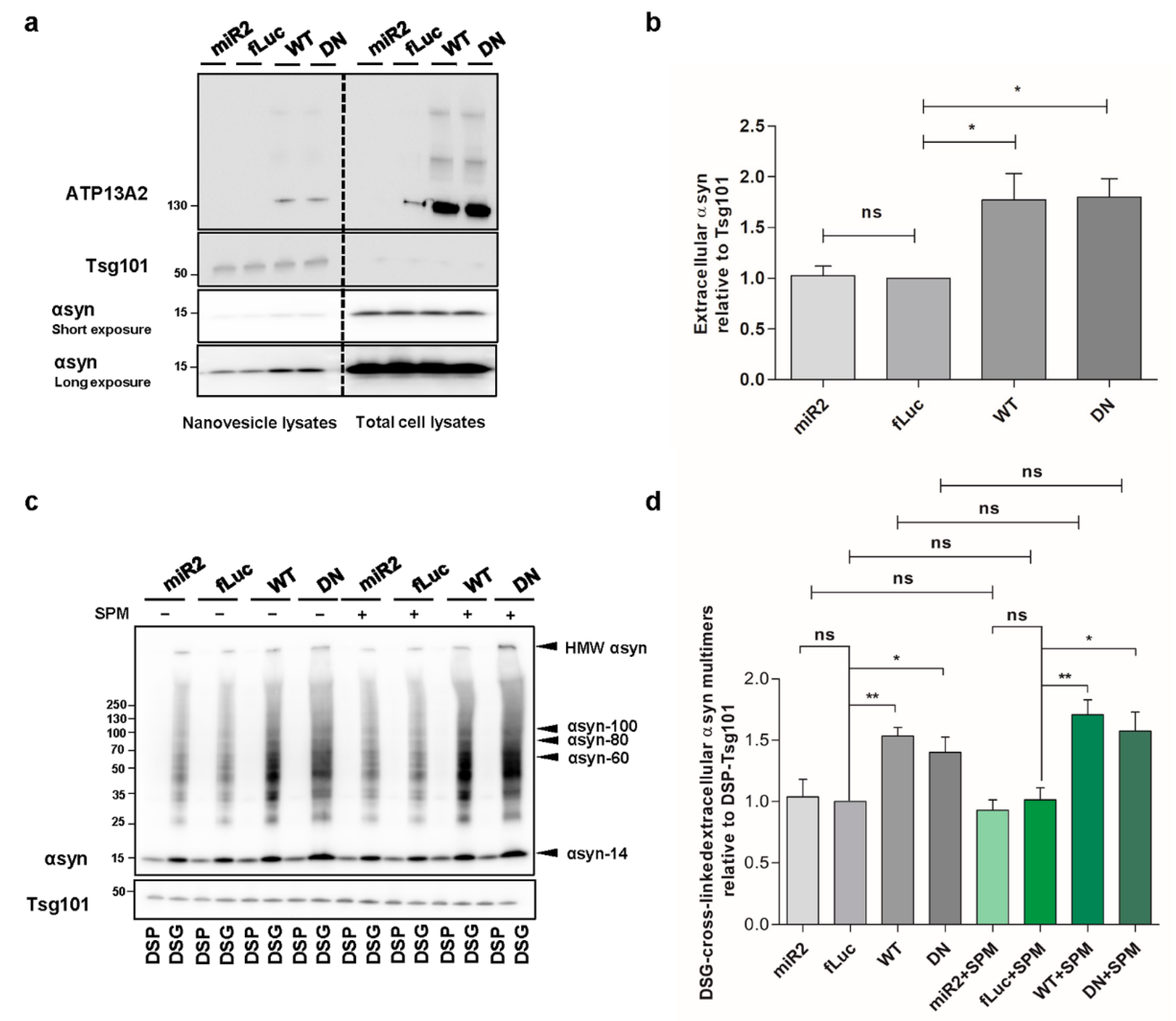
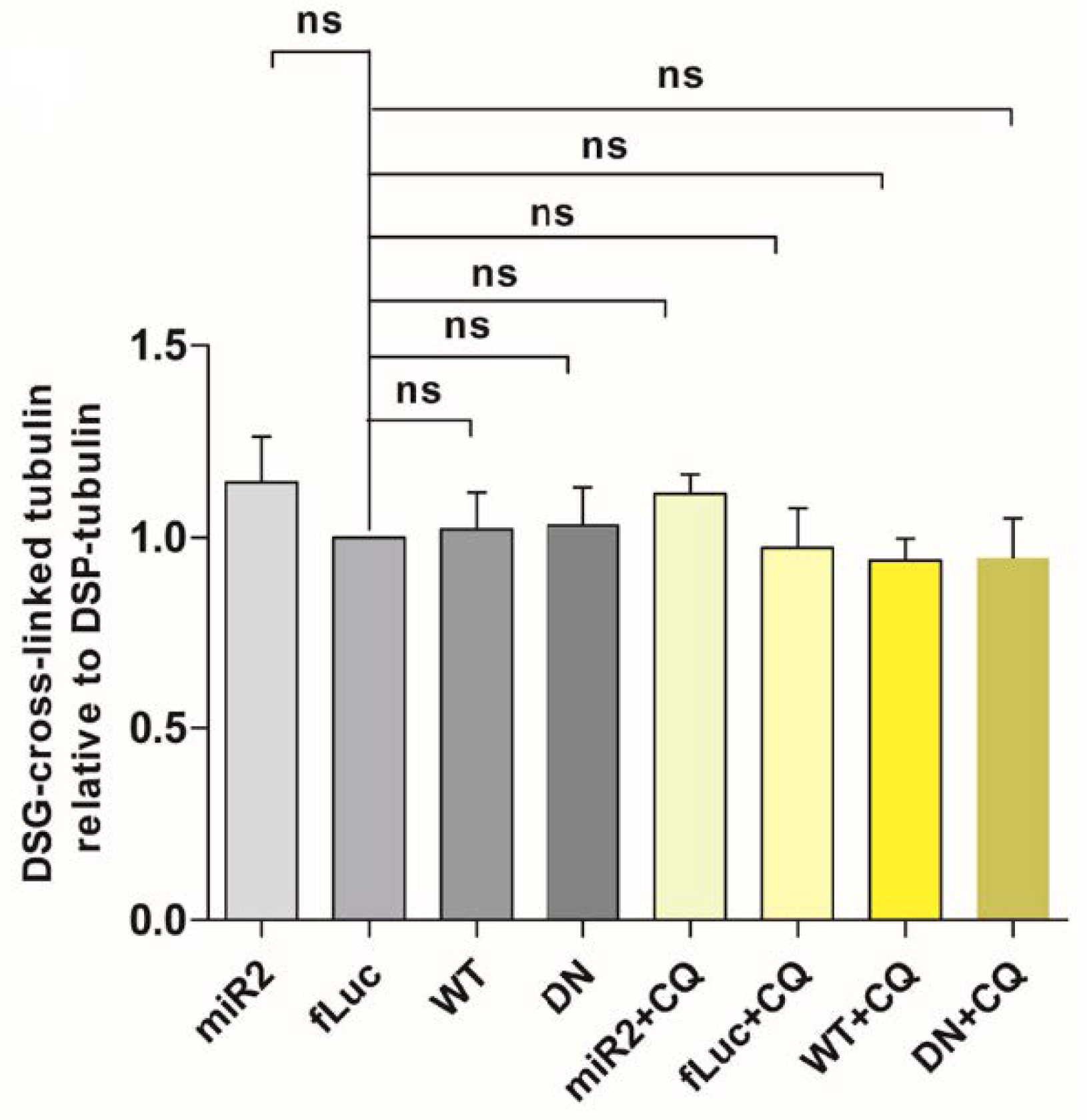
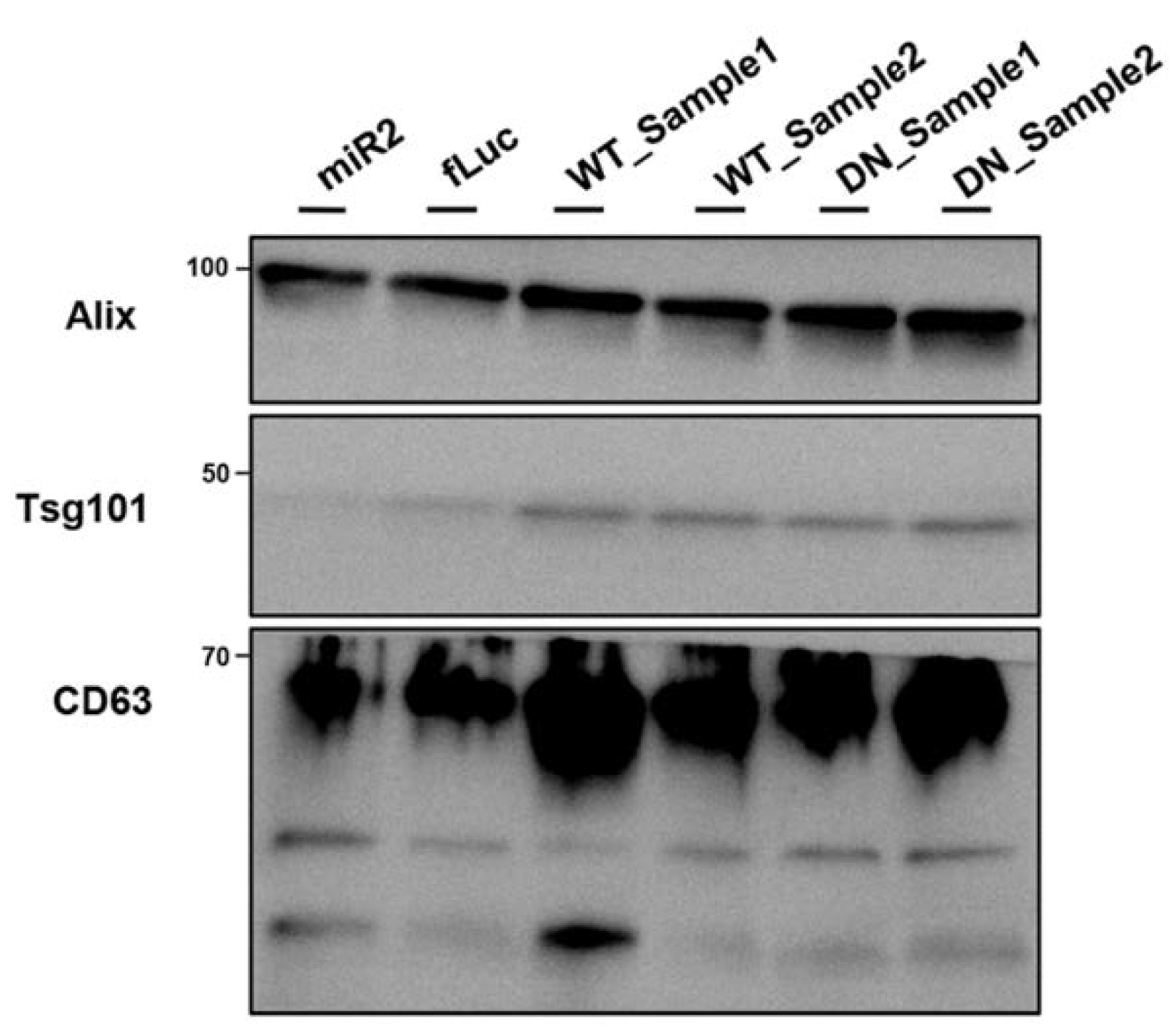
2.7. ATP13A2 Promotes Secretion of αsyn Multimers
3. Discussion
4. Materials and Methods
4.1. Generation of Stable Overexpression Cell Lines
4.2. Oxidative Stress Cell Model and SPM Addition
4.3. Intact Cell Cross-Linking
4.4. Membrane Fractionation
4.5. Nanovesicle Isolation
4.6. Lysosomal Membrane Integrity
4.7. Immunoblotting
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Boassa, D.; Berlanga, M.L.; Yang, M.A.; Terada, M.; Hu, J.; Bushong, E.A.; Hwang, M.; Masliah, E.; George, J.M.; Ellisman, M.H. Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson’s disease pathogenesis. J. Neurosci. 2013, 33, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Emanuele, M.; Chieregatti, E. Mechanisms of alpha-synuclein action on neurotransmission: Cell-autonomous and non-cell autonomous role. Biomolecules 2015, 5, 865–892. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Choi, C.; Lee, S.-J. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Necula, M.; Chirita, C.N.; Kuret, J. Rapid Anionic Micelle-mediated α-Synuclein Fibrillization in Vitro. J. Biol. Chem. 2003, 278, 46674–46680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchi, F.; Deleersnijder, A.; Van den Haute, C.; Munck, S.; Pottel, H.; Michiels, A.; Debysere, Z.; Gerard, M.; Baekelandt, V. High-content analysis of α-synuclein aggregation and cell death in a cellular model of Parkinson’s disease. J. Neurosci. Methods 2015, 261, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.-L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Kong, S.M.; Chan, B.K.; Park, J.-S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiberg, H.; Hansen, L.; Korbo, L.; Nielsen, I.; Svenstrup, K.; Bech, S.; Pinborg, L.; Friberg, L.; Hjermind, L.; Olsen, O.; et al. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9). Clin. Genet. 2011, 82, 256–263. [Google Scholar] [CrossRef]
- Podhajska, A.; Musso, A.; Trancikova, A.; Stafa, K.; Moser, R.; Sonnay, S.; Glauser, L.; Moore, D.J. Common Pathogenic Effects of Missense Mutations in the P-Type ATPase ATP13A2 (PARK9) Associated with Early-Onset Parkinsonism. PLoS ONE 2012, 7, e39942. [Google Scholar] [CrossRef] [Green Version]
- Jamil, M.; Siddiqui, A.W.; Sollinger, A.; Ferrara, J. ATP13A2-Related Heriditary Spastic Paraplegia (HSP) (P6.044). Neurology 2018, 90, P6.044. [Google Scholar]
- Bras, J.; Verloes, A.; Schneider, S.A.; Mole, S.E.; Guerreiro, R.J. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 2012, 21, 2646–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spataro, R.; Kousi, M.; Farhan, S.M.K.; Willer, J.R.; Ross, J.P.; Dion, P.A.; Rouleau, G.A.; Daly, M.J.; Neale, B.M.; La Bella, V.; et al. Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum. Genom. 2019, 13, 1–10. [Google Scholar] [CrossRef]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Sato, F.; Sato, S.; Koike, M.; Taruno, Y.; Saiki, S.; Funayama, M.; Ito, H.; Taniguchi, Y.; Uemura, N.; et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. 2013, 587, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1–1843.e7. [Google Scholar]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–997. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited α-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 Regulates Secretion of Exosomes and -Synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.-C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Dirr, E.R.; Ekhator, O.R.; Blackwood, R.; Holden, J.G.; Masliah, E.; Schultheis, P.J.; Fleming, S.M. Exacerbation of sensorimotor dysfunction in mice deficient in Atp13a2 and overexpressing human wildtype alpha-synuclein. Behav. Brain Res. 2018, 343, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Paolo, G.D.; Przedborski, S.; et al. α-Synuclein-Independent Histopathological and Motor Deficits in Mice Lacking the Endolysosomal Parkinsonism Protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef] [Green Version]
- Daniel, G.; Musso, A.; Tsika, E.; Fiser, A.; Glauser, L.; Pletnikova, O.; Schneider, B.L.; Moore, D.J. α-Synuclein-induced dopaminergic neurodegeneration in a rat model of Parkinson’s disease occurs independent of ATP13A2 (PARK9). Neurobiol. Dis. 2015, 73, 229–243. [Google Scholar] [CrossRef]
- van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.-P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [Green Version]
- Vrijsen, S.; Besora-Casals, L.; Van Veen, S.; Zielich, J.; Haute, C.V.D.; Hamouda, N.N.; Fischer, C.; Ghesquière, B.; Tournev, I.; Agostinis, P.; et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef]
- Grabenauer, M.; Bernstein, S.L.; Lee, J.C.; Wyttenbach, T.; Dupuis, N.F.; Gray, H.B.; Winkler, J.R.; Bowers, M.T. Spermine Binding to Parkinson’s Protein α-Synuclein and Its Disease-Related A30P and A53T Mutants. J. Phys. Chem. B 2008, 112, 11147–11154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnoslobodtsev, A.V.; Peng, J.; Asiago, J.M.; Hindupur, J.; Rochet, J.-C.; Lyubchenko, Y.L. Effect of Spermidine on Misfolding and Interactions of Alpha-Synuclein. PLoS ONE 2012, 7, e38099. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and Protein Degradation Systems: A Reciprocal Relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Lopes da Fonseca, T.; Pinho, R.; Outeiro, T.F. A familial ATP13A2 mutation enhances alpha-synuclein aggregation and promotes cell death. Hum. Mol. Genet. 2016, 25, 2959–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In Vivo Cross-linking Reveals Principally Oligomeric Forms of α-Synuclein and β-Synuclein in Neurons and Non-neural Cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuber, F. Influence of polyamines on membrane functions. Biochem. J. 1989, 260, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirsoy, S.; Martin, S.; Motamedi, S.; van Veen, S.; Holemans, T.; Van den Haute, C.; Jordanova, A.; Baekelandt, V.; Vangheluwe, P.; Agostinis, P. ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum. Mol. Genet. 2017, 26, 1656–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, T.; Hoyer, W.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Cellular Polyamines Promote the Aggregation of α-Synuclein. J. Biol. Chem. 2003, 278, 3235–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holemans, T.; Sørensen, D.M.; van Veen, S.; Martin, S.; Hermans, D.; Kemmer, G.C.; Van den Haute, C.; Baekelandt, V.; Pomorski, T.G.; Agostinis, P.; et al. A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc. Natl. Acad. Sci. USA 2015, 112, 9040–9045. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, J.; Van den Haute, C.; Lobbestael, E.; Martin, S.; van Veen, S.; Vangheluwe, P.; Baekelandt, V. ATP13A2 Regulates Cellular α-Synuclein Multimerization, Membrane Association, and Externalization. Int. J. Mol. Sci. 2021, 22, 2689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052689
Si J, Van den Haute C, Lobbestael E, Martin S, van Veen S, Vangheluwe P, Baekelandt V. ATP13A2 Regulates Cellular α-Synuclein Multimerization, Membrane Association, and Externalization. International Journal of Molecular Sciences. 2021; 22(5):2689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052689
Chicago/Turabian StyleSi, Jianmin, Chris Van den Haute, Evy Lobbestael, Shaun Martin, Sarah van Veen, Peter Vangheluwe, and Veerle Baekelandt. 2021. "ATP13A2 Regulates Cellular α-Synuclein Multimerization, Membrane Association, and Externalization" International Journal of Molecular Sciences 22, no. 5: 2689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052689