
Adaptive Changes in the Central Control of Energy Homeostasis Occur in Response to Variations in Energy Status

 ,
, 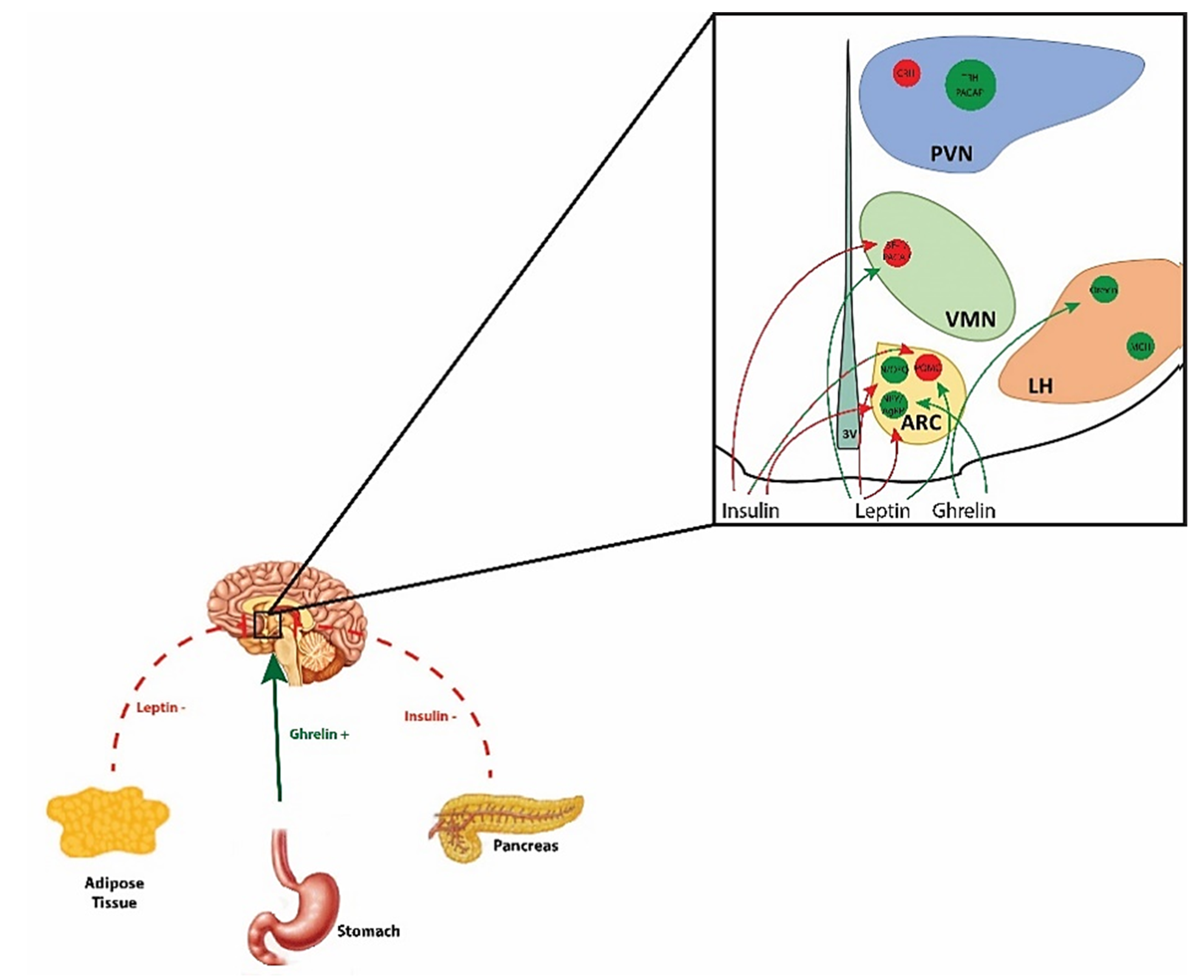{kind=link}
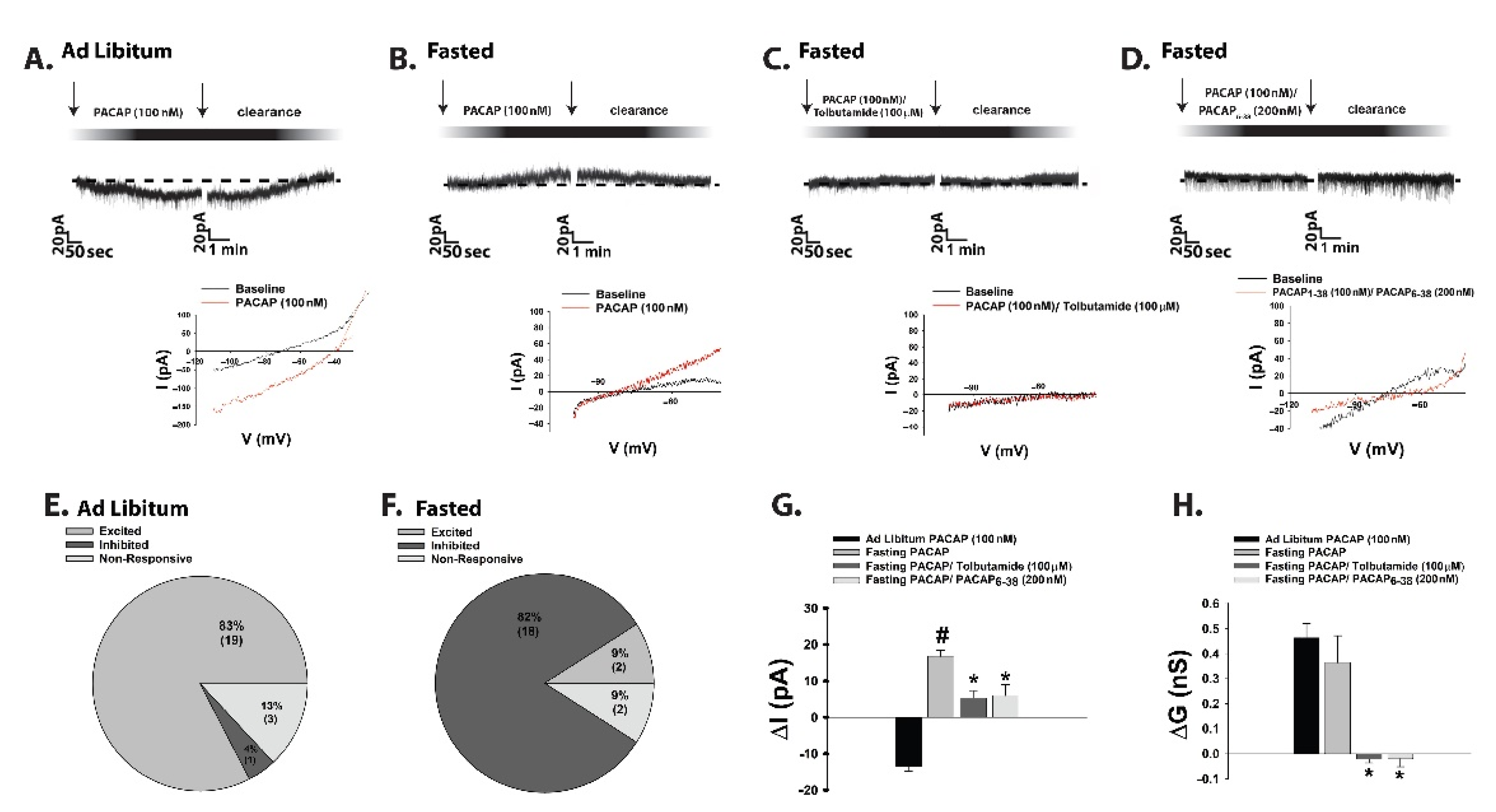{kind=link}
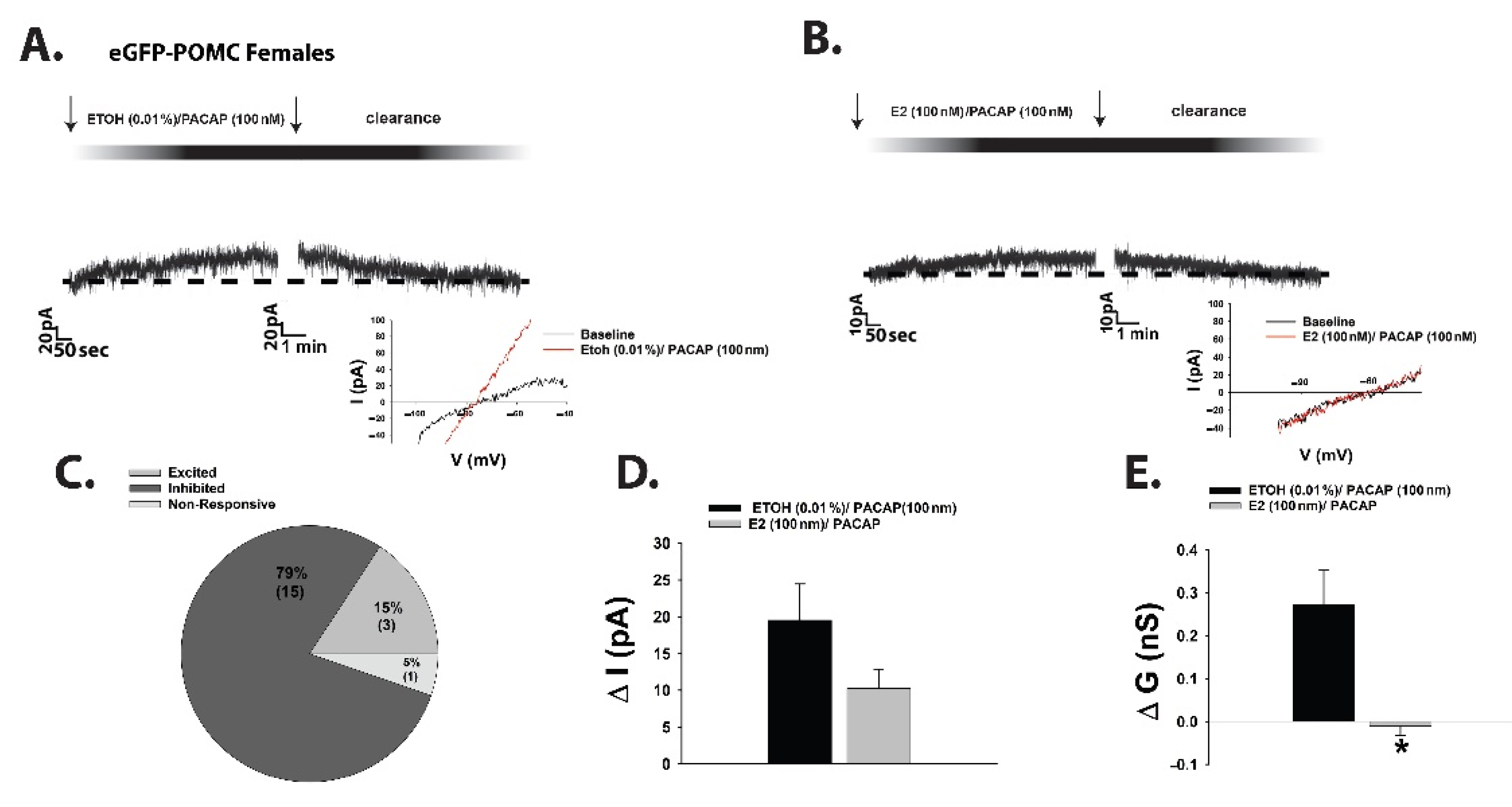{kind=link}
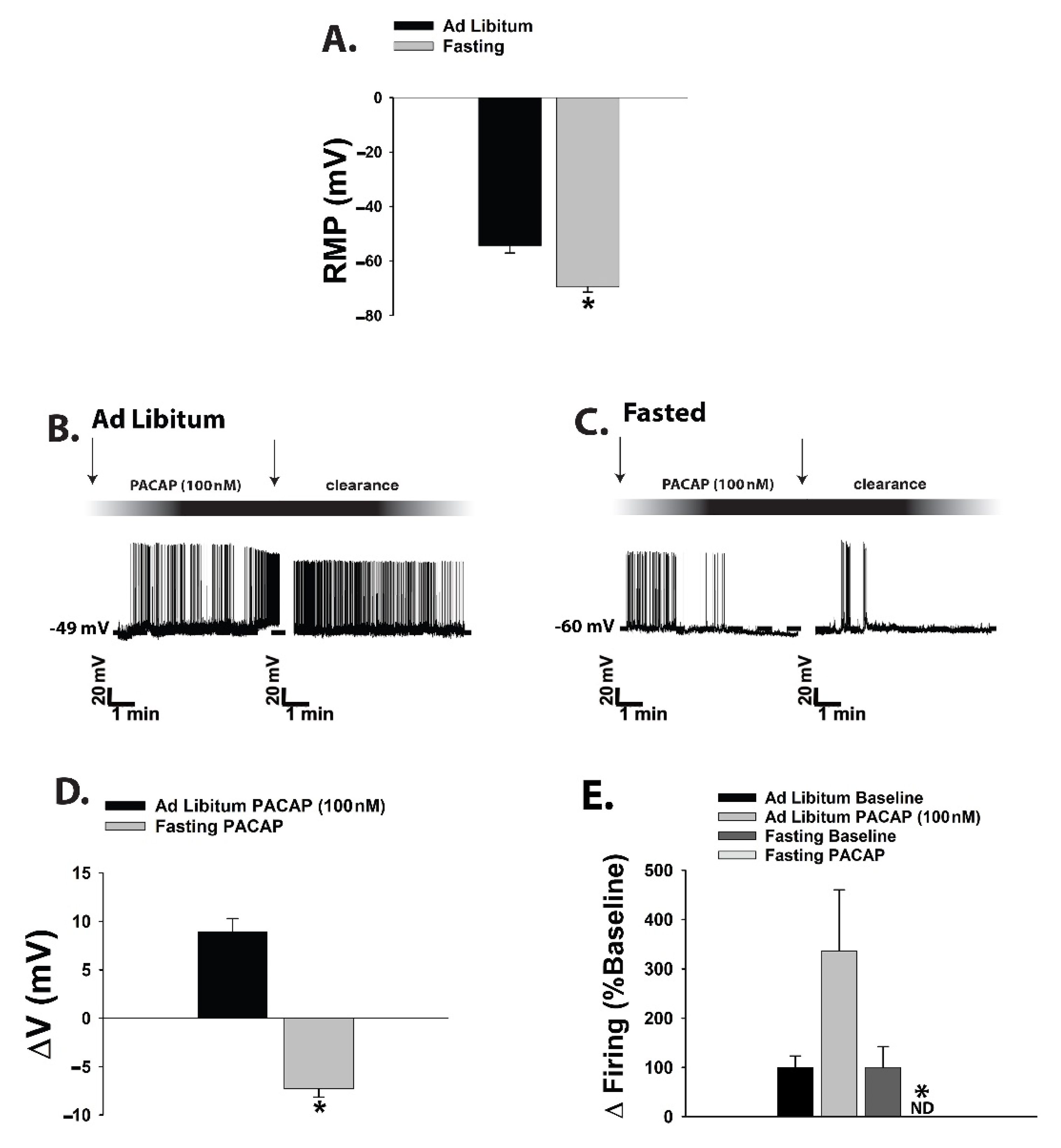{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Hypothalamic Energy Balance Circuit in Homeostatic Feeding
2. The Mesolimbic Dopamine Network and Hedonic Feeding Behavior
3. Influences of Sex and Diet on Central Energy Balance Circuits
4. Nociceptin/Orphanin FQ Regulation of Homeostatic and Hedonic Energy Balance Circuits
5. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Regulation of Homeostatic and Hedonic Energy Balance Circuits
6. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berthoud, H.-R. The neurobiology of food intake in an obesogenic environment. Proc. Nutr. Soc. 2012, 71, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majdic, G.; Young, M.; Gomez-Sanchez, E.; Anderson, P.; Szczepaniak, L.S.; Dobbins, R.L.; McGarry, J.D.; Parker, K.L. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology 2002, 143, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Fordahl, S.C.; Jones, S.R. High-fat-diet induced deficits in dopamine terminal function are reversed by restoring insulin signaling. ACS Chem. Neurosci. 2017, 8, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Wise, R.A.; Baler, R. The dopamine motive system: Implications for drug and food addiction. Nat. Rev. Neurosci. 2017, 18, 741–752. [Google Scholar] [CrossRef]
- Hernandez, J.; Fabelo, C.; Perez, L.; Moore, C.; Chang, R.; Wagner, E.J. Nociceptin/orphanin FQ modulates energy homeostasis through inhibition of neurotransmission at VMN SF-1/ARC POMC synapses in a sex- and diet-dependent manner. Biol. Sex Diff. 2019, 10. [Google Scholar] [CrossRef]
- Browning, K.N.; Kalyuzhny, A.E.; Travagli, R.A. Opioid peptides inhibit excitatory but not inhibitory synaptic transmission in the rat dorsal motor nucleus of the vagus. J. Neurosci. 2002, 22, 2998–3004. [Google Scholar] [CrossRef]
- Sobrino Crespo, C.; Perianes Cachero, A.; Puebla Jimenez, L.; Barrios, V.; Arilla Ferreiro, E. Peptides and food intake. Front. Endocrinol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Sternson, S.M.; Shepherd, G.M.G.; Friedman, J.M. Topographic mapping of VMH-arcuate nucleus microcirsuits and their reoganization by fasting. Nat. Neurosci. 2005, 8, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Shah, B.P.; Madara, J.C.; Olson, D.P.; Strochlic, D.E.; Garfield, A.S.; Vong, L.; Pei, H.; Watabe-Uchida, M.; Uchida, N.; et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 2014, 507, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Conde, K.; Fabelo, C.; Krause, W.C.; Propst, R.; Goethel, J.; Fischer, D.; Hur, J.; Meza, C.; Ingraham, H.A.; Wagner, E.J. Testosterone rapidly augments retrograde endocannabinoid signaling in proopiomelanocortin neurons to suppress glutamatergic input from steroidogenic factor 1 neurons via upregulation of diacylglycerol lipase-α. Neuroendocrinology 2017, 105, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Fabelo, C.; Hernandez, J.; Chang, R.S.S.; Alicea, N.; Tian, S.; Conde, K.; Wagner, E.J. Endocannabinoid signaling at hypothalamic steroidogenic factor-1/proopiomelanocortin synapses is sex- and diet-sensitive. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Hernandez, J.; Gastelum, C.; Guadagno, K.; Perez, L.; Wagner, E.J. Pituitary adenylate cyclase-activating polypeptide excites proopiomelanocortin neurons: Implications for the regulation of energy homeostasis. Neuroendocrinology 2020. [Google Scholar] [CrossRef] [PubMed]
- Jacobowitz, D.M.; O’Donohue, T.L. α-Melanocyte stimulating hormone: Immunolohistochemical identification and mapping in neurons of rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 6300–6304. [Google Scholar] [CrossRef] [Green Version]
- Kiss, J.Z.; Cassell, M.D.; Palkovits, M. Analysis of the ACTH/β-end/α-MSH-immunoreactive afferent input to the hypothalamic paraventricular nucleus of rat. Brain Res. 1984, 324, 91–99. [Google Scholar] [CrossRef]
- Fan, W.; Boston, B.A.; Kesterson, R.A.; Hruby, V.J.; Cone, R.D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997, 385, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Yaswen, L.; Diehl, N.; Brennan, M.B.; Hochgeschwender, U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. 1999, 5, 1066–1070. [Google Scholar] [CrossRef]
- Bomberg, E.M.; Grace, M.K.; Levine, A.S.; Olszewski, P.K. Functional interaction between nociceptin/orphanin FQ and α-melanocyte-stimulating hormone in the regulation of feeding. Peptides 2006, 27, 1827–1834. [Google Scholar] [CrossRef]
- Appleyard, S.M.; Hayward, M.; Young, J.I.; Butler, A.A.; Cone, R.D.; Rubinstein, M.; Low, M.J. A role for endogenous β-endorphin in energy homeostasis. Endocrinology 2003, 144, 1753–1760. [Google Scholar] [CrossRef] [Green Version]
- Horvath, T.L. The hardship of obesity: A soft-wired hypothalamus. Nat. Neurosci. 2005, 8, 561–565. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.; Choi, B.; Zhang, C.-Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose sensing by POMC neurons regulates glucose homestasis and is impaired in obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Burdakov, D.; Luckman, S.M.; Verkhratsky, A. Glucose-sensing neurons of the hypothalamus. Phil. Trans. R. Soc. B 2005, 360, 2227–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgardt, B.F.; Okamura, T.; Brüning, J.C. Hormone and glucose signalling in POMC and AgRP neurons. J. Physiol. 2009, 587, 5305–5314. [Google Scholar] [CrossRef] [PubMed]
- Stricker, E.M. Hyperphagia. N. Engl. J. Med. 1978, 298, 1010–1013. [Google Scholar] [CrossRef]
- Chronwall, B.M. Anatomy and physiology of the neuroendocrine arcuate nucleus. Peptides 1985, 6, 1–11. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Krashes, M.J.; Koda, S.; Ye, C.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Q.; Ye, C.-P.; Jones, J.E.; Elmquist, J.K.; Lowell, B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 2008, 11, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.M.B.; Abusnana, S.; Sunter, D.; Murphy, K.G.; Ghatei, M.A.; Bloom, S.R. The effect of the orexins on food intake: Comparison with neuropeptide Y, melanin-concentrating hormone and galanin. J. Endocrinol. 1999, 160, R7–R12. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.J. Sex differences in cannabinoid-regulated biology: A focus on energy homeostasis. Front. Neuroendocrinol. 2016, 40, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zubcevic, L.; Brüning, J.C.; Ashcroft, F.M.; Burdakov, D. Electrical inhibition of identified anorexigenic POMC neurons by orexin/hypocretin. J. Neurosci. 2007, 27, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.-H.; Chen, Y.-J.L.; Chua, S.C.; Talmage, D.A.; Role, L.W. Integration of endocannabinoid and leptin signaling in an appetite-related neural circuit. Neuron 2005, 48, 1055–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullier, A.; Bouret, S.G.; Prevot, V.; Dehouck, B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J. Comp. Neurol. 2010, 518, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef]
- Zigman, J.M.; Elmquist, J.K. Minireview: From anorexia to obesity—The yin and yang of body weight control. Endocrinology 2003, 144, 3749–3756. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.A.; Cone, R.D.; Enriori, P.; Louiselle, I.; Williams, S.M.; Evans, A.E. Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann. N. Y. Acad. Sci. 2003, 994, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Fang, Y.; Rønnekleiv, O.K.; Kelly, M.J. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J. Neurosci. 2010, 30, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Van den Top, M.; Lee, K.; Whyment, A.D.; Blanks, A.M.; Spanswick, D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat. Neurosci. 2004, 5, 493–494. [Google Scholar] [CrossRef]
- Wei, W.; Pham, K.; Gammons, J.W.; Sutherland, D.; Liu, Y.; Smith, A.; Kaczorowski, C.C.; O’Connell, K.M. Diet composition, not calorie intake, rapidly alters intrinsic excitability of hypothalamic AgRP/NPY neurons in mice. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Wagner, E.J.; Ronnekleiv, O.K.; Kelly, M.J. Insulin and leptin excite anorexigenic pro-opiomelanocortin neurones via activation of TRPC5 channels. J. Neuroendocrinol. 2017, 30, e12501. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Goparahu, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014, 19, 682–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.-R.; Chuang, J.-C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J. Neurosci. 2010, 30, 2472–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, G.T.; Michael, N.J.; Lee-Young, R.S.; Mangiafico, S.P.; Pryor, J.T.; Munder, A.C.; Simonds, S.E.; Bruning, J.C.; Zhang, Z.-Y.; Cowley, M.A.; et al. Insulin regulates POMC neuronal plasticity to control glucose metabolism. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.-Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, G.T.; Andrews, Z.B.; Simonds, S.E.; Michael, N.J.; DeVeer, M.; Bruning, J.C.; Spanswick, D.; Cowley, M.A.; Tiganis, T. A Hypothalamic Phosphatase Switch Coordinates Energy Expenditure with Feeding. Cell Metab. 2017, 26, 375–393. [Google Scholar] [CrossRef]
- Dodd, G.T.; Xirouchaki, C.E.; Eramo, M.; Mitchell, C.A.; Andrews, Z.B.; Henry, B.A.; Cowley, M.A.; Tiganis, T. Intranasal targeting of hypothalamic PTP1B and TCPTP reinstates leptin and insulin sensitivity and promotes weight loss in obesity. Cell Rep. 2019, 28, 2905–2922. [Google Scholar] [CrossRef] [PubMed]
- Kola, B.; Farkas, I.; Christ-Crain, M.; Wittmann, G.; Lolli, F.; Amin, F.; Harvey-White, J.; Liposits, Z.; Kunos, G.; Grossman, A.B.; et al. The orexigenic effect of ghrelin is mediated through central activation of the endogenous cannabinoid system. PLoS ONE 2008, 3, e1797. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Kola, B.; Feltrin, D.; Perez-Tilve, D.; Tschöp, M.H.; Grossman, A.B.; Korbonits, M. Ghrelin and cannabinoids require the ghrelin receptor to affect cellular energy metabolism. Mol. Cell. Endocrinol. 2013, 365, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Scott, J.W.; Pan, D.A.; Hudson, E.R. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003, 546, 113–120. [Google Scholar] [CrossRef]
- Hardie, D.G. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [Green Version]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Kola, B.; Hubina, E.; Tucci, S.A.; Kirkham, T.C.; Garcia, E.A.; Mitchell, S.E.; Williams, L.M.; Hawley, S.A.; Hardie, D.G.; Grossman, A.B.; et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 25196–25201. [Google Scholar] [CrossRef] [Green Version]
- Yavari, A.; Stocker, C.J.; Ghaffari, S.; Wargent, E.T.; Steeples, V.; Czibik, G.; Pinter, K.; Bellahcene, M.; Woods, A.; de Morentin, P.B.M.; et al. Chronic Activation of γ2 AMPK Induces Obesity and Reduces β Cell Function. Cell Metab. 2016, 23, 821–836. [Google Scholar] [CrossRef] [Green Version]
- DiLeone, R.J.; Georgescu, D.; Nestler, E.J. Lateral hypothalamic neuropeptides in reward and drug addiction. Life Sci. 2003, 73, 759–768. [Google Scholar] [CrossRef]
- Georgescu, D.; Sears, R.M.; Hommel, J.D.; Barrot, M.; Bolaños, C.A.; Marsh, D.J.; Bednarek, M.A.; Bibb, J.A.; Maratos-Flier, E.; Nestler, E.J.; et al. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J. Neurosci. 2005, 25, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- McClung, C.A.; Nestler, E.J.; Zachariou, V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J. Neurosci. 2005, 25, 6005–6015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalivas, P.W.; Duffy, P. Sensitization to repeated morphine injection in the rat: Possible involvement of A10 dopamine neurons. J. Pharmacol. Exp. Ther. 1987, 241, 204–212. [Google Scholar]
- Wise, R.A. Role of brain dopamine in food reward and reinforcement. Phil. Trans. R. Soc. B 2006, 361, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durst, M.; Konczol, K.; Balazsa, T.; Eyre, M.D.; Toth, Z.E. Reward-representing D1-type neurons in the medial shell of the accumbens nucleus regulate palatable food intake. Int. J. Obes. 2019, 43, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.E. Hypothalamic dopaminergic neuronal systems. In Psychopharmacology: The Third Generation of Progress; Meltzer, H.Y., Ed.; Raven Press: New York, NY, USA, 1987; pp. 127–139. [Google Scholar]
- Tsai, H.-C.; Zhang, F.; Adamantidis, A.; Stuber, G.D.; Bonci, A.; De Lecea, L.; Deisseroth, K. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science 2009, 324, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- Witten, I.B.; Steinberg, E.E.; Lee, S.Y.; Davidson, T.J.; Zalocusky, K.A.; Brodsky, M.; Yizhar, O.; Cho, S.L.; Gong, S.; Ramakrishnan, C.; et al. Recombinase-driver rat lines: Tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron 2011, 72, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Ilango, A.; Kesner, A.J.; Keller, K.L.; Stuber, G.D.; Bonci, A.; Ikemoto, S. Similar roles of substantia nigra and ventral tegmental dopamine neurons in reward and aversion. J. Neurosci. 2014, 34, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, E.E.; Boivin, J.R.; Saunders, B.T.; Witten, I.B.; Deisseroth, K.; Janak, P.H. Positive reinforcement mediated by midbrain dopamine neurons requires D1 and D2 receptor activation in the nucleus accumbens. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.R.; Yvon, C.; Turiault, M.; Mirzabekov, J.J.; Doehner, J.; Labouebe, G.; Deisseroth, K.; Tye, K.M.; Luscher, C. GABA neurons of the VTA drive conditioned place aversion. Neuron 2012, 73, 1173–1183. [Google Scholar] [CrossRef]
- Jennings, J.H.; Sparta, D.R.; Stamatakis, A.M.; Ung, R.L.; Pleil, K.E.; Kash, T.L.; Stuber, G.D. Distinct extended amygdala circuits for divergent motivational states. Nature 2013, 496, 224–228. [Google Scholar] [CrossRef]
- Carlezon, W.A., Jr.; Chartoff, E.H. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat. Protoc. 2007, 2, 2987–2995. [Google Scholar] [CrossRef]
- Wang, H.-L.; Qi, J.; Zhang, S.; Wang, H.; Morales, M. Rewarding Effects of Optical Stimulation of Ventral Tegmental Area Glutamatergic Neurons. J. Neurosci. 2015, 35, 15948–15954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.E.; Berridge, K.C. The incentive sensitization theory of addiction: Some current issues. Phil. Trans. R. Soc. B 2008, 363, 3137–3146. [Google Scholar] [CrossRef]
- Avena, N.M.; Bocarsly, M.E. Dysregulation of brain reward systems in eating disorders: Neurochemical information from animal models of binge eating, bulimia nervosa, and anorexia nervosa. Neuropharmacology 2012, 63, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, J.L.; McKee, S.E.; Hill-Smith, T.; Grissom, N.M.; George, R.; Lucki, I.; Reyes, T.M. Removal of high-fat diet after chronic exposure drives binge behavior and dopaminergic dysregulation in female mice. Neuroscience 2016, 326, 170–179. [Google Scholar] [CrossRef]
- Berridge, K.C. Food reward: Brain substrates of wanting and liking. Neurosci. Biobehav. Rev. 1996, 20, 1–25. [Google Scholar] [CrossRef]
- Berridge, K.C. ‘Liking’ and ‘wanting’ food rewards: Brain substrates and roles in eating disorders. Physiol. Behav. 2009, 97, 537–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, K.C. Measuring hedonic impact in animals and infants: Microstructure of affective taste reactivity patterns. Neurosci. Biobehav. Rev. 2000, 24, 173–198. [Google Scholar] [CrossRef]
- Grill, H.J.; Norgren, R. The taste reactivity test. I. Mimetic responses to gustatory stimuli in neurologically normal rats. Brain Res. 1978, 143, 263–279. [Google Scholar] [CrossRef]
- Steiner, J.E.; Glaser, D.; Hawilo, M.E.; Berridge, K.C. Comparative expression of hedonic impact: Affective reactions to taste by human infants and other primates. Neurosci. Biobehav. Rev. 2001, 25, 53–74. [Google Scholar] [CrossRef]
- Smith, K.S.; Berridge, K.C. The ventral pallidum and hedonic reward: Neurochemical maps of sucrose “liking” and food intake. J. Neurosci. 2005, 25, 8637–8649. [Google Scholar] [CrossRef] [Green Version]
- Mahler, S.V.; Smith, K.S.; Berridge, K.C. Endocannabinoid hedonic hotspot for sensory pleasure: Anandamide in nucleus accumbens shell enhances ‘liking’ of a sweet reward. Neuropsychopharmacology 2007, 32, 2267–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Le Moal, M. Drug abuse: Hedonic homeostatic dysregulation. Science 1997, 278, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Smith, J.; Mirenowicz, J. Dissociation of Pavlovian and instrumental incentive learning under dopamine antagonists. Behav. Neurosci. 2000, 114, 468–483. [Google Scholar] [CrossRef]
- Everitt, B.J.; Robbins, T.W. Neural systems of reinforcement for drug addiction: From actions to habits to compulsion. Nat. Neurosci. 2005, 8, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.M.; Palmiter, R.D. Reward without dopamine. J. Neurosci. 2003, 23, 10827–10831. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Dou, Y.-N.; Sun, Y.-G. Topography of Reward and Aversion Encoding in the Mesolimbic Dopaminergic System. J. Neurosci. 2019, 39, 6472–6481. [Google Scholar] [CrossRef]
- De Jong, J.W.; Afjei, S.A.; Dorocic, I.P.; Peck, J.R.; Liu, C.; Kim, C.K.; Tian, L.; Deisseroth, K.; Lammel, S. A Neural Circuit Mechanism for Encoding Aversive Stimuli in the Mesolimbic Dopamine System. Neuron 2019, 101, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Soares-Cunha, C.; de Vasconelos, N.A.P.; Coimbra, B.; Domingues, A.V.; Silva, J.M.; Loureiro-Campos, E.; Gaspar, R.; Sotiropoulos, I.; Sousa, N.; Rodrigues, A.J. Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol. Psychiatry 2020, 25, 3241–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figlewicz, D.P.; Benoit, S.C. Insulin, leptin, and food reward: Update 2008. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R9–R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, S.; Pissios, P.; Manchon, R.P.; Stiles, L.; Frank, L.; Pothos, E.N.; Maratos-Flier, E.; Flier, J.S. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 2006, 51, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Hommel, J.D.; Trinko, R.; Sears, R.M.; Georgescu, D.; Liu, Z.-W.; Gao, X.-B.; Thurmon, J.J.; Marinelli, M.; DiLeone, R.J. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 2006, 51, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Leinninger, G.M.; Jo, Y.-H.; Leshan, R.L.; Louis, G.W.; Yang, H.; Barrera, J.G.; Wilson, H.; Opland, D.M.; Faouzi, M.A.; Gong, Y.; et al. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Neuron 2009, 10, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Abizaid, A.; Liu, Z.-W.; Andrews, Z.B.; Shanabrough, M.; Borok, E.; Elsworth, J.D.; Roth, R.H.; Sleeman, M.W.; Picciotto, M.R.; Tschöp, M.H.; et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Investig. 2006, 116, 3229–3239. [Google Scholar] [CrossRef] [PubMed]
- Kalafateli, A.L.; Vallöf, D.; Jörnulf, J.W.; Heilig, M.; Jerlhag, E. A cannabinoid receptor antagonist attenuates ghrelin-induced activation of the mesolimbic dopamine system in mice. Physiol. Behav. 2018, 184, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Hoebel, B.G. Food reward and cocaine increase extracellular dopamine in the nucleus accumbens as measured by microdialysis. Life Sci. 1988, 42, 1705–1712. [Google Scholar] [CrossRef]
- Martel, P.; Fantino, M. Mesolimbic dopaminergic system activity as a function of food reward: A microdialysis study. Pharmacol. Biochem. Behav. 1996, 53, 221–226. [Google Scholar] [CrossRef]
- Bassareo, V.; Di Chiara, G. Modulation of feeding-induced activation of mesolimbic dopamine transmission by appetitive stimuli and its relation to motivational state. Eur. J. Neurosci. 1999, 11, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Avena, N.M.; Hoebel, B.G. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 2005, 134, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Avena, N.M.; Barson, J.R.; Hoebel, B.G.; Leibowitz, S.F. A high-fat meal, or intraperitoneal administration of a fat emulsion, increases extracellular dopamine in the nucleus accumbens. Brain Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Di Ciano, P.; Coury, A.; Depoortere, R.Y.; Egilmez, Y.; Lane, J.D.; Emmett-Oglesby, M.W.; Lepiane, F.G.; Phillips, A.G.; Blaha, C.D. Comparison of changes in extracellular dopamine concentrations in the nucleus accumbens during intravenous self-administration of cocaine or d-amphetamine. Behav. Pharmacol. 1995, 6, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Kiyatkin, E.A. Functional significance of mesolimbic dopamine. Neurosci. Biobehav. Rev. 1995, 19, 573–598. [Google Scholar] [CrossRef]
- Di Chiara, G.; Acquas, E.; Tanda, G. Ethanol as a neurochemical surrogate of conventional reinforcers: The dopamine-opioid link. Alcohol 1996, 13, 13–17. [Google Scholar] [CrossRef]
- Tanda, G.; Pontieri, F.E.; Di Chiara, G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a μ1 opioid receptor mechanism. Science 1997, 276, 2048–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheer, J.F.; Wassum, K.M.; Sombers, L.A.; Heien, M.L.A.V.; Ariansen, J.L.; Aragona, B.J.; Phillips, P.E.M.; Wightman, R.M. Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J. Neurosci. 2007, 27, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Stewart, R.B.; Thompson, S.C.; Meisch, R.A. Food-deprivation increases cocaine-induced conditioned place preference and locomotor activity in rats. Psychopharmacology 1997, 131, 1–8. [Google Scholar] [CrossRef]
- Colantuoni, C.; Schwenker, J.; McCarthy, J.; Rada, P.; Ladenheim, B.; Cadet, J.L.; Schwartz, G.J.; Moran, T.H.; Hoebel, B.G. Excessive sugar intake alters binding to dopamine and mu-opioid receptors in the brain. Neuroreport 2001, 12, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Hoebel, B.G. A diet promoting sugar dependency causes behavioral cross-sensitization to a low dose of amphetamine. Neuroscience 2003, 122, 17–20. [Google Scholar] [CrossRef]
- Avena, N.M.; Hoebel, B.G. Amphetamine-sensitized rats show sugar-induced hyperactivity (cross-sensitization) and sugar hyperphagia. Pharmacol. Biochem. Behav. 2003, 74, 635–639. [Google Scholar] [CrossRef]
- Bello, N.T.; Sweigart, K.L.; Lakoski, J.M.; Norgren, R.; Hajnal, A. Restricted feeding with scheduled sucrose access results in an upregulation of the rat dopamine transporter. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1260–R1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshi, V.P.; Kelley, A.E. Sensitization and conditioning of feeding following multiple morphine microinjections into the nucleus accumbens. Brain Res. 1994, 648, 342–346. [Google Scholar] [CrossRef]
- Nocjar, C.; Panksepp, J. Chronic intermittent amphetamine pretreatment enhances future appetitive behavior for drug- and natural-reward: Interaction with environmental variables. Behav. Brain Res. 2002, 128, 189–203. [Google Scholar] [CrossRef]
- Valdivia, S.; Cornejo, M.P.; Reynaldo, M.; De Francesco, P.N.; Perello, M. Escalation in high fat intake in a binge eating model differentially engages dopamine neurons of the ventral tegmental area and requires ghrelin signaling. Psychoneuroendocrinology 2015, 60, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.J.; Volkow, N.D.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusil, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef]
- Di Segni, M.; Patrono, E.; Patella, L.; Puglisi-Allegra, S.; Ventura, R. Animal models of compulsive eating behavior. Nutrients 2014, 6, 4591–4609. [Google Scholar] [CrossRef] [Green Version]
- Jauch-Chara, K.; Oltmanns, K.M. Obesity—A neuropsychological disease? Systematic review and neuropsychological model. Prog. Neurobiol. 2014, 114, 84–101. [Google Scholar] [CrossRef]
- Kreisler, A.D.; Garcia, M.G.; Spierling, S.R.; Hui, B.E.; Zorilla, E.P. Extended vs. brief intermittent access to palatable food differently promote binge-like intake, rejection of less preferred food, and weight cycling in female rats. Physiol. Behav. 2017, 177, 305–316. [Google Scholar] [CrossRef]
- Hoek, H.W. Incidence, prevalence and mortality of anorexia nervosa and other eating disorders. Curr. Opin. Psychiatry 2006, 19, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Colditz, G.A. Prevalence of Overweight and Obesity in the United States, 2007–2012. JAMA Intern. Med. 2015, 175, 1412–1413. [Google Scholar] [CrossRef] [Green Version]
- Hallam, J.; Boswell, R.G.; DeVito, E.E.; Kober, H. Gender-related Differences in Food Craving and Obesity. Yale J. Biol. Med. 2016, 89, 161–173. [Google Scholar] [PubMed]
- Weingarten, H.P.; Elston, D. Food cravings in a college population. Appetite 1991, 17, 167–175. [Google Scholar] [CrossRef]
- Zellner, D.A.; Garriga-Trillo, A.; Rohm, E.; Centeno, S.; Parker, S. Food liking and craving: A cross-cultural approach. Appetite 1999, 33, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Volkow, N.D.; Telang, F.; Jayne, M.; Ma, J.; Rao, M.; Zhu, W.; Wong, C.T.; Pappas, N.R.; Geliebter, A.; et al. Exposure to appetitive food stimuli markedly activates the human brain. Neuroimage 2004, 21, 1790–1797. [Google Scholar] [CrossRef]
- Uher, R.; Treasure, J.; Heining, M.; Brammer, M.J.; Campbell, I.C. Cerebral processing of food-related stimuli: Effects of fasting and gender. Behav. Brain Res. 2006, 169, 111–119. [Google Scholar] [CrossRef]
- Wang, G.J.; Volkow, N.D.; Telang, F.; Jayne, M.; Ma, Y.; Pradhan, K.; Zhu, W.; Wong, C.T.; Thanos, P.K.; Geliebter, A.; et al. Evidence of gender differences in the ability to inhibit brain activation elicited by food stimulation. Proc. Natl. Acad. Sci. USA 2009, 106, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Asarian, L.; Geary, N. Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 2007, 148, 5656–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammacharoen, S.; Lutz, T.A.; Geary, N.; Asarian, L. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 2008, 149, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Borgquist, A.; Meza, C.; Wagner, E.J. Role of neuronal nitric oxide synthase in the estrogenic attenuation of cannabinoid-induced changes in energy homeostasis. J. Neurophysiol. 2015, 113, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mela, V.; Vargas, A.; Meza, C.; Kachani, M.; Wagner, E.J. Modulatory influences of estradiol and other anorexigenic hormones on metabotropic, Gi/o-coupled receptor function in the hypothalamic control of energy homeostasis. J. Steroid Biochem. Mol. Biol. 2016, 160, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellert, B.A.; Nguyen, M.C.; Nguyen, C.; Nguyen, Q.H.; Wagner, E.J. Estrogen rapidly attenuates cannabinoid-induced changes in energy homeostasis. Eur. J. Pharmacol. 2009, 622, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Washburn, N.; Borgquist, A.; Wang, K.; Jeffery, G.S.; Kelly, M.J.; Wagner, E.J. Receptor subtypes and signal transduction mechanisms contributing to the estrogenic attenuation of cannabinoid-induced changes in energy homeostasis. Neuroendocrinology 2013, 97, 160–175. [Google Scholar] [CrossRef] [Green Version]
- Ihara, H.; Kitamura, A.; Kasamatsu, S.; Ida, T.; Kakihana, Y.; Tsutsuki, H.; Sawa, T.; Watanabe, Y.; Akaike, T. Superoxide generation from nNOS splice variants and its potential involvement in redox signal regulation. Biochem. J. 2017, 474, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Roseberry, A.G.; Liu, H.; Diano, S.; Shanabrough, M.; Cai, X.; Friedman, J.M.; Horvath, T.L. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 2004, 304, 110–115. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Deveaux, V.; Cadoudal, T.; Ichigotani, Y.; Teixeira-Clerc, F.; Louvet, A.; Manin, S.; Nhieu, J.T.-V.; Belot, M.P.; Zimmer, A.; Even, P.; et al. Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS ONE 2009, 4, e5844. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Konner, A.C.; Bruning, J.C. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012, 16, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgquist, A.; Meza, C.; Wagner, E.J. The role of AMP-activated protein kinase in the androgenic potentiation of cannabinoid-induced changes in energy homeostasis. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E482–E495. [Google Scholar] [CrossRef]
- Qiu, J.; Bosch, M.A.; Meza, C.; Navarro, U.-V.; Nestor, C.C.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Estradiol protects proopiomelanocortin neurons against insulin resistance. Endocrinology 2018, 159, 647–664. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Bosch, M.A.; Zhang, C.; Ronnekleiv, O.K.; Kelly, M.J. Estradiol protects neuropeptide Y/agouti-related peptide neurons against insulin resistance in females. Neuroendocrinology 2020, 110, 105–118. [Google Scholar] [CrossRef]
- Hu, M.; Becker, J.B. Effects of sex and estrogen on behavioral sensitization to cocaine in rats. J. Neurosci. 2003, 23, 693–699. [Google Scholar] [CrossRef]
- Evans, S.M.; Foltin, R.W. Does the response to cocaine differ as a function of sex or hormonal status in human and non-human primates? Horm. Behav. 2009, 58, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandegrift, B.J.; Hilderbrand, E.R.; Satta, R.; Tai, R.; He, D.; You, C.; Chen, H.; Xu, P.; Coles, C.; Brodie, M.S.; et al. Estrogen Receptor a Regulates Ethanol Excitation of Ventral Tegmental Area Neurons and Binge Drinking in Female Mice. J. Neurosci. 2020, 40, 5196–5207. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.C.; Bennett, S.A.; Vickers, G.J. The estrous cycle affects cocaine self-administration on a progressive ratio schedule in rats. Psychopharmacology 1989, 98, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.E.; Morgan, A.D.; Lynch, W.J.; Campbell, U.C.; Dess, N.K. Intravenous cocaine and heroin self-administration in rats selectively bred for differential saccharin intake: Phenotype and sex differences. Psychopharmacology 2002, 161, 304–313. [Google Scholar] [CrossRef]
- Becker, J.B.; Hu, M. Sex differences in drug abuse. Front. Neuroendocrinol. 2008, 29, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Richard, J.E.; Lopez-Ferreras, L.; Anderberg, R.H.; Olandersson, K.; Skibicka, K.P. Estradiol is a critical regulator of food-reward behavior. Psychoneuroendocrinology 2017, 78, 193–202. [Google Scholar] [CrossRef]
- Contini, A.; Sanna, F.; Maccioni, P.; Colombo, G.; Argiolas, A. Comparison between male and female rats in a model of self-administration of a chocolate-flavored beverage: Behavioral and neurochemical studies. Behav. Brain Res. 2018, 344, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.-L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.-C. ORL1, a novel member of the opioid receptor family. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Meunier, J.-C.; Mollereau, C.; Toll, L.; Suaudeau, C.; Moisand, C.; Alvinerie, P.; Butour, J.-L.; Guillemot, J.-C.; Ferrara, P.; Monsarrat, B.; et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 1995, 377, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Reinscheid, R.K.; Nothacker, H.-P.; Bourson, A.; Ardati, A.; Henningsen, R.A.; Bunzow, J.R.; Grandy, D.K.; Langen, H.; Monsma, F.J.; Civelli, O. Orphanin FQ: A neuropeptide that activates an opioidlike G protein-coupled receptor. Science 1995, 270, 792–794. [Google Scholar] [CrossRef]
- Bunzow, J.R.; Saez, C.; Mortrud, M.; Bouvier, C.; Williams, J.T.; Low, M.; Grandy, D.K. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a μ, δ or kappa opioid receptor type. FEBS Lett. 1994, 347, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Mogil, J.S.; Grisel, J.E.; Reinscheid, R.K.; Civelli, O.; Belknap, J.K.; Grandy, D.K. Orphanin FQ is a functional anti-opioid peptide. Neuroscience 1996, 75, 333–337. [Google Scholar] [CrossRef]
- Sim, L.J.; Xiao, R.Y.; Childers, S.R. Identification of opioid receptor-like (ORL1) peptide-stimulated [35S]GTPgammaS binding in rat brain. Neuroreport 1996, 7, 729–733. [Google Scholar] [CrossRef]
- Ikeda, K.; Watanabe, M.; Ichikawa, T.; Kobayashi, T.; Yano, R.; Kumanishi, T. Distribution of prepro-nociceptin/orphanin FQ mRNA and its receptor mRNA in developing and adult mouse central nervous systems. J. Comp. Neurol. 1998, 399, 139–151. [Google Scholar] [CrossRef]
- Toll, L.; Bruchas, M.R.; Calo, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.R.; Planer, W.; Siuda, E.R.; Zhao, H.-C.; Stickler, L.; Chang, S.D.; Baird, M.A.; Cao, Y.-Q.; Bruchas, M.R. Serine 363 is required for nociceptin/orphanin FQ opioid receptor (NOPR) desensitization, internalization and arrestin signaling. J. Biol. Chem. 2012, 287, 42019–42030. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.; Vaughan, C.W.; Chieng, B.; Christie, M.J. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurones in vitro. Br. J. Pharmacol. 1996, 119, 1614–1618. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.; Yeo, A.; Henderson, G. The effect of nociceptin on Ca2+ channel current and intracellular Ca2+ in the SH-SY5Y human neuroblastoma cell line. Br. J. Pharmacol. 1996, 118, 205–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, M.; Christie, M.J. Modulation of Ca2+ channel currents of acutely dissociated rat periaqueductal grey neurons. J. Physiol. 1998, 509, 47–58. [Google Scholar] [CrossRef]
- Fukuda, K.; Shoda, T.; Morikawa, H.; Kato, S.; Mima, H.; Mori, K. Activation of phospholipase A2 by the nociceptin receptor expressed in Chinese hamster ovary cells. J. Neurochem. 1998, 71, 2186–2192. [Google Scholar] [CrossRef]
- Yung, L.Y.; Joshi, S.A.; Chan, R.Y.; Chan, J.S.; Pei, G.; Wong, Y.H. GalphaL1 (Galpha14) couples the opioid receptor-like1 receptor to stimulation of phospholipase C. J. Pharmacol. Exp. Ther. 1999, 288, 232–238. [Google Scholar] [PubMed]
- Armstead, W. NOC/oFQ activates PKC and generates superoxide to impair hypotensive cerebrovasodilation after hypoxia/ischemia. Med. Sci. Monit. 2002, 8, BR0. [Google Scholar]
- Norton, C.S.; Neal, C.R.; Kumar, S.; Akil, H.; Watson, S.J. Nociceptin/orphanin FQ and opiod receptor-like receptor mRNA expression in dopamine systems. J. Comp. Neurol. 2002, 444, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Sinchak, K.; Romeo, H.E.; Micevych, P.E. Site-specific estrogen and progestin regulation of orphanin FQ/nociceptin and nociceptin opioid receptor mRNA expression in the female rat limbic hypothalamic system. J. Comp. Neurol. 2006, 496, 252–268. [Google Scholar] [CrossRef]
- Pomonis, J.D.; Billington, C.J.; Levine, A.S. Orphanin FQ, agonist of the orphan opioid receptor, stimulates feeding in rats. Neuroreport 1996, 8, 369–371. [Google Scholar] [CrossRef]
- Polidori, C.; Calo’, G.; Ciccocioppo, R.; Guerrini, R.; Regoli, D.; Massi, M. Pharmacological characterization of the nociceptin mediating hyperphagia: Identification of a selective antagonist. Psychopharmacology 2000, 148, 430–437. [Google Scholar] [CrossRef]
- Polidori, C.; de Caro, G.; Massi, M. The hyperphagic effect of nociceptin/orphanin FQ in rats. Peptides 2000, 21, 1051–1062. [Google Scholar] [CrossRef]
- Olszewski, P.K.; Levine, A.S. Minireview: Characterization of influence of central nociceptin/orphanin FQ on consummatory behavior. Endocrinology 2004, 145, 2627–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Economidou, D.; Policani, F.; Angellotti, T.; Massi, M.; Terada, T.; Ciccocioppo, R. Effect of novel NOP receptor ligands in food intake in rats. Peptides 2006, 27, 775–783. [Google Scholar] [CrossRef]
- Matsushita, H.; Ishihara, A.; Mashiko, S.; Tanaka, T.; Kanno, T.; Iwaasa, H.; Ohta, H.; Kanatani, A. Chronic intracerebroventricular infusion of nociceptin/orphanin FQ produces body weight gain by affecting both feeding and energy metabolism. Endocrinology 2009, 150, 2668–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhang, B.; Pietruszewski, L.; Lutfy, K.; Wagner, E.J. The role of the NOP receptor in regulating food intake, meal pattern, and the excitability of proopiomelanocortin neurons. Neuropharmacology 2010, 59, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.J.; Rønnekleiv, O.K.; Grandy, D.K.; Kelly, M.J. The peptide orphanin FQ inhibits β-endorphin neurons and neurosecretory cells in the hypothalamic arcuate nucleus by activating an inwardly-rectifying K+ conductance. Neuroendocrinology 1998, 67, 73–82. [Google Scholar] [CrossRef]
- Borgquist, A.; Kachani, M.; Tavitian, N.; Sinchak, K.; Wagner, E.J. Estradiol negatively modulates the pleiotropic actions of orphanin FQ/nociceptin at proopiomelanocortin synapses. Neuroendocrinology 2013, 98, 60–72. [Google Scholar] [CrossRef]
- Hernandez, J.; Perez, L.; Soto, R.; Le, N.; Gastelum, C.; Wagner, E.J. Nociceptin/orphanin FQ neurons in the Arcuate Nucleus and Ventral Tegmental Area Act via Nociceptin Opioid Peptide Receptor Signaling to Inhibit Proopiomelanocortin and A 10 Dopamine Neurons and Thereby Modulate Ingestion of Palatable Food. Physiol. Behav. 2020, 228. [Google Scholar] [CrossRef]
- Bewick, G.A.; Dhillo, W.S.; Darch, S.J.; Murphy, K.G.; Gardiner, J.V.; Jethwa, P.H.; Kong, W.M.; Ghatei, M.A.; Bloom, S.R. Hypothalamic cocaine- and amphetamine-regulated transcript (CART) and agouti-related protein (AgRP) neurons coexpress the NOP1 receptor and nociceptin alters CART and AgRP release. Endocrinology 2005, 146, 3526–3534. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Paeger, L.; Sotelo-Hitschfeld, T.; Bremser, S.; Prinzensteiner, M.; Klemm, P.; Mykytiuk, V.; Widdershooven, P.J.M.; Vesting, A.J.; Grzelka, K.; et al. PNOCARC neurons promote hyperphagia and obesity upon high-fat feeding. Neuron 2020, 106, 1009–1025. [Google Scholar] [CrossRef]
- Parsons, M.P.; Burt, J.; Cranford, A.; Alberto, C.; Zipperlen, K.; Hirasawa, M. Nociceptin Induces Hypophagia in the Perifornical and Lateral Hypothalamic Area. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.P.; Ly, N.T.; Maidment, N.T. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience 1996, 75, 1–4. [Google Scholar] [CrossRef]
- Murphy, N.P.; Maidment, N.T. Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. J. Neurochem. 1999, 73, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.P.; Lee, Y.; Maidment, N.T. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999, 832, 168–170. [Google Scholar] [CrossRef]
- Di Giannuario, A.; Pieretti, S.; Catalani, A.; Loizzo, A. Orphanin FQ reduces morphine-induced dopamine release in the nucleus accumbens: A microdialysis study in rats. Neurosci. Lett. 1999, 272, 183–186. [Google Scholar] [CrossRef]
- Zheng, F.; Grandy, D.K.; Johnson, S.W. Actions of orphanin FQ/nociceptin on rat ventral tegmental area neurons in vitro. Br. J. Pharmacol. 2002, 136, 1065–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olianas, M.C.; Dedoni, S.; Boi, M.; Onali, P. Activation of nociceptin/orphanin FQ-NOP receptor system inhibits tyrosine hydroxylase phosphorylation, dopamine synthesis, and dopamine D(1) receptor signaling in rat nucleus accumbens and dorsal striatum. J. Neurochem. 2008, 107, 544–556. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Miller, R.J. Pre- and postsynaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. J. Physiol. 1999, 517, 431–445. [Google Scholar] [CrossRef]
- Chee, M.J.; Price, C.J.; Statnick, M.A.; Colmers, W.F. Nociceptin/orphanin FQ suppresses the excitability of neurons in the ventromedial nucleus of the hypothalamus. J. Physiol. 2011, 589, 3103–3114. [Google Scholar] [CrossRef] [PubMed]
- Conde, K.; Meza, C.; Kelly, M.J.; Sinchak, K.; Wagner, E.J. Estradiol rapidly attenuates ORL-1 receptor-mediated inhibition of proopiomelanocortin neurons via Gq-coupled, membrane-initiated signaling. Neuroendocrinology 2016, 103, 787–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgquist, A.; Rivas, V.M.; Kachani, M.; Sinchak, K.; Wagner, E.J. Gonadal steroids differentially modulate the actions of orphanin FQ/nociceptin at a physiologically relevant circuit controlling female sexual receptivity. J. Neuroendocrinol. 2014, 26, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, M.; Cagniard, B.; Murphy, N.P. Endogenous nociceptin modulates diet preference independent of motivation and reward. Physiol. Behav. 2009, 97, 1–13. [Google Scholar] [CrossRef]
- Toledo, M.A.; Pedregal, C.; Lafuente, C.; Diaz, N.; Martinez-Grau, M.A.; Jiménez, A.; Benito, A.; Torrado, A.; Mateos, C.; Joshi, E.M.; et al. Discovery of a novel series of orally active nociceptin/orphanin FQ (NOP) receptor antagonists based on a dihydrospiro(piperidine-4,7′-thieno[2,3-c]pyran) scaffold. J. Med. Chem. 2014, 57, 3418–3429. [Google Scholar] [CrossRef]
- Statnick, M.A.; Chen, Y.; Ansonoff, M.A.; Witkin, J.M.; Rorick-Kehn, L.; Suter, T.M.; Song, M.; Hu, C.; Lafuente, C.; Jiménez, A.; et al. A Novel Nociceptin Receptor Antagonist LY2940094 Inhibits Excessive Feeding Behavior in Rodents: A Possible Mechanism for the Treatment of Binge Eating Disorder. J. Pharmacol. Exp. Ther. 2016, 356, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardaway, J.A.; Jensen, J.; Kim, M.; Mazzone, C.M.; Sugam, J.A.; Diberto, J.F.; Lowery-Gionta, E.G.; Hwa, L.S.; Pleil, K.E.; Bulik, C.M.; et al. Nociceptin receptor antagonist SB 61211 decreases high fat diet binge eating. Behav. Brain Res. 2016, 307, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Rudecki, A.P.; Gray, S.L. PACAP in the defense of energy homeostasis. Trends Endocrinol. Metab. 2016, 27, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Arimura, A.; Somogyvari-Vigh, A.; Miyata, A.; Mizuno, K.; Coy, D.H.; Kitada, C. Tissue Distribution of PACAP as Determined by RIA: Highly Abundant in the Rat Brain and Testes. Endocrinology 1991, 129, 2787–2789. [Google Scholar] [CrossRef] [PubMed]
- Ghatei, M.A.; Takahashi, K.; Suzuki, Y.; Gardiner, J.; Jones, P.M.; Bloom, S.R. Distribution, molecular characterization of pituitary adenylate cyclase-activating polypeptide and its precursor encoding messenger RNA in human and rat tissues. J. Endocrinol. 1993, 136, 159–166. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef]
- Gottschall, P.E.; Tatsuno, I.; Miyata, A.; Arimura, A. Characterization and distribution of binding sites for the hypothalamic peptide, pituitary adenylate cyclase-activating polypeptide. Endocrinology 1990, 127, 272–277. [Google Scholar] [CrossRef]
- Lam, H.C.; Takahashi, K.; Ghatei, M.A.; Kanse, S.M.; Polak, J.M.; Bloom, S.R. Binding sites of a novel neuropeptide pituitary-adenylate-cyclase-activating polypeptide in the rat brain and lung. Eur. J. Biochem. 1990, 193, 725–729. [Google Scholar] [CrossRef]
- Nishimoto, M.; Furuta, A.; Aoki, S.; Kudo, Y.; Miyakawa, H.; Wada, K. PACAP/PAC1 autrocrine system promotes proliferation and astrogenesis in neural progenitor cells. Glia 2007, 55, 317–327. [Google Scholar] [CrossRef]
- Lutz-Bucher, B.; Monnier, D.; Koch, B. Evidence for the presence of receptors for pituitary adenylate cyclase-activating polypeptide in the neurohypophysis that are positively coupled to cyclic AMP formation and neurohypophyseal hormone secretion. Neuroendocrinology 1996, 64, 153–161. [Google Scholar] [CrossRef]
- Uchimura, D.; Katafuchi, T.; Hori, T.; Yanaihara, N. Facilitatory effects of pituitary adenylate cyclase activating polypeptide (PACAP) on neurons in the magnocellular portion of the rat hypothalamic paraventricular nucleus (PVN) in vitro. J. Neuroendocrinol. 1996, 8, 137–143. [Google Scholar] [CrossRef]
- Shibuya, I.; Kabashima, N.; Tanaka, K.; Setiadji, V.S.; Noguchi, J.; Harayama, N.; Ueta, Y.; Yamashita, H. Patch-clamp analysis of the mechanism of PACAP-induced excitation in rat supraoptic neurones. J. Neuroendocrinol. 1998, 10, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, I.; Noguchi, J.; Tanaka, K.; Harayama, N.; Inoue, U.; Kabashima, N.; Ueta, Y.; Hattori, Y.; Yamashita, H. PACAP increases the cytosolic Ca2+ concentration and stimulates somatodendritic vasopressin release in rat supraoptic neurons. J. Neuroendocrinol. 1998, 10, 31–42. [Google Scholar] [CrossRef]
- Ichinose, M.; Asai, M.; Sawada, M. Activation of outward current by pituitary adenylate cyclase activating polypeptide in mouse microglial cells. J. Neurosci. Res. 1998, 51, 382–390. [Google Scholar] [CrossRef]
- Delgado, M.; De la Fuente, M.; Martinez, C.; Gomariz, R.P. Pituitary adenylate cyclase-activating polypeptides (PACAP27 and PACAP38) inhibit the mobility of murine thymocytes and splenic lymphocytes: Comparison with VIP and implication of cAMP. J. Neuroimmunol. 1995, 62, 137–146. [Google Scholar] [CrossRef]
- Garrido, E.; Delgado, M.; Martinez, C.; Gomariz, R.P.; De la Fuente, M. Pituitary adenylate cyclase-activating polypeptide (PACAP38) modulates lymphocyte and macrophage functions: Stimulation of adherence and opposite effect on mobility. Neuropeptides 1996, 30, 583–595. [Google Scholar] [CrossRef]
- Martinez, C.; Delgado, M.; Pozo, D.; Leceta, J.; Calvo, J.R.; Ganea, D.; Gomariz, R.P. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced IL-6 production by murine peritoneal macrophages. J. Leukoc. Biol. 1998, 63, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Martinez, C.; Pozo, D.; Calvo, J.R.; Leceta, J.; Ganea, D.; Gomariz, R.P. Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activation polypeptide (PACAP) protect mice from lethal endotoxemia through the inhibition of TNF-alpha and IL-6. J. Immunol. 1999, 162, 1200–1205. [Google Scholar]
- Delgado, M.; Munoz-Elias, E.J.; Gomariz, R.P.; Ganea, D. VIP and PACAP inhibit IL-12 production in LPS-stimulated macrophages. Subsequent effect on IFNgamma synthesis by T cells. J. Neuroimmunol. 1999, 96, 167–181. [Google Scholar] [CrossRef]
- Delgado, M.; Munoz-Elias, E.J.; Gomariz, R.P.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide enhance IL-10 production by murine macrophages: In vitro and in vivo studies. J. Immunol. 1999, 162, 1707–1716. [Google Scholar] [PubMed]
- Villalba, M.; Bockaert, J.; Journot, L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J. Neurosci. 1997, 17, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Vaudry, D.; Basille, M.; Anouar, Y.; Fournier, A.; Vaudry, H.; Gonzalez, B.J. The neurotrophic activity of PACAP on rat cerebellar granule cells is associated with activation of the protein kinase A pathway and c-fos gene expression. Ann. N. Y. Acad. Sci. 1998, 865, 92–99. [Google Scholar] [CrossRef]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Anouar, Y.; Fournier, A.; Vaudry, H. Pituitary adenylate cyclase-activating polypeptide stimulates both c-fos gene expression and cell survival in rat cerebellar granule neurons through activation of the protein kinase A pathway. Neuroscience 1998, 84, 801–812. [Google Scholar] [CrossRef]
- Tatsuno, I.; Gottschall, P.E.; Arimura, A. Inhibition of mitogen-stimulated proliferation of murine splenocytes by a novel neuropeptide, pituitary adenylate cyclase activating polypeptide: A comparative study with vasoactive intestinal peptide. Endocrinology 1991, 128, 728–734. [Google Scholar] [CrossRef]
- Mei, Y.A. High-voltage-activated calcium current and its modulation by dopamine D4 and pituitary adenylate cyclase activating polypeptide receptors in cerebellar granule cells. Zhongguo Yao Li Xue Bao 1999, 20, 3–9. [Google Scholar]
- Roy, A.; Derakhshan, F.; Wilson, R.J.A. Stress peptide PACAP engages multiple signaling pathways within the carotid body to initiate excitatory responses in respiratory and sympathetic chemosensory afferents. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1070–R1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, R.P.; Kadunganattil, S.; Shepherd, A.J.; Merrill, R.; Planer, W.; Bruchas, M.R.; Strack, S.; Mohapatra, D.P. Convergent phosphomodulation of the major neuronal dendritic potassium channel Kv4.2 by pituitary adenylate cyclase-activating polypeptide. Neuropharmacology 2016, 101, 291–308. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Schermerhorn, T.; Straub, S.G.; Sharp, G.W. Pituitary adenylate cyclase-activating peptide, carbachol, and glucose stimulate insulin release in the absence of an increase in intracellular Ca2+. Mol. Pharmacol. 1996, 50, 1047–1054. [Google Scholar] [PubMed]
- Yada, T.; Sakurada, M.; Ishihara, H.; Nakata, M.; Shioda, S.; Yaekura, K.; Hamakawa, N.; Yanagida, K.; Kikuchi, M.; Oka, Y. Pituitary adenylate cyclase-activating polypeptide (PACAP) is an islet substance serving as an intra-islet amplifier of glucose-induced insulin secretion in rats. J. Physiol. 1997, 505, 319–328. [Google Scholar] [CrossRef]
- Yada, T.; Sakurada, M.; Nakata, M.; Yaekura, K.; Kikuchi, M. PACAP as low as 10(-13) M raises cytosolic Ca2+ activity in pancreatic B-cells by augmenting Ca2+ influx through L-type Ca2+ channels to trigger insulin release. Adv. Exp. Med. Biol. 1997, 426, 165–171. [Google Scholar]
- Filipsson, K.; Sundler, F.; Ahren, B. PACAP is an islet neuropeptide which contributes to glucose-stimulated insulin secretion. Biochem. Biophys. Res. Commun. 1999, 256, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Vu, J.P.; Goyal, D.; Luong, L.; Oh, S.; Sanghu, R.; Norris, J.; Parsons, W.; Pisegna, J.R.; Germano, P.M. PACAP intraperitoneal treatment suppresses appetite and food intake via PAC1 receptor in mice by inhibiting ghrelin and increasing GLP-1 and leptin. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G816–G825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [Green Version]
- McTaggart, J.S.; Clark, R.H.; AShcroft, F.M. The role of the KATP channel in glucose homeostasis in health and disease: More than meets the islet. J. Physiol. 2010, 588, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Pan, J.; Tan, Y.-Y.; Sun, X.-K.; Zhang, Y.-F.; Zhou, H.-Y.; Ren, R.-J.; Wang, X.-J.; Chen, S.-D. Neuroprotective effects of PACAP27 in mice model of Parkinson’s disease involved in the modulation of K(ATP) subunits and D2 receptors in the striatum. Neuropeptides 2008, 42, 267–276. [Google Scholar] [CrossRef]
- Koide, M.; Syed, A.U.; Braas, K.M.; May, V.; Wellman, G.C. Pituitary adenylate cyclase activating polypeptide (PACAP) dilates cerebellar arteries through activation of large-conductance Ca(2+)-activated (BK) and ATP-sensitive (K ATP) K (+) channels. J. Mol. Neurosci. 2014, 54, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Tanida, M.; Hayata, A.; Shintani, N.; Yamamoto, N.; Kurata, Y.; Shibamoto, T.; Morgan, D.A.; Rahmouni, K.; Hashimoto, H. Central PACAP mediates the sympathetic effects of leptin in a tissue-specific manner. Neuroscience 2013, 238, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Mungan, Z.; Hammer, R.A.; Akarca, U.S.; Komaki, G.; Ertan, A.; Arimura, A. Effect of PACAP on gastric acid secretion in rats. Peptides 1995, 16, 1051–1056. [Google Scholar] [CrossRef]
- Mirfendereski, S.; Tobin, G.; Hakanson, R.; Ekstrom, J. Pituitary adenylate cyclase activating peptide (PACAP) in salivary glands of the rat: Origin, and secretory and vascular effects. Acta Physiol. Scand. 1997, 160, 15–22. [Google Scholar] [CrossRef]
- Li, P.; Chang, T.M.; Coy, D.; Chey, W.Y. Inhibition of gastric acid secretion in rat stomach by PACAP is mediated by secretin, somatostatin, and PGE(2). Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G121–G127. [Google Scholar] [CrossRef]
- Kuwahara, A.; Kuwahara, Y.; Mochizuki, T.; Yanaihara, N. Action of pituitary adenylate cyclase-activating polypeptide on ion transport in guinea pig distal colon. Am. J. Physiol. 1993, 264, G433–G441. [Google Scholar] [CrossRef]
- Takeuchi, K.; Takehara, K.; Kato, S.; Yagi, K. PACAPs stimulate duodenal bicarbonate secretion at PACAP receptors in the rat. Am. J. Physiol. 1997, 272, G646–G653. [Google Scholar] [CrossRef]
- Katsoulis, S.; Schmidt, W.E.; Schwarzhoff, R.; Folsch, U.R.; Jin, J.G.; Grider, J.R.; Makhlouf, G.M. Inhibitory transmission in guinea pig stomach mediated by distinct receptors for pituitary adenylate cyclase-activating peptide. J. Pharmacol. Exp. Ther. 1996, 278, 199–204. [Google Scholar] [PubMed]
- Robberecht, P.; De Neef, P.; Lefebvre, R.A. Influence of selective VIP receptor agonists in the rat gastric fundus. Eur. J. Pharmacol. 1998, 359, 77–80. [Google Scholar] [CrossRef]
- Ozawa, M.; Aono, M.; Moriga, M. Central effects of pituitary adenylate cyclase activating polypeptide (PACAP) on gastric motility and emptying in rats. Dig. Dis. Sci. 1999, 44, 735–743. [Google Scholar] [CrossRef]
- Schworer, H.; Katsoulis, S.; Creutzfeldt, W.; Schmidt, W.E. Pituitary adenylate cyclase activating peptide, a novel VIP-like gut-brain peptide, relaxes the guinea-pig taenia caeci via apamin-sensitive potassium channels. Naunyn-Schmiedebergs Arch. Pharmacol. 1992, 346, 511–514. [Google Scholar] [CrossRef]
- Katsoulis, S.; Clemens, A.; Schworer, H.; Creutzfeldt, W.; Schmidt, W.E. Pituitary adenylate cyclase activating polypeptide (PACAP) is a potent relaxant of the rat ileum. Peptides 1993, 14, 587–592. [Google Scholar] [CrossRef]
- Grider, J.R.; Katsoulis, S.; Schmidt, W.E.; Jin, J.G. Regulation of the descending relaxation phase of intestinal peristalsis by PACAP. J. Auton. Nerv. Syst. 1994, 50, 151–159. [Google Scholar] [CrossRef]
- Ekblad, E.; Sundler, F. Distinct receptors mediate pituitary adenylate cyclase-activating peptide- and vasoactive intestinal peptide-induced relaxation of rat ileal longitudinal muscle. Eur. J. Pharmacol. 1997, 334, 61–66. [Google Scholar] [CrossRef]
- Yokota, C.; Kawai, K.; Ohashi, S.; Watanabe, Y.; Yamashita, K. PACAP stimulates glucose output from the perfused rat liver. Peptides 1995, 16, 55–60. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Kasai, K.; Hasegawa, K.; Suzuki, Y.; Shimoda, S. Glycogenolytic activity of pituitary adenylate cyclase activating polypeptide (PACAP) in vivo and in vitro. Life Sci. 1994, 55, 1219–1228. [Google Scholar] [CrossRef]
- Masuo, Y.; Ohtaki, T.; Masuda, Y.; Nagai, Y.; Suno, M.; Tsuda, M.; Fujino, M. Autoradiographic distribution of pituitary adenylate cyclase activating polypeptide (PACAP) binding sites in the rat brain. Neurosci. Lett. 1991, 126, 103–106. [Google Scholar] [CrossRef]
- Hawke, Z.; Ivanov, T.R.; Bechtold, D.A.; Dhillon, H.; Lowell, B.B.; Luckman, S.M. PACAP neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J. Neurosci. 2009, 29, 14828–14835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomimoto, S.; Ojika, T.; Shintani, N.; Hashimoto, H.; Hamagami, K.-I.; Ikeda, K.; Nakata, M.; Yada, T.; Sakurai, Y.; Shimada, T.; et al. Markedly reduced white adipose tissue and increased insulin sensitivity in adcyap1-deficient mice. J. Pharmacol. Sci. 2008, 107, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Segal, J.P.; Stallings, N.R.; Lee, C.E.; Zhao, L.; Socci, N.; Viale, A.; Harris, T.M.; Soares, M.B.; Childs, G.; Elmquist, J.K.; et al. Use of laser-capture microdissection for the identification of marker genes for the ventromedial hypothalamic nucleus. J. Neurosci. 2005, 25, 4181–4188. [Google Scholar] [CrossRef] [Green Version]
- Mounien, L.; Do Rego, J.-C.; Bizet, P.; Boutelet, I.; Gourcerol, G.; Fournier, A.; Brabet, P.; Costentin, J.; Vaudry, H.; Jegou, S. Pituitary adenylate cyclase-activating polypeptide inhibits food intake in mice through activation of the hypothalamic melanocortin system. Neuropsychopharmacol 2009, 34, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodai, T.; Nunn, N.; Worth, A.A.; Feetham, C.H.; Belle, M.D.C.; Piggins, H.D.; Luckman, S.M. PACAP neurons in the ventromedial hypothalamic nucleus are glucose inhibited and their selective activation induces hyperglycemia. Front. Endocrinol. 2018, 9, 632. [Google Scholar] [CrossRef]
- Lindberg, D.; Chen, P.; Li, C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to the autonomic centers of the hypothalamus and hindbrain. J. Comp. Neurol. 2013, 521, 3167–3190. [Google Scholar] [CrossRef]
- Resch, J.M.; Boisvert, J.P.; Hourigan, A.E.; Muller, C.R.; Yi, S.S.; Choi, S. Stimulation of the hypothalamic ventromedial nuclei by pituitary adenylate cyclase-activating polypeptide induces hypophagia and thermogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1625–R1634. [Google Scholar] [CrossRef] [Green Version]
- Resch, J.M.; Maunze, B.; Gerhardt, A.K.; Magnuson, S.K.; Phillips, K.A.; Choi, S. Intrahypothalamic pituitary adenylate cyclase-activating polypeptide regulates energy balance via site-specific actions on feeding and metabolism. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1452–E1463. [Google Scholar] [CrossRef] [Green Version]
- Mounien, L.; Bizet, P.; Boutelet, I.; Gourcerol, G.; Fournier, A.; Vaudry, H.; Jégou, S. Pituitary adenylate cyclase-activating polypeptide directly modulates the activity of proopiomelanocortin neurons in the rat arcuate nucleus. Neuroscience 2006, 143, 155–163. [Google Scholar] [CrossRef]
- Ramikie, T.S.; Ressler, K.J. Stress-related disorders, pituitary adenylate cyclase-activating peptide (PACAP)ergic system, and sex differences. Dialogues Clin. Neurosci. 2016, 18, 403–413. [Google Scholar] [PubMed]
- Hurley, M.M.; Maunze, B.; Block, M.E.; Frenkel, M.M.; Reily, M.J.; Kim, E.; Chen, Y.; Li, Y.; Baker, D.A.; Liu, Q.-S.; et al. Pituitary adenylate-cyclase activating polypeptide regulates hunger- and palatability-induced binge eating. Front. Neurosci. 2016, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.M.; Robble, M.R.; Callan, G.; Choi, S.; Wheeler, R.A. Pituitary adenylate cyclase-activiating polypeptide (PACAP) acts in the nucleus accumbens to reduce hedonic drive. Int. J. Obes. 2019, 43, 928–932. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gastelum, C.; Perez, L.; Hernandez, J.; Le, N.; Vahrson, I.; Sayers, S.; Wagner, E.J. Adaptive Changes in the Central Control of Energy Homeostasis Occur in Response to Variations in Energy Status. Int. J. Mol. Sci. 2021, 22, 2728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052728
Gastelum C, Perez L, Hernandez J, Le N, Vahrson I, Sayers S, Wagner EJ. Adaptive Changes in the Central Control of Energy Homeostasis Occur in Response to Variations in Energy Status. International Journal of Molecular Sciences. 2021; 22(5):2728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052728
Chicago/Turabian StyleGastelum, Cassandra, Lynnea Perez, Jennifer Hernandez, Nikki Le, Isabella Vahrson, Sarah Sayers, and Edward J. Wagner. 2021. "Adaptive Changes in the Central Control of Energy Homeostasis Occur in Response to Variations in Energy Status" International Journal of Molecular Sciences 22, no. 5: 2728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052728