1. Introduction
Microglia are resident macrophages of the central nervous system (CNS), constitute 5–10% of total brain cells, maintain CNS homeostasis, and protect the CNS by phagocytosing pathogens [
1,
2,
3,
4]. Microglial heterogeneity, defined in relation to key regulators, markers, and drug targets, is categorized by homeostatic, pro-inflammatory, and anti-inflammatory microglial subtypes [
5]. In addition, microglia play important disease-modifying roles in neurodegenerative diseases, including Alzheimer’s disease (AD), as innate immune cells of the CNS [
5,
6,
7]. Transcriptome studies have indicated that homeostatic microglia gradually adopt a unique phagocytic disease-associated microglia (DAM) phenotype in neurodegenerative disease, chronic inflammation, and advanced aging [
5]. In contrast, researchers have not yet determined the effects of microbial pathogens, which induce microglial proliferation, on AD progression using the CNS infection model; additionally, detailed studies on the effect of microbial infection in advanced AD are limited.
Toxoplasma gondii, a protozoan parasite that commonly infects humans and animals, chronically infects the brain, which acquires immunity against toxoplasmosis [
8,
9]. In the brain,
T. gondii infection induces the expression of anti-inflammatory cytokines and negative regulators of toxoplasmic encephalitis, namely suppressor of cytokine signaling 1 (SOCS1) and Arg1, to reduce the inflammatory response [
8,
9]. Through a series of immunomodulatory processes, microglia increase significantly from three to nine weeks after
T. gondii infection and slightly decrease after 9 weeks [
9]. In our previous study, we demonstrated that neuroinflammation was suppressed and homeostasis restored by increasing anti-inflammatory cytokine production in the state of disease-associated inflammatory response [
8,
9]. However, the proliferation and role of microglia in chronic
T. gondii infection has not been sufficiently discussed. Given that microglia continuously monitor the surrounding parenchyma to sense alterations in brain function and are involved in controlling neuronal excitability, synaptic activity, neurogenesis, and clearance of apoptotic cells in the healthy adult brain [
10], it would be interesting to determine how microglia affect chronic neurodegenerative diseases, such as AD, after
T. gondii infection.
Microglia are categorized by their phenotypes in states of health or disease [
1,
2,
3,
4,
5,
6,
7,
10]. Microglial function in the normal brain has been described as that of surveillance and limiting of the over-production of neurons, as well as excitatory synapses, through microglia-mediated phagocytosis to maintain homeostasis [
2]. However, microglia are activated by acute insults and chronic disease states; reactive microglia are motile and destructive as they physically surround or target structures, such as dying cells, neurons, dendrites, blood vessels, and amyloid plaques, and therefore defend against CNS diseases [
2]. In
T. gondii infection, the microglial phenotype is significant for the disease-associated immune response [
9].
T. gondii infection induces M1 polarization of microglia; however, at the same time, it reduces detrimental inflammatory immune responses through the induction of SOCS1, reduction of phosphorylation of signal transducer and activator of transcription 1 (pSTAT1), and the induction of anti-inflammatory cytokines [
9]. In addition, the immune environment induced by
T. gondii infection in the CNS has been determined to play a role in the inhibition of neurodegeneration and clearance of β-amyloid plaques in Tg2576 AD mice [
8]. Nevertheless, little is known regarding the functional role of
T. gondi-induced proliferation of microglia in AD pathology. In other words, the role of
T. gondi-induced proliferation of microglia in the phagocytosis of β-amyloid plaques has not yet been reported.
The 5XFAD mouse, an experimental animal model of early-onset AD, in which β-amyloid plaques are first observed at two months of age and cognitive deficits at four to six months, is used to study the age-dependent progression of AD-like pathology [
11].
T. gondii infection ameliorated β-amyloidosis by activating the immune response, including increased Aβ phagocytosis by Ly6C
hi monocytes, on day 28 after
T. gondii infection in eight-week-old 5XFAD mice [
12]. Although the microglial population increased in the brain and microglia localized to the vicinity of Aβ plaques, the role of microglia received less attention, due to their lower ex vivo phagocytic capacity, than Ly6C
hi monocytes [
12]. However, in that study, the infection period of
T. gondii for the ex vivo phagocytic capacity experiment was only one month, which is the early stage of infection and, thus, insufficient to evaluate the effects of infection. Given that
T. gondii infection persists for a long time in the brain and the pathophysiology of AD is regulated over the long term, the phagocytic role of microglia in the brain has to be observed in mice older than six months with progression of AD. In that respect, we found reduced β-amyloidosis at six months after
T. gondii infection in nine-month-old Tg2576 AD mice; however, at that time, no study on the role of microglia in ameliorating AD existed [
8].
Regarding therapeutic approaches for AD, drug development is very important. Medications for AD provide temporary relief for memory loss and cognitive disorder [
13] but cannot stop or reverse AD progression [
13]. Therefore, the discovery of new medications and cure strategies for AD therapy is required. Activated microglia are found surrounding and phagocytosing Aβ plaques; however, microglia are also implicated in the etiology of the disease through oxidative damage and pro-inflammatory signaling [
14,
15]. Despite differences of opinion in defining the role of activated microglia for treating AD, targeting microglia for treating AD remains an attractive prospect [
13,
14,
15]. The goal of a therapeutic approach targeting microglia in AD may be viewed as an attempt to return the spectrum of microglial phenotypes from the disease state (mainly synaptic pruning, reactive oxygen species generation, and inflammatory cytokine production) to the cognitively normal state (mainly, trophic support and phagocytosis) [
15]. Therefore, it is important to focus on the biological role of microglial proliferation in
T. gondii-infected AD model mice. Notably, we demonstrated that
T. gondii infection induces M1 polarization and Th1 inflammatory responses in response to microbial infection but does not induce inflammatory responses through immune regulation during chronic infection [
8,
9,
16]. Given that chronic
T. gondii infection reduced amyloid burden in aged Tg2576 mice [
8], the increase in microglial proliferation after
T. gondii infection may contribute to Aβ clearance [
9]. As both activation and phagocytic activity of microglia are important for disease modification in AD, this study highlights the role of microglia in reducing amyloid burden considering homeostasis and activation of microglia, close association between microglia and Aβ plaques, switch to the DAM phenotype, and phagocytic capacity and apoptosis of microglia. Simultaneously, we emphasize the importance of continuous microglial proliferation as a therapeutic target for AD.
3. Discussion
Therapies for AD include targeting Aβ plaque formation, neurofibrillary tangle formation, and neuroinflammation [
17]. However, based on results from investigating mechanism-based treatments for AD, the questions of which of the targeted processes are critical for disease progression and how best to inhibit AD remain controversial [
17]. Rational translational research reveals that molecular targeting for treating AD includes the use of inhibitors against amyloid aggregation, β-secretase, and γ-secretase [
13,
17]. In addition, antibodies against amyloid (passive immunization); inhibitors of glycogen synthase kinase (GSK)3β, which phosphorylates tau; and nonsteroidal anti-inflammatory drugs (NSAIDs) targeting neuroinflammation can be used as AD treatment [
13,
17]. However, treatment results have been unsatisfactory [
17]. In recent years, embryonic stem cells, brain-derived neural stem cells, and induced pluripotent stem cells have been used as cell therapy for AD treatment; however, problems persist, including the requirement for a neurosurgical procedure, immunosuppression, and tumor formation [
18]. As a novel therapeutic strategy for AD, a nano-based drug delivery system was suggested [
19]. Despite extensive efforts, treating AD clinically has remained a challenge. The important thing is to consider the multifactorial character of AD, and development of a single drug is unlikely to lead to universal AD therapy [
19]. In sum, the development of alternative therapies is needed in order to suppress all the complications of AD by anticipating the intrinsic role of cells that can function for a long time in the brain [
19,
20,
21].
Glial targets for effective therapies of AD were introduced as an implicative strategy for AD therapy [
20]. However, glial-mediated inflammation is a “double-edged sword”, performing both detrimental and beneficial functions in AD [
20,
21]. In other words, although it is still debatable whether the glial-mediated inflammatory response in AD is a consequence or cause of neurodegeneration, regarding immune surveillance, microglia, as resident macrophages, can preferentially provide the beneficial functions of an immunological first defense against invading pathogens and other types of brain injury [
20,
21]. Studies have demonstrated the presence of activated microglia at sites of Aβ deposition, suggesting that glial cells interact with Aβ plaques and regulate plaque levels in the brain [
20,
22,
23]. A recent study suggested that most plaque-associated myeloid cells responsible for Aβ plaque clearance are resident microglia [
24].
When AD mice were induced to overexpress IL-1β in the hippocampus through adenoviral transduction, microglia number increased and microglial capacity to facilitate Aβ plaque clearance also increased [
23]. Similarly, our findings revealed that
T. gondii infection induced continuous proliferation of microglia in AD mice, resulting in Aβ plaque clearance. Despite large-scale microglial proliferation, we found no evidence that microglia proliferation increased the number of pyramidal neurons associated with neurodegeneration in the brain tissue of the AD + Toxo group. Inflammation may be the key neuropathological event leading to neurodegeneration in AD [
20]. Therefore, the activation of glia, microglia, and astrocytes plays an important role in inducing the inflammatory signaling pathway during neurodegeneration [
20]. By contrast, although
T. gondii infection induced M1 polarization and microglial proliferation during 12 weeks of chronic brain infection, significant induction of inflammatory responses was not observed [
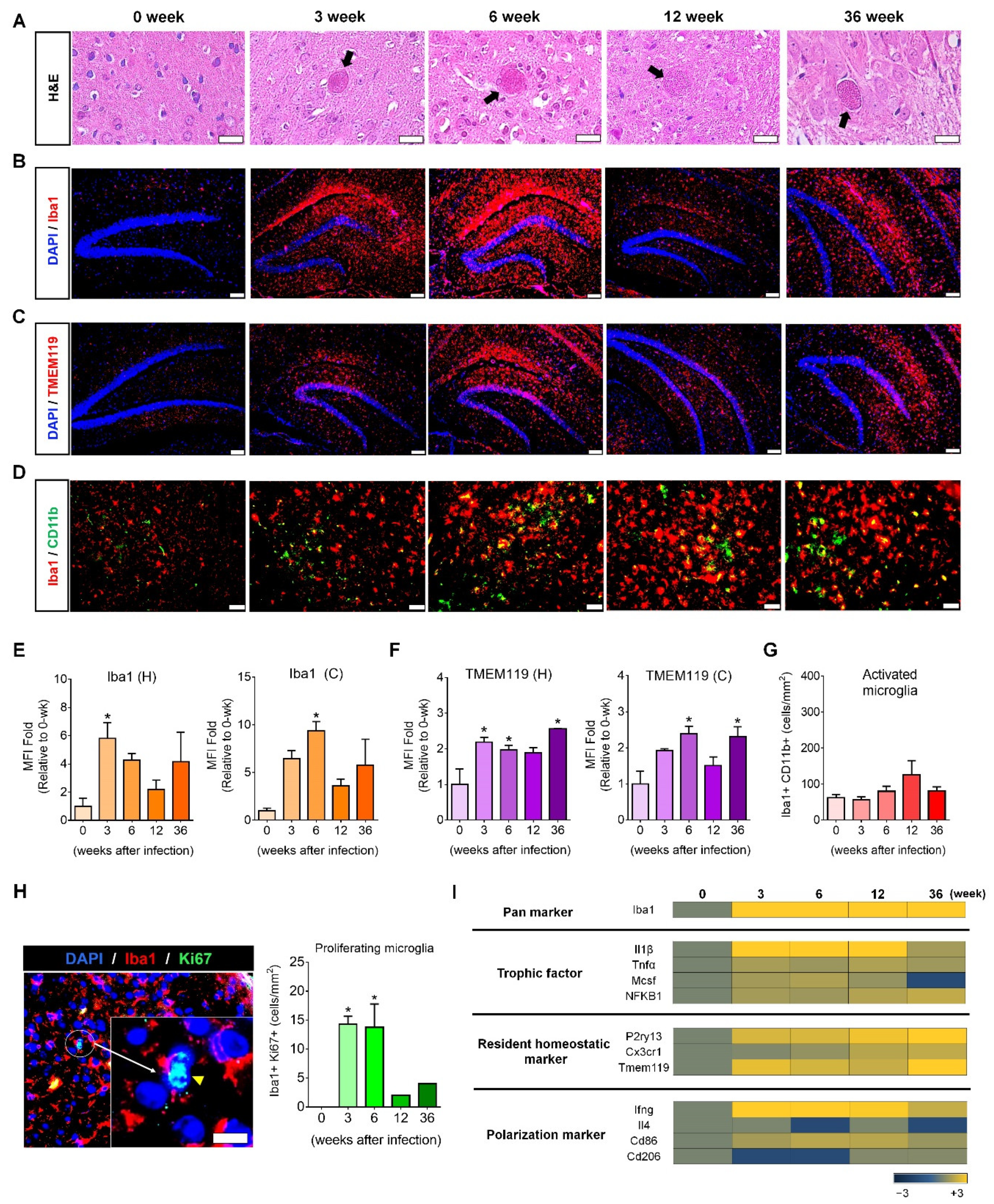
9]. In this study, microglial proliferation was visualized histologically during the 36-week chronic
T. gondii infection period, which was molecularly supported by Ki67
+-mitosis and trophic factor gene expression, indicating the presence of newly dividing homeostatic microglia. Regarding the inflammatory response, although we have not analyzed inflammatory cytokines and inflammatory immune responses, results from our previous study revealed that the increase of SOCS1 and Arginase1 and the decrease of pSTAT1 and nitric oxide were negative regulators of toxoplasmic encephalitis [
9]. Therefore,
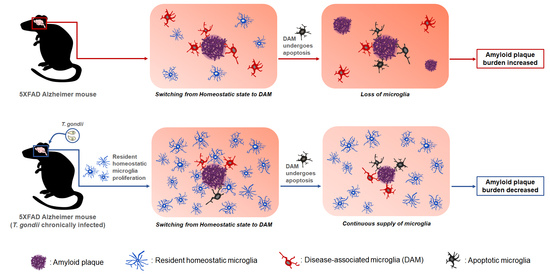
T. gondii infection is a new model that proves the beneficial effect of microglia on AD progression.
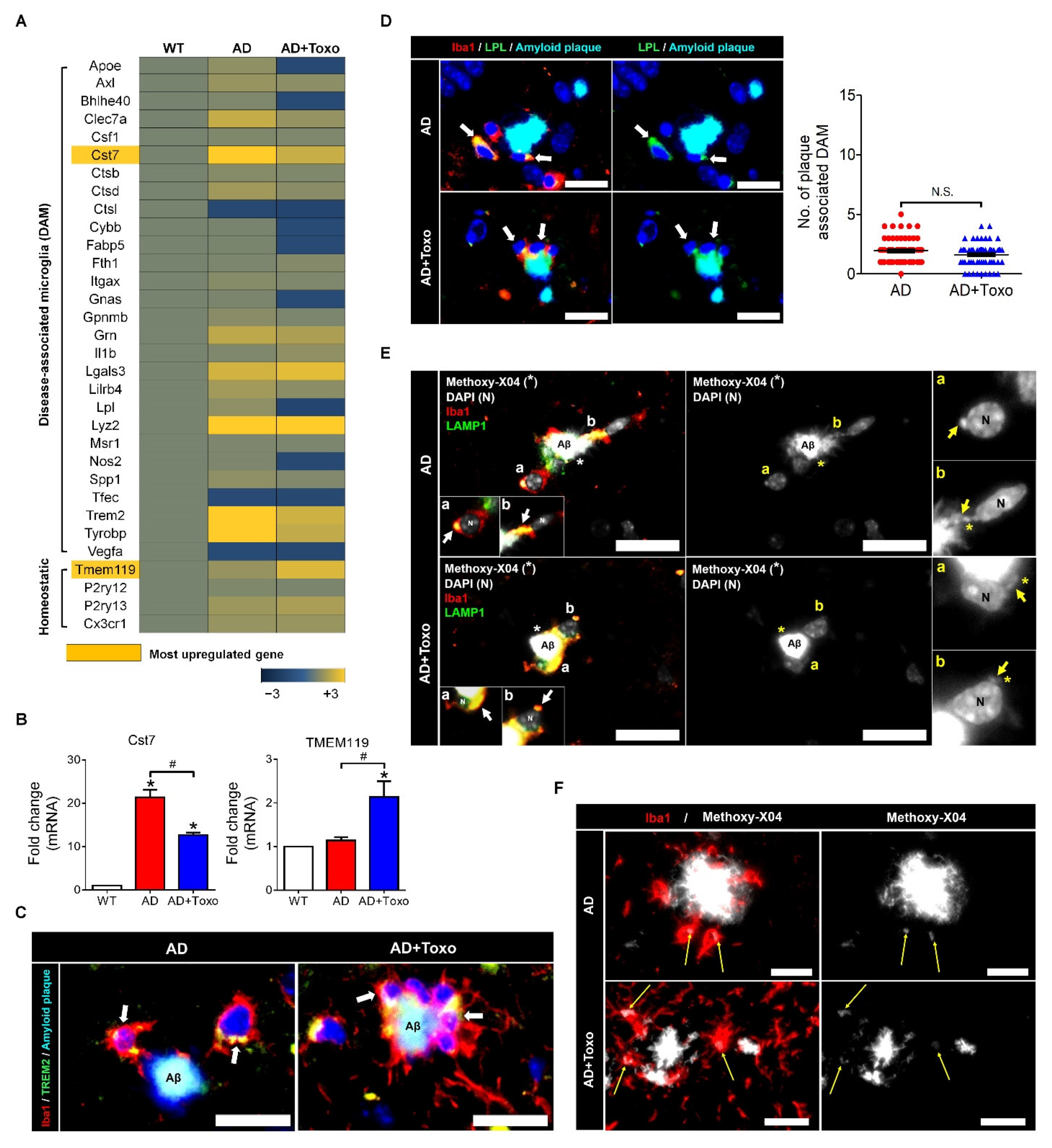
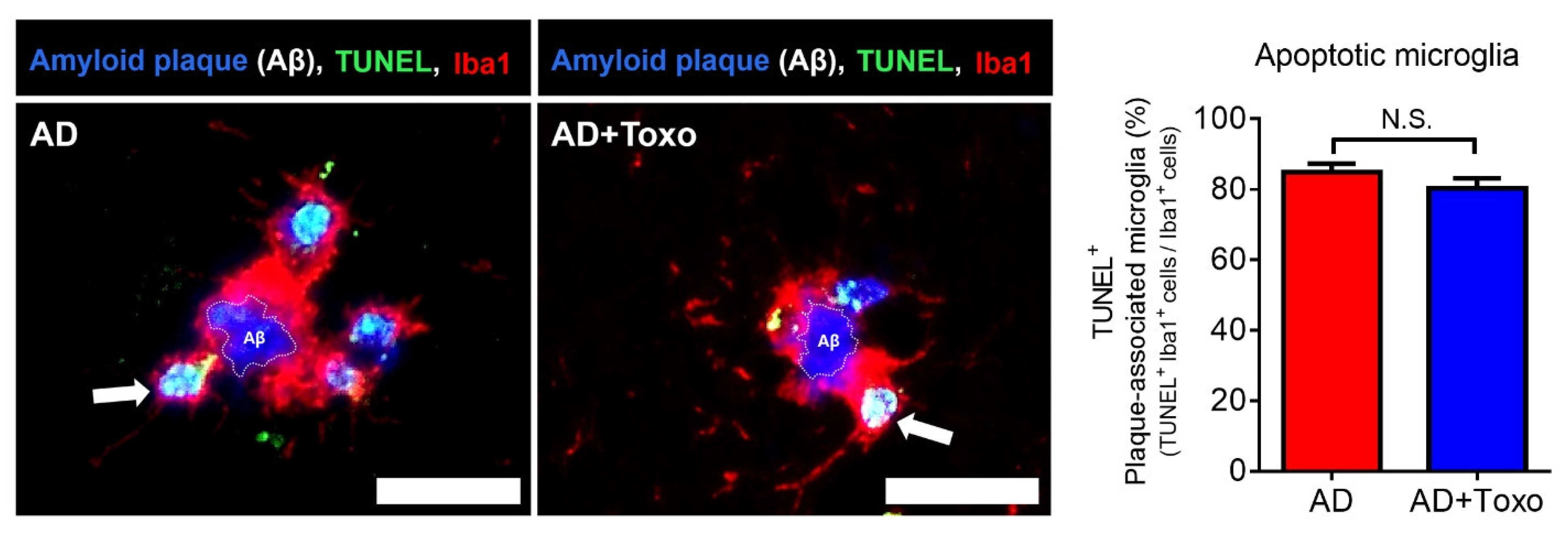
TREM2 expression in plaque-associated microglia is required for microglia-mediated Aβ phagocytosis [
21]. In this study, the appearance of TREM2-positive microglia around Aβ plaques and the increase in lysosomal digestion of Aβ plaques were related to Aβ clearance in the AD + Toxo group. However, because the fate of DAM-switched microglia interacting with Aβ was cell death, the presence of stimuli, such as
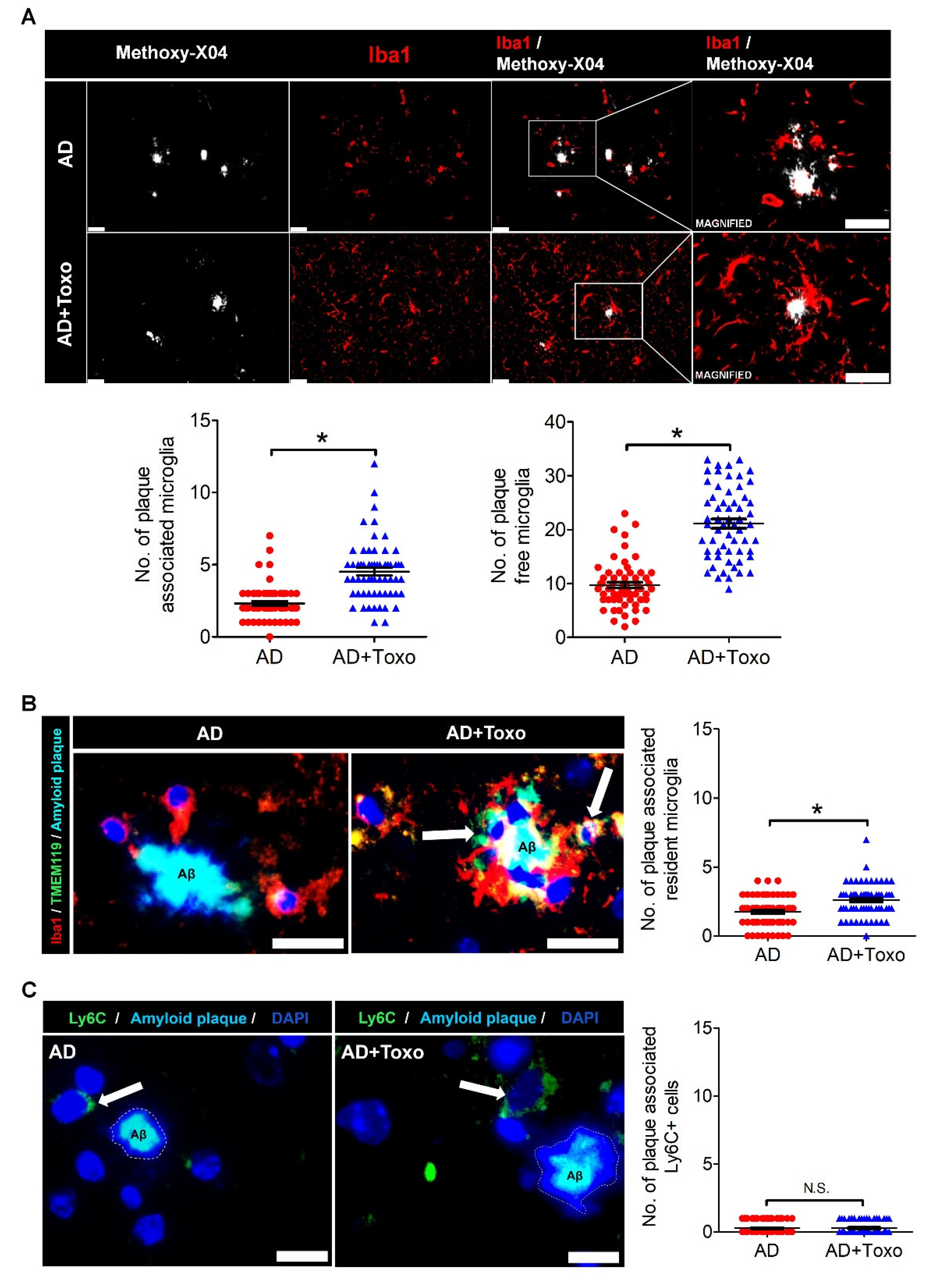
T. gondii, which continuously influence the proliferation of homeostatic microglia, is an important factor for Aβ clearance, as seen for the AD + Toxo group. In other words, it seems obvious that an increased number of homeostatic microglia underwent cell death after migrating from the periphery to plaque border. Our results are supported by the microglia turnover rate (newly appearing cells after disappearance of microglial cells), which increased in AD mice in the presence of amyloid lesions compared with wild-type mice [
25]. A highlight of our study was that microglial proliferation was followed up for 36 weeks after
T. gondii infection. Considering that chronic
T. gondii infection is defined from approximately eight to nine weeks after infection and maintains the infection state over a lifelong period, our study is significant in that proliferation kinetics of microglia over a long period after infection was evaluated [
9,
12,
26]. Most proliferated microglia were TMEM119
+/Iba-1
+-stained homeostatic microglia, and both plaque-associated and plaque-free microglia increased in the AD + Toxo group compared with the AD group. In AD mice, microglia migrate to amyloid plaques, phagocytose the amyloid plaques, and undergo apoptosis after lysosomal degradation of Aβ [
20,
27,
28,
29]. This process of microglia participating in Aβ degradation is also explained by microglial activation, which is triggered through highly expressed pattern recognition receptors, including CD14, CD36, and toll-like receptors on microglia surface [
20,
28]. In this study, microglia functioned as neuroprotective cells, clearing the pathological Aβ aggregates through lysosomal degradation. This suggestion was also supported by previous our data obtained from behavioral studies using Morris’ water maze, where AD mice infected with
T. gondii (AD + Toxo group) exhibited amelioration of symptoms of loss of cognitive function. By contrast, Green and colleagues reported that microglial depletion impairs parenchymal plaque development []. When microglia, which depend on colony-stimulating factor 1 receptor (CSF1R) signaling for survival, were treated with a CSF1R inhibitor, their population was depleted and plaques failed to form in the parenchymal space [
30]. The authors asserted that microglia play a role in the onset and development of AD pathology [
30]. However, considering that microglia maintain homeostasis in the brain and play various roles, such as adult neurogenesis and neuronal circuit formation [
2,
3,
10,
31], it is reasonable to think that microglial depletion plays a role in eliminating the non-resolving inflammatory response during AD pathogenesis. However, whether this depletion is sufficient to prevent sustainably AD progression is debatable. In this context, our study highlights the role of microglia in inhibiting AD progression. In other words, our findings suggest that the characteristics of immunity induced by
T. gondii infection, namely simultaneous increase in homeostatic microglia and suppression of inflammatory immunity, are important. In the brain, this immune environment induces an increase in Aβ phagocytosis of microglia while suppressing the non-resolving inflammatory response.
Regarding clearance of Aβ in
T. gondii-infected 5XFAD mice, Ly6C
hi monocytes were shown to be associated with increased phagocytosis and degradation of soluble Aβ [
12]. However, in that study, Iba1-labeled microglia but not Ly6C
hi monocytes interacted more closely with plaques; histological analysis of
T. gondii-infected brains also exhibited proliferation and activation of resident microglia [
12]. Nevertheless, that study concluded that Ly6C
hi monocytes had more phagocytic capacity than microglia when evaluated for their ability to specifically phagocytose Aβ
42 in an ex vivo phagocytosis assay and insisted that chronic
T. gondii infection enhances β-amyloid phagocytosis and clearance by recruited monocytes [
12]. Comparing that study with our study, the biggest difference is the infection period of
T. gondii. In the above study, 5XFAD mice were sacrificed at eight weeks after infecting eight-week-old mice with
T. gondii, while in this study, mice were sacrificed at 40 weeks after infecting eight-week-old mice with
T. gondii. With the passage of
T. gondii infection time, the early inflammatory immune response is gradually converted to an anti-inflammatory immune response, and the acute
T. gondii infection becomes a chronic infection [
8,
9]. The early inflammatory response of
T. gondii in the brain results in the infiltration of Ly6C
+ monocytes in the brain; however, chronic
T. gondii infection exhibited an increase in microglial population [
12,
16,
32,
33]. Therefore, in this study, the interaction between microglia and Aβ and the reduction of amyloid burden in chronic
T. gondii infection suggests that microglia are effectively inhibitors of Aβ deposition.
Taken together, T. gondii infection induced the proliferation of homeostatic microglia during chronic infection, and plaque-associated microglia in T. gondii-infected AD mice were significantly increased compared with those in uninfected AD mice. Considering that plaque-associated microglia undergo apoptosis after phagocytosing Aβ plaques, the continuous increase of microglia during T. gondii infection may be an important factor in reducing Aβ deposition. Notably, results from our previous study revealed that neuroinflammation and neurodegeneration were not induced during chronic T. gondii infection even after microglial proliferation. Therefore, the reduction in neuroinflammation should be considered when considering using microglia as a target for AD treatment. In this study, we demonstrated the potential of T. gondii in maintaining long-term microglial proliferation in the brain. Further studies should determine what factors can proliferate microglia in T. gondii itself.
4. Materials and Methods
4.1. T. gondii Infection and Experimental Design
For infecting 5XFAD mice, T. gondii ME49 cysts were obtained from brain tissues of C57BL/6 mice (Orient Bio Animal Center, Seongnam, South Korea) infected with 10 cysts. Seven-week-old C57BL/6 mice were infected with ten cysts for the histopathological examination and microarray analysis of the brain at 0, 3, 6, 12, and 36 weeks after infection (n = 2–4 per group). Eight-week-old 5XFAD mice were orally infected with ten cysts and euthanized using CO2 asphyxiation for histopathological examination of the brain at ten months after infection (n = 6).
4.2. 5XFAD Transgenic Mice
5XFAD mice (The Jackson Laboratory, Bar Harbor, ME, USA) were kindly provided by Dr. Inhee Mook-Jung at Seoul National University and bred by mating male mice with wild-type females (C57BL6/SJL). Genotyping for confirming the presence of human
APP and
PSEN1 transgenes was performed through PCR analysis of tail genomic DNA. PCR products (350 bp for
APP and 608 bp for
PSEN1) were analyzed using 1% agarose gel electrophoresis and detected through ethidium bromide staining. Primer sequences are listed in the
Supplementary Materials (Table S1).
4.3. Ethics Statement
All animal experiments were approved by the Institutional Animal Care and Use Committee at Seoul National University (Permit Number: SNU-110315-5). Mice were maintained in an animal facility according to the standards of the Animal Protection Act and the Laboratory Animal Act in Korea. Mice experiments were performed according to global standards, such as those established by the Association for Assessment and Accreditation of Laboratory Animal Care International. All efforts were made to minimize animal suffering (Approved Number: SNUIBC-R110302-1-1).
4.4. Hematoxylin and Eosin Staining and Congo Red Staining of Brain Tissue
Brain tissues were fixed in 10% formalin and embedded in paraffin after dehydration through an ethanol gradient. Brain tissues, which were coronally sectioned at 10-µm thick, were stained with Harris’ hematoxylin and eosin (H&E) to detect T. gondii tissue cysts and neuronal degeneration, indicated by acidophilic neurons in layer V of the cerebral cortex (n = 6 per group). Observations were performed using light microscopy (CKX 41, Olympus, Tokyo, Japan) and a color digital camera (DP72, Olympus). For Congo red staining, sectioned brain tissues were incubated in 0.4% aqueous Congo red solution (Sigma-Aldrich, St Louis, MO) for 10 min at room temperature (RT), counterstained in hematoxylin for 10 min, and dipped in acid alcohol (1% HCl in EtOH) for differentiation. Finally, after the sections were dehydrated and mounted, dense core plaques in the cortex and hippocampus were counted using a color digital camera attached to a light microscope and evaluated using ImageJ (Version 1.45, National Institute of Health, Bethesda, MD, USA).
4.5. Immunohistochemistry
Immunohistochemistry (IHC) was performed after Congo red staining. Briefly, tissue sections were deparaffinized in xylene, rehydrated in a graded series of ethanol, and rinsed with distilled water. After antigen retrieval, endogenous peroxidase activity was blocked using H2O2 in blocking buffer (1% fetal bovine serum in PBS) for 30 min. Then the slides were incubated with rabbit anti-mouse Iba-1 (Wako, VA, USA) as the primary antibody (Ab) and UltraMap anti-Rb horseradish peroxidase (HRP) (Ventana Medical Systems, Tucson, AZ, USA) as the secondary Ab. The ChromoMap DAB detection kit (Ventana Medical System) was used for detecting the DAB signal and hematoxylin was used for counterstaining. Slides were observed using a light microscope equipped with a color digital camera.
4.6. Immunofluorescence Staining and Quantitative Analysis
Brain sections with antigen retrieval were permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 10 min at RT and blocked in 2% bovine serum albumin (BSA)/PBS or 10% donkey serum for double-staining. Antibodies used for immunofluorescence (IF) staining are listed in the
Supplementary Materials (Table S2). Samples were stained with the corresponding secondary Ab and nuclei were stained with 4′,6-diamino-2-phenylindole, dihydrochloride (DAPI; Sigma-Aldrich, St. Louis, MO, USA). For detecting amyloid plaque, methoxy-X04 (Abcam, Pittsburgh, PA, USA) staining was performed before DAPI counterstaining. Tissue sections were stained with 100-μM methoxy-X04 in 40% ethanol (adjusted to pH 10 with 0.1 N NaOH) for 10 min and then incubated with 0.2% NaOH in 80% ethanol for 2 min. Immunostained slides were observed using fluorescence microscopy (Leica DMI6000B, Wetzlar, Germany). For quantifying area covered by microglia in the cortex and hippocampus, slides stained with Iba1 and TMEM119 Abs were used for calculating mean fluorescence intensity (MFI) using ImageJ in four identical regions (non-overlapped images at 20× magnification) captured by fluorescence microscopy (
n = 5–6 per group). For counting immunolabeled cells, the number of cells per defined tissue area was counted at 40× and 20× magnifications. Plaque-associated cells (microglia and Ly6C
+ monocytes) included cells at distances <30 µm from the center of the amyloid plaque. Plaque-free cells included cells at distances >30 µm from the center of the amyloid plaque. For counting the number of cells around plaques, microglia were selected based on DAPI-labeled nuclei. The result was presented as the total number of immunolabeled cells around plaques.
4.7. ELISA for Amyloid β (1–42) Quantification
The Amyloid β (1–42) Assay Kit (#27711, IBL, Tokyo, Japan) was used to measure amounts of Aβ42 (n = 7 per group). The test sample included 100 µL brain lysate, and all procedures for the assay were performed according to the manufacturer’s guidance. Reagents were prepared at RT approximately 30 min before use.
4.8. Multiplex Cytokine Immunoassay
Expression levels of IL-1β, TNF-α, IFN-γ, and IL-4 in 5XFAD mouse brains were examined using multiplex immunoassay (Bio-Plex mouse cytokine assay kit, Bio-Rad Laboratories, Hercules, CA, USA) (n = 5–6 per group). Brain tissues were lysed using the MicroRotofor™ Cell Lysis Kit (Bio-Rad Laboratories), and proteins in the homogenate were quantified using the bicinchoninic acid (BCA) assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). The assay was performed according to the manufacturer’s instructions and results were analyzed using the Bio-Plex Manager Software and Bio-Plex Data Pro™ software.
4.9. qRT-PCR
Total RNA from brain tissue was isolated using the HiGene Total RNA Prep Kit (BIOFACT, Daejeon, Korea) according to the manufacturer’s protocol and reverse-transcribed using the RT-PCR premix kit (Elpis Biotech Inc., Daejeon, Korea). Quantitative real-time PCR was performed using CFX96 (Bio-Rad) and SYBR green (Enzynomics™, Daejeon, Korea) (
n = 4–5 per group). Primer sequences used for real-time PCR are presented in the
Supplementary Materials (Table S1).
4.10. Microarray
Total RNA from brain tissue was extracted and pooled for microarray analysis (n = 3), which was performed using the Illumina MouseRef-8 v2 Expression BeadChip array (Illumina, Inc., San Diego, CA, USA) by Macrogen Inc. (Seoul, Korea). Export processing and analysis of arrayed data were performed using Illumina GenomeStudio v2011.1 (Gene Expression Module v1.9.0), and data were analyzed with the R v. 2.15.1 statistical software. Hierarchical cluster analysis was performed using Permute Matrix. Heat maps were created using the Excel Spreadsheet Software (Microsoft Corporation, Redmond, WA, USA) with conditional formatting. Gene expression rates in AD and AD + Toxo groups were compared with that in the wild-type (WT) group. Positive correlations are depicted in yellow (increased expression) and negative (decreased expression) in blue. The color scale of the heat map represents the relative minimum (−3) and maximum (+3) values of each gene.
4.11. TUNEL Staining for Plaque-Associated Apoptotic Microglia
TUNEL staining was performed on paraffin-embedded sections using the Cell Meter™ Fixed Cell and Tissue TUNEL Apoptosis Assay Kit (AAT Bio., Sunnyvale, CA, USA) according to the manufacturer’s protocol. Briefly, sections were deparaffinized, rehydrated, and immersed in 4% paraformaldehyde for 20 min at RT. Then, sections were incubated in 20 μg/mL proteinase K for 10 min and fixed with 4% paraformaldehyde for 20 min. Subsequently, sections were incubated with 25 μL TUNEL reaction mixture for 60 min and immunostained with Iba1 Ab (Abcam) as the primary Ab and anti-goat Alexa Fluor 647 (Invitrogen, Carlsbad, CA, USA) as the secondary Ab. Finally, DAPI-stained sections were mounted. TUNEL positive (apoptotic) cells were observed using fluorescence microscopy at 40× magnification (n = 6 per group).
4.12. Statistics
All statistical analyses were performed using the GraphPad Prism 5 software (GraphPad, La Jolla, CA, USA). Data are presented as the mean ± standard error of the mean. For MFI fold change data in
Figure 1, one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test was used for statistical evaluation. To compare experimental groups, namely WT, AD, and AD + Toxo, one-way ANOVA followed by Tukey’s multiple-comparison test was performed. Significant differences for two groups were assessed using Student’s
t-test with Welch’s correction. An asterisk (*) indicates a significant difference compared with the control (
p < 0.05) and a sharp (#) indicates a significant difference among experimental groups (
p < 0.05).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




