
Synthesis and Pharmacological In Vitro Investigations of Novel Shikonin Derivatives with a Special Focus on Cyclopropane Bearing Derivatives

 ,
,
Abstract
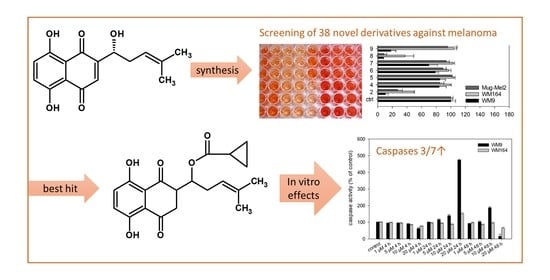
:
1. Introduction
2. Results and Discussion
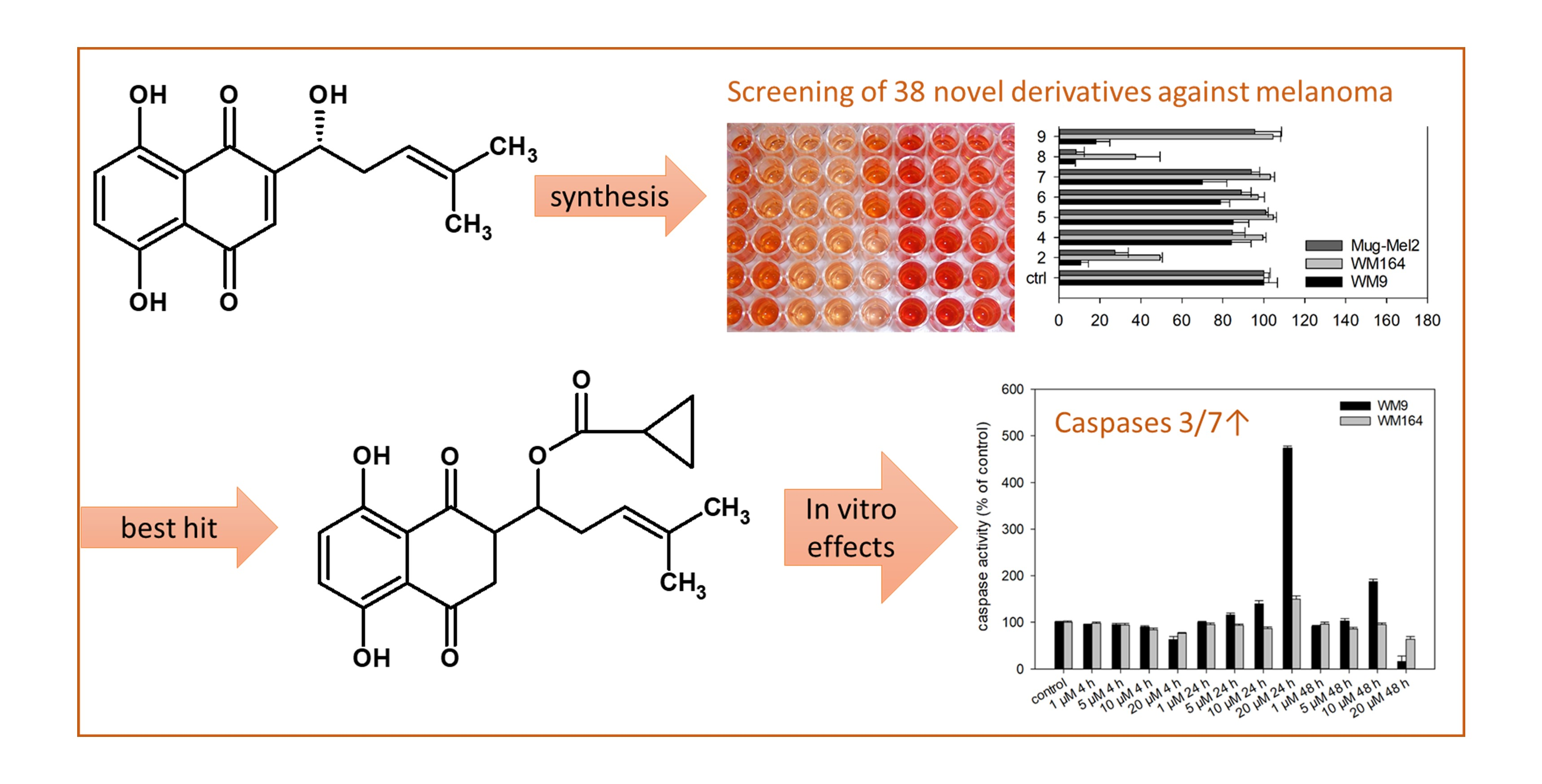
2.1. Syntheses of Shikonin Derivatives
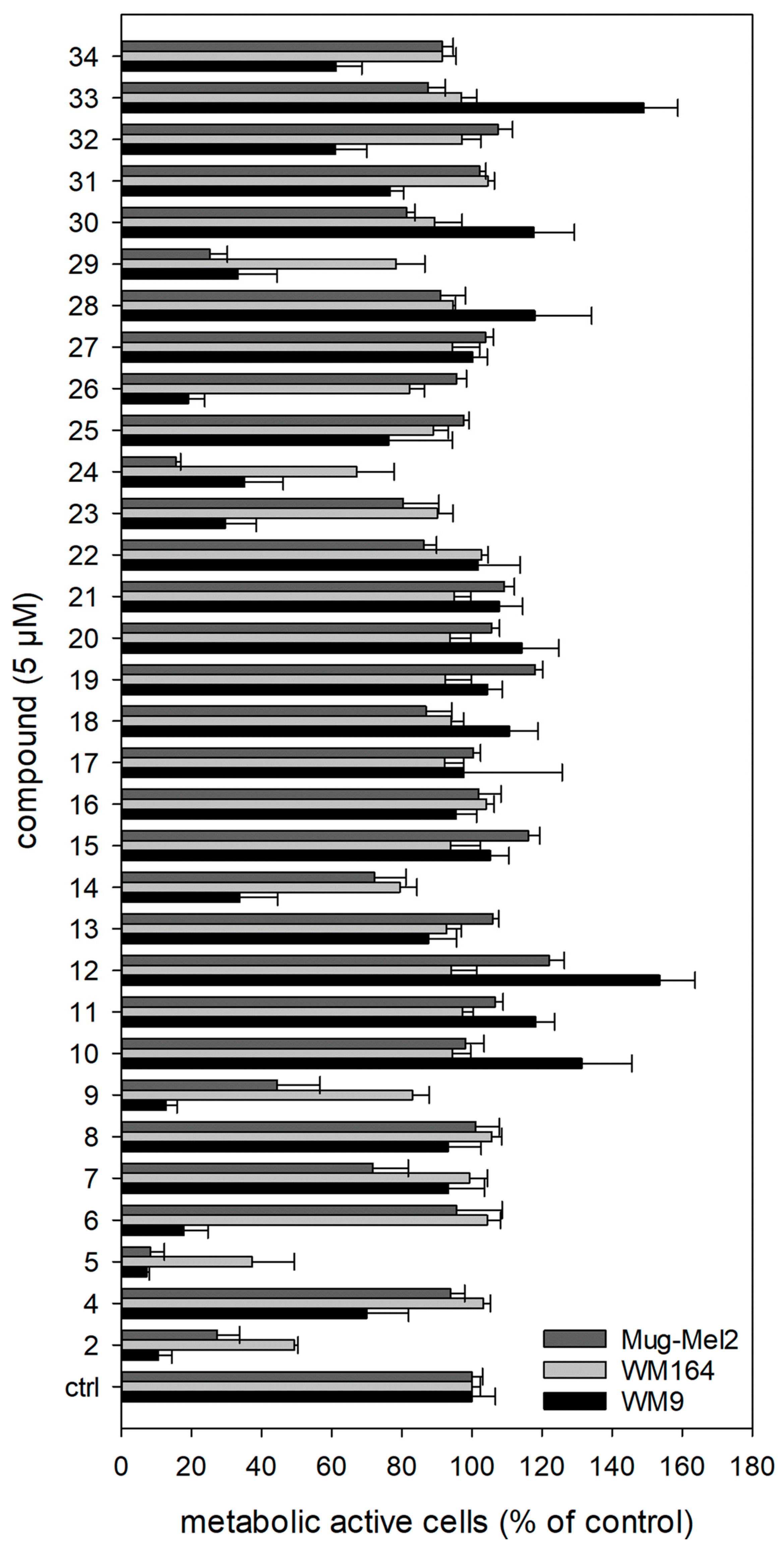
2.2. Results of the XTT Screening
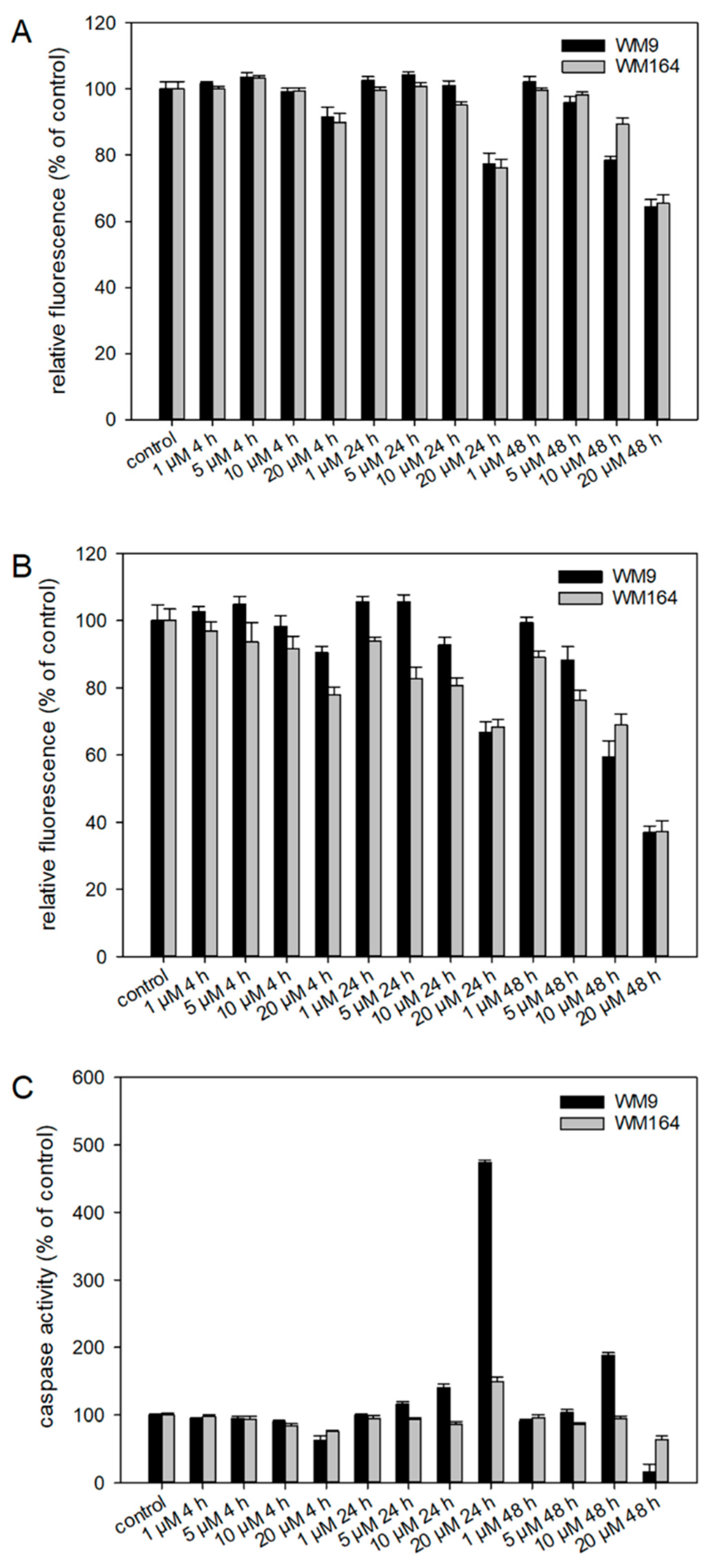
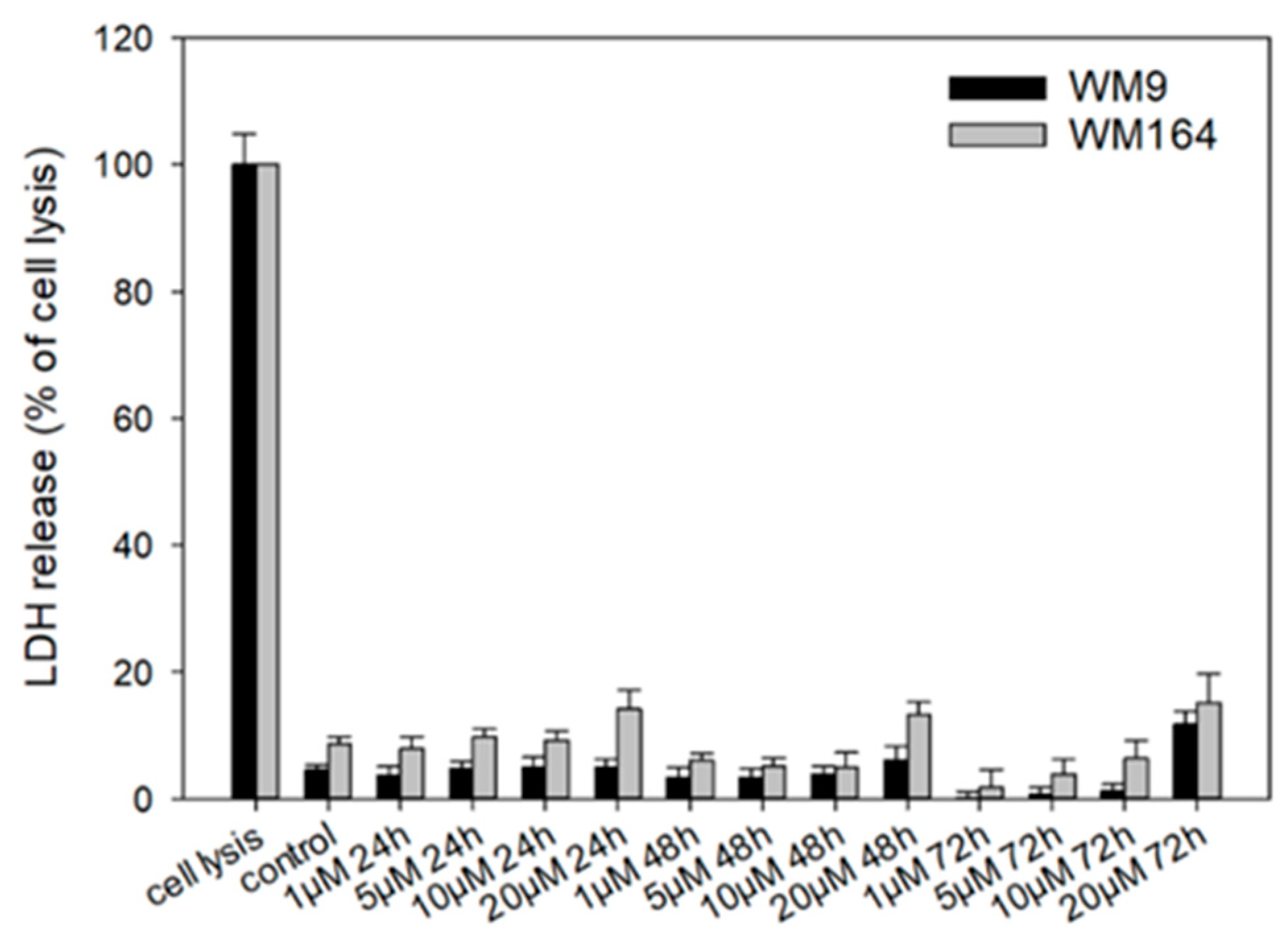
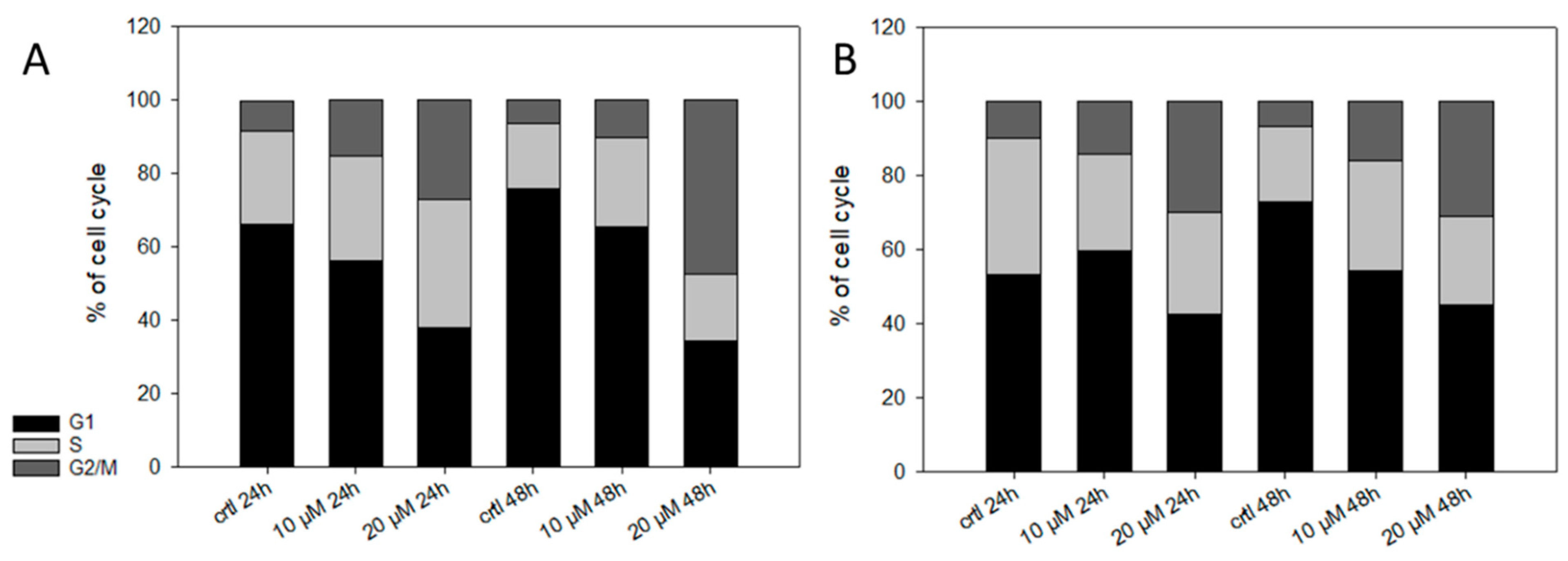
2.3. Pharmacological Effects of Cyclopropyloxoacetate 5
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis of Shikonin Derivatives
3.3. General Procedure for the Acylation of Shikonin
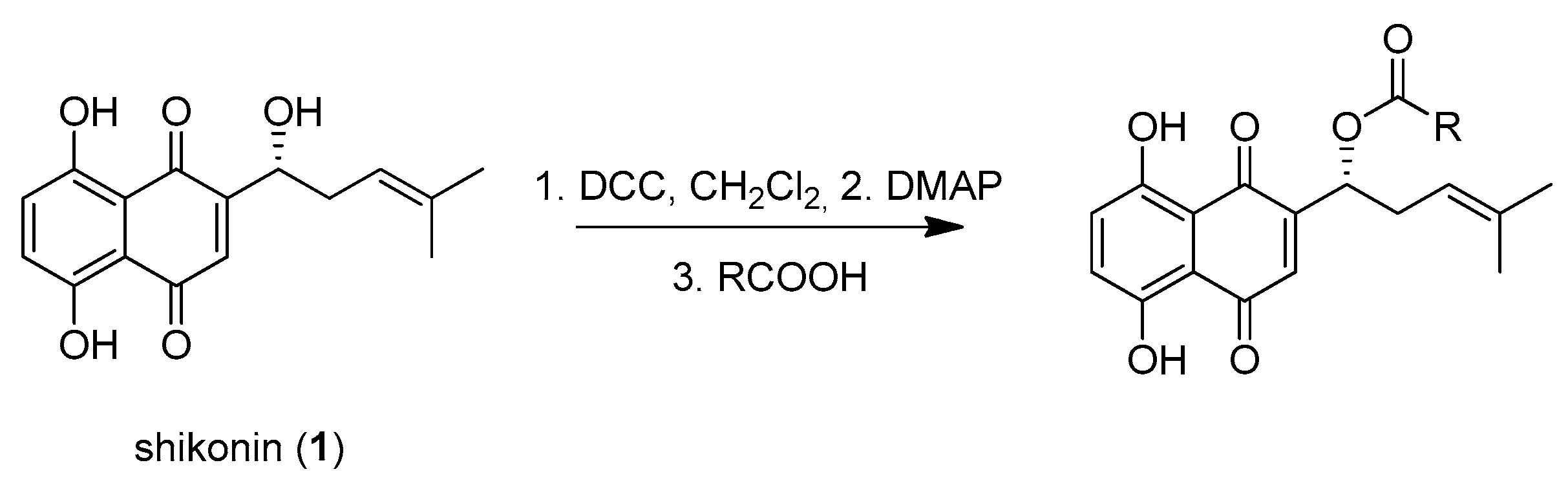
3.4. Synthesis of Precursor Acids
3.4.1. 2-(1-Bicyclo[4.1.0]heptyl)acetic Acid (p1)
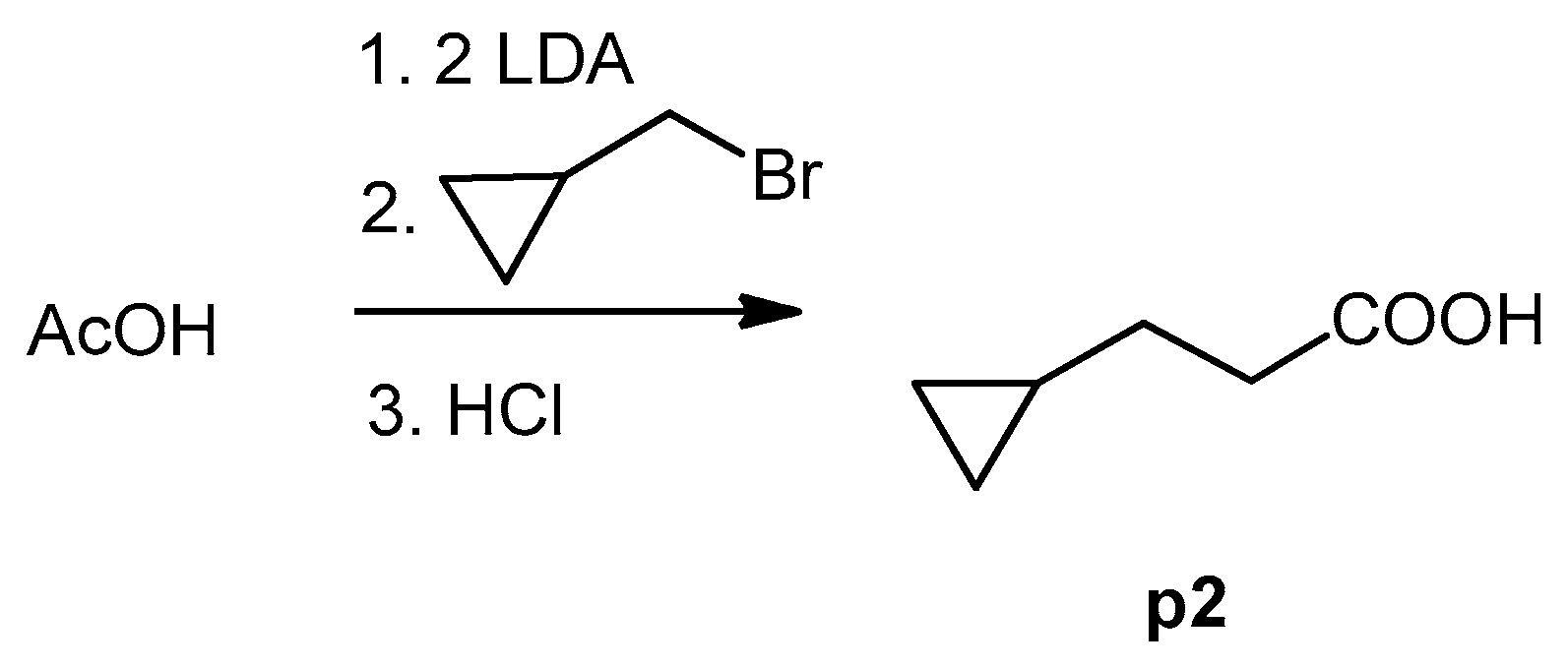
3.4.2. 3-Cyclopropylpropanoic Acid (p2)
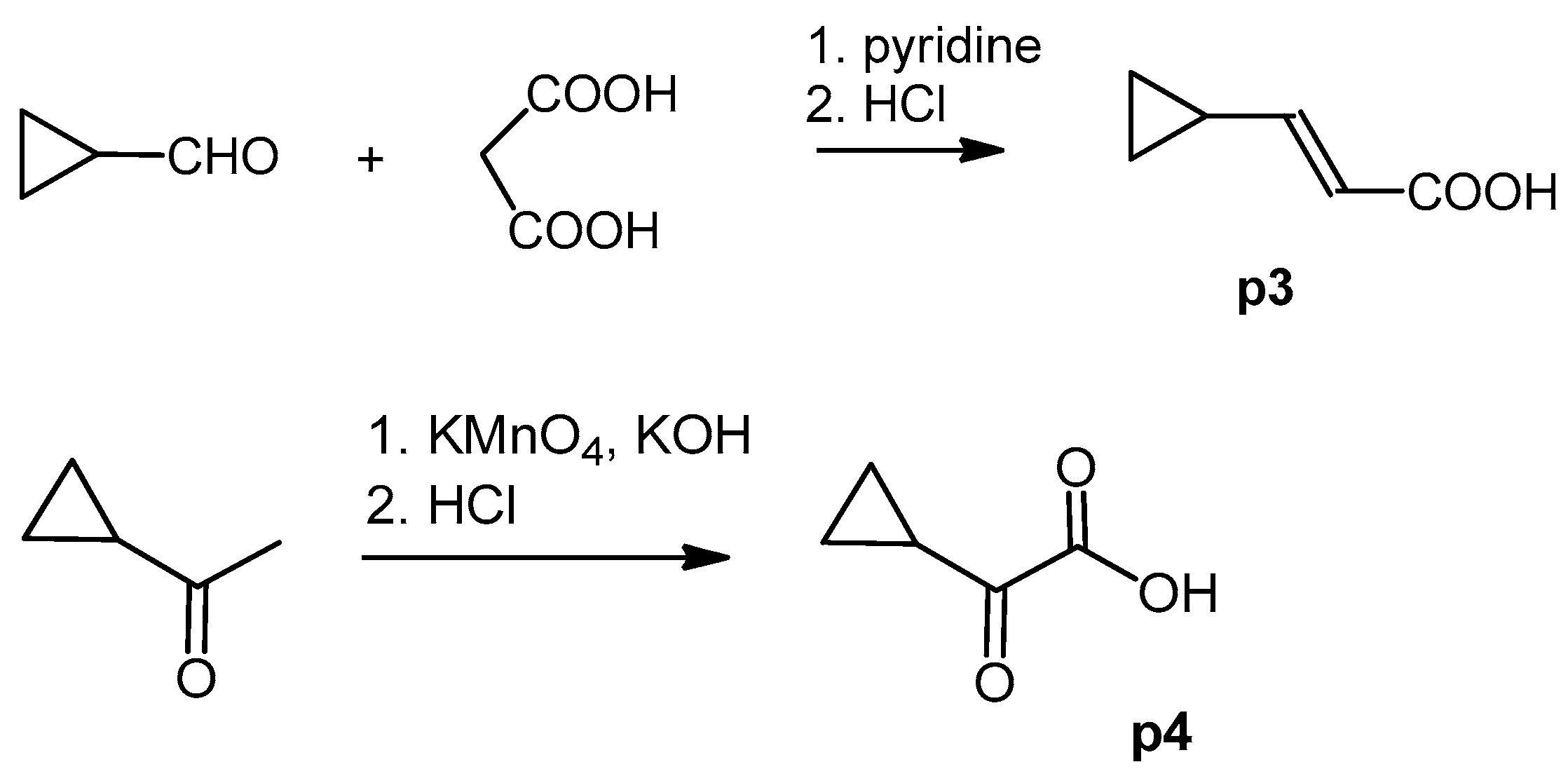
3.4.3. (E) 3-Cyclopropylpropenoic Acid (p3)
3.4.4. 2-Cyclopropyl-2-Oxoacetic Acid (p4)
3.5. Cell Culture
3.6. XTT Viability Assay
3.7. ApoToxGloTM Triplex Assay
3.8. LDH Assay
3.9. Cell Cycle Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shain, A.H.; Bastian, B.C. From melanocytes to melanoma. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Corrie, P.; Hategan, M.; Fife, K.; Parkinson, C. Management of Melanoma. Br. Med. Bull. 2014, 111, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Scolyer, R.A.; Rawson, R.V.; Gershenwald, J.E.; Ferguson, P.M.; Prieto, V.G. Melanoma pathology reporting and staging. Mod. Pathol. 2020, 33 (Suppl. 1), 15–24. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Cragg, M.G.; Pezzuto, J.M. Natural products as vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med. Princ. Pract. 2016, 25 (Suppl. 2), 41–59. [Google Scholar] [CrossRef]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. The chemistry and biology of alkannin, shikonin and related naphthazarin natural products. Angew. Chem. Int. Ed. Engl. 1999, 38, 270–301. [Google Scholar] [CrossRef]
- Chen, X.; Yang, L.; Oppenheim, J.J.; Howard, M.Z. Cellular pharmacology studies of shikonin derivatives. Phytother. Res. 2002, 16, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Andújar, I.; Rios, J.L.; Giner, R.M.; Recio, M.C. Pharmacological properties of shikonin—A review of literature since 2002. Planta Med. 2013, 79, 1685–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Vong, C.T.; Chen, H.; Gao, Y.; Lyu, P.; Qiu, L.; Zhao, M.; Liu, Q.; Cheng, Z.; Zou, J.; et al. Naturally occurring anti-cancer compounds: Shining from Chinese herbal medicine. Chin. Med. 2019, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duke, J.A.; Ayensu, S. Medicinal Plants of China; Reference Publications Inc.: Algonac, MI, USA, 1985; Volume 1. [Google Scholar]
- Zhao, Q.; Kretschmer, N.; Bauer, R.; Efferth, T. Shikonin and its derivatives inhibit the epidermal growth factor receptor signaling and synergistically kill glioblastoma cells in combination with erlotinib. Int. J. Cancer 2015, 137, 1446–1456. [Google Scholar] [CrossRef]
- Rinner, B.; Kretschmer, N.; Knausz, H.; Mayer, A.; Boechzelt, H.; Hao, X.J.; Heubl, G.; Efferth, T.; Schaider, H.; Bauer, R. A petrol ether extract of the roots of Onosma paniculatum induces cell death in a caspase dependent manner. J. Ethnopharmacol. 2010, 129, 182–188. [Google Scholar] [CrossRef]
- Kretschmer, N.; Rinner, B.; Deutsch, A.J.; Lohberger, B.; Knausz, H.; Kunert, O.; Blunder, M.; Boechzelt, H.; Schaider, H.; Bauer, R. Naphthoquinones from Onosma paniculata induce cell-cycle arrest and apoptosis in melanoma Cells. J. Nat. Prod. 2012, 75, 865–869. [Google Scholar] [CrossRef]
- Kretschmer, N.; Deutsch, A.; Durchschein, C.; Rinner, B.; Stallinger, A.; Higareda-Almaraz, J.C.; Scheideler, M.; Lohberger, B.; Bauer, R. Comparative Gene Expression Analysis in WM164 Melanoma Cells Revealed That β,β-Dimethylacrylshikonin Leads to ROS generation, Loss of Mitochondrial Membrane Potential, and Autophagy Induction. Molecules 2018, 23, 2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallinger, A.; Kretschmer, N.; Kleinegger, F.; Brvar, L.; Liegl-Atzwanger, B.; Prokesch, A.; Durchschein, C.; Bauer, R.; Deutsch, A.; Rinner, B. β,β-Dimethylacrylshikonin Induces Apoptosis in Melanoma Cell Lines by NOXA Upregulation. J. Nat. Prod. 2020, 83, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Durchschein, C.; Hufner, A.; Rinner, B.; Stallinger, A.; Deutsch, A.; Lohberger, B.; Bauer, R.; Kretschmer, N. Synthesis of novel shikonin derivatives and pharmacological effects of cyclopropylacetylshikonin on melanoma cells. Molecules 2018, 23, 2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damianakos, H.; Kretschmer, N.; Syklowska-Baranek, K.; Pietrosiuk, A.; Bauer, R.; Chinou, I. Antimicrobial and cytotoxic isohexenylnapththazarins from Arnebia euchroma (Royle) Johnst. (Boraginaceae) callus and cell suspension culture. Molecules 2012, 17, 14310–14322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papageorgiou, V.P. Naturally occurring isohexenylnaphthazarin pigments: A new class of drugs. Planta Med. 1980, 38, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Rao, Z.; Liu, X.; Zhou, W.; Yi, J.; Li, S.S. Synthesis and antitumour activity of β-hydroxyisovalerylshikonin analogues. Eur. J. Med. Chem. 2001, 46, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-M.; Lin, H.-Y.; Kong, W.-Y.; Guo, J.; Shi, J.; Huang, S.-C.; Qi, J.-L.; Yang, R.-W.; Gu, H.-W.; Yang, Y.-H. Synthesis and biological evaluation of heterocyclic carboxylic acyl shikonin derivatives. Chem. Biol. Drug Res. 2014, 83, 334–343. [Google Scholar] [CrossRef]
- Zhao, L.M.; Cao, F.X.; Jin, H.S.; Zhang, J.H.; Szwaya, J.; Wang, G. One-pot synthesis of 1,4-dihydroxy-2-(E-1-hydroxy-4-phenylbut-3-enyl)anthracene-9,10-diones as novel shikonin analogs and evaluation of their antiproliferative activities. Bioorg. Med. Chem. Lett. 2016, 26, 2691–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.Z.; Baik, K.U. Process for Preparing 5,8-Dihydronaphthoquinone Derivatives, Novel 5,8-Dihydronaphthoquinone Derivatives and Their Use as Anticancer Agent. KR. Patent WO 95/02572, 26 January 1995. [Google Scholar]
- Baik, K.-U.; Song, G.Y.; Kim, Y.; Sok, D.-E.; Ahn, B.-Z. 2-Substituted naphthazarins; synthesis and antitumor activity. Arch. Pharm. Pharm. Med. Chem. 1997, 330, 377–382. [Google Scholar] [CrossRef]
- Shen, C.C.; Syu, W.J.; Li, S.Y.; Lin, C.H.; Lee, G.H.; Sun, C.M. Antimicrobial activities of naphthazarins from Arnebia euchroma. J. Nat. Prod. 2002, 65, 1857–1862. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, W.; Shi, J.; Kong, W.Y.; Qi, J.L.; Wang, X.M.; Yang, Y.H. Design, synthesis and biological evaluation of cinnamic acyl shikonin derivatives. Chem. Biol. Drug Des. 2013, 81, 275–283. [Google Scholar] [CrossRef]
- Baloch, S.K.; Ling, L.J.; Qiu, H.Y.; Ma, L.; Lin, H.Y.; Huang, S.C.; Qi, J.L.; Wang, X.M.; Lu, G.H.; Yang, Y.H. Synthesis and biological evaluation of novel shikonin ester derivatives as potential anti-cancer agents. RSC Adv. 2014, 4, 35588–35596. [Google Scholar] [CrossRef]
- Renaud, P.; Fox, M.A. Reaction of Dilithiated Carboxylic Acids with Iodine: Evidence for the Formation of a Radical Anion Intermediate. J. Org. Chem. 1988, 53, 3745–3752. [Google Scholar] [CrossRef]
- Barczak, N.T.; Jarvo, E.R. Regioselective Silver-Mediated Kondakov–Darzens Olefin Acylation. Chem. Eur. J. 2011, 17, 12912–12916. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, S.; Cooper, C.S.; Fung, A.K.L.; Lynch, J.K.; Plagge, F.; Chu, D.T.W. Asymmetric dipolar cycloaddition reaction: A practical, convergent synthesis of chiral pyrrolidines. Tetrahedron Asymmetry 1997, 8, 883–887. [Google Scholar] [CrossRef]
- Prokopenko, V.V.; Okonnishnikova, G.P.; Klimenko, I.P.; Shulishov, E.V.; Tomilov, Y.V. Synthesis and chemical transformations of 2-cyclopropyl-2-diazoacetates. Russ. Chem Bull. Int. Ed. 2007, 56, 1515–1521. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, W.; Shu, D.; Werness, J.B.; Tang, W. Synthesis of Cyclobutenes via Transition Metal Catalyzed Highly Selective Ring Expansion of Cyclopropanes. Angew. Chem. Int. Ed. 2008, 47, 8933–8936. [Google Scholar] [CrossRef]
- Kim, S.-H.; Song, G.-Y.; Sok, D.-E.; Ahn, B.-Z. Anti-cell adhesive effect of phenylacetylshikonin analogs related to their cytotoxicity in A549 cells. Arch. Pharm Res. 1997, 20, 155–157. [Google Scholar] [CrossRef]
- Sun, W.-X.; Han, H.-W.; Yang, M.-K.; Wen, Z.-L.; Wang, Y.-S.; Fu, J.-Y.; Lu, Y.-T.; Wang, M.-Y.; Bao, J.-X.; Lu, G.-H.; et al. Design, synthesis and biological evaluation of benzoylacrylic acid shikonin ester derivatives as irreversible dual inhibitors of tubulin and EGFR. Bioorg. Med. Chem. 2019, 27, 115153. [Google Scholar] [CrossRef]
- Yang, M.D.; Shen, X.B.; Hu, Y.S.; Chen, Y.Y.; Liu, X.H. Novel naphthalene-enoates: Design and anticancer activity through regulation cell autophagy. Biomed. Pharmacother. 2019, 113, 108747. [Google Scholar] [CrossRef]
- Shen, X.B.; Wang, Y.; Han, X.-Z.; Sheng, L.-Q.; Wu, F.; Liu, X. Design, synthesis, and anticancer activity of naphthoquinone derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 773–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.R.; Tsukada, M.; Suzuki, N.; Shimamura, T.; Gao, L.; Koyanagi, J.; Komada, F.; Saito, S. Comparison of the cytotoxic activities of naturally occurring hydroxyanthraquinones and hydroxynaphthoquinones. Eur. J. Med. Chem. 2008, 43, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.Z.; Baik, K.U.; Kweon, G.R.; Kyu, L.; Hwang, B.D. Acylshikonin analogues: Synthesis and inhibition of DNA topoisomerase-I. J. Med. Chem. 1995, 38, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.; Al-Arif, M.T. Cyclisation of Substituted 2-(4-Methyl-pent-3-enyl)-5,8-dihydroxy-1,4-naphthoquinones. J. Prakt Chem. 1982, 324, 865–869. [Google Scholar] [CrossRef]
- Kim, S.-H.; Song, G.-Y.; Jin, G.-Z.; Ahn, B.-Z. Antitumor activity of arylacetylshikonin analogs. Arch. Pharm. Res. 1996, 19, 416–422. [Google Scholar] [CrossRef]
- Thangapazham, R.L.; Singh, A.K.; Seth, P.; Misra, N.; Shafali, M.; Vijayavitthal, T.; Raj, K.; Maheshwari, R.K. Shikonin analogue (SA) 93/637 induces apoptosis by activation of caspase-3 in U937 cells. Front. Biosci. 2008, 13, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, X.; Lin, H.; Shi, J.; Liu, H.; Kong, W.; Haori, Q.; Huang, S. Shikonin Octyl Methoxycinnamate Derivant as Well as Synthesis Method and Application Thereof. Patent CN102531893A, 4 July 2012. [Google Scholar]
- Baloch, S.K.; Ma, L.; Xu, G.H.; Bai, L.F.; Zhao, H.; Tang, C.Y.; Pang, Y.J.; Yang, R.W.; Wang, X.M.; Lu, G.H.; et al. A potent anticancer agent of shikonin derivative targeting tubulin. Chirality 2015, 27, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Liu, W.; Ding, J.; Cai, J.; Duan, W. Shikonin derivatives: Synthesis and inhibition of human telomerase. Bioorg. Med. Chem. Lett. 2002, 12, 1375–1378. [Google Scholar] [CrossRef]
- Yang, F.; Chen, Y.; Duan, W.; Zhang, C.; Zhu, H.; Ding, J. SH-7, a new synthesized shikonin derivative, exerting its potent antitumor activities as a topoisomerase inhibitor. Int. J. Cancer 2006, 119, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-G.; Jin, G.-Z.; Song, G.-Y.; Hoon, C.; Ahn, B.-Z. Haloacetylshikonin Derivatives: Synthesis and Evaluation of Antitumor Activity. Yakhak Hoechi 1998, 42, 159–164. [Google Scholar]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar] [PubMed]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic Significance of BRAF and NRAS Mutations in Melanoma: A German Study from Routine Care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef]
- Mackiewicz, J.; Mackiewicz, A. BRAF and MEK Inhibitors in the Era of Immunotherapy in Melanoma Patients. Contemp. Oncol. (Poznan Poland) 2018, 22, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-Mutant Melanoma: Current Challenges and Future Prospect. Oncol. Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.J.; Zhang, X.; Cheng, C.; Wang, F.; Wang, X.K.; Liang, Y.J.; To, K.K.W.; Zhou, W.; Huang, H.B.; Fu, L.W. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br. J. Pharmacol. 2012, 166, 1669–1683. [Google Scholar] [CrossRef] [Green Version]
- Akiyode, O.; George, D.; Getti, G.; Boateng, J. Systematic comparison of the functional physico-chemical characteristics and biocidal activity of microbial derived biosurfactants on blood-derived and breast cancer cells. J. Colloid Interface Sci. 2016, 479, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Fayez, H.; El-Motaleb, M.A.; Selim, A.A. Synergistic Cytotoxicity of Shikonin-Silver Nanoparticles as an Opportunity for Lung Cancer. J. Label. Comp. Radiopharm. 2020, 63, 25–32. [Google Scholar] [CrossRef]
- Wang, H.; Tang, Y.; Fang, Y.; Zhang, M.; Wang, H.; He, Z.; Wang, B.; Xu, Q.; Huang, Y. Reprogramming Tumor Immune Microenvironment (TIME) and Metabolism via Biomimetic Targeting Codelivery of Shikonin/JQ1. Nano Lett. 2019, 19, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhao, H.R.; Meng, Q.Q.; Zhang, Q.J.; Dong, J.Y.; Zhu, B.Q.; Li, S.S. Synthesis and biological evaluation of sulfur-containing shikonin oxime derivatives as potential antineoplastic agents. Eur. J. Med. Chem. 2018, 143, 166–181. [Google Scholar] [CrossRef]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Liu, T.J.; Lai, H.C. Shikonin Induces Apoptosis, Necrosis, and Premature Senescence of Human A549 Lung Cancer Cells through Upregulation of p53 Expression. Evid. Based Complement. Alternat. Med. 2015, 2015, 620383. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kao, S.H.; Hunag, J.E.; Sheu, G.T.; Yeh, C.W.; Hseu, Y.C.; Wang, C.J.; Hsu, L.S. Shikonin time-dependently induced necrosis or apoptosis in gastric cancer cells via generation of reactive oxygen species. Chem. Biol. Interact. 2014, 211, 44–53. [Google Scholar] [CrossRef]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, X.; Cao, Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. Oncol. Targets Ther. 2019, 12, 2605–2614. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.Y.; Yin, Y.; Lian, B.P.; Leng, J.F.; Xia, Y.Z.; Kong, L.Y. Synthesis and biological evaluation of novel shikonin-benzo[b]furan derivatives as tubulin polymerization inhibitors targeting the colchicine binding site. Eur. J. Med. Chem. 2020, 190, 112105. [Google Scholar] [CrossRef] [PubMed]
- Albreht, A.; Vovk, I.; Simonovska, B.; Srbinoska, M. Identification of shikonin and its ester derivatives from the roots of Echium italicum L. J. Chromatogr. A 2009, 1216, 3156–3162. [Google Scholar] [CrossRef]
- Kim, J.Y.; Jeong, H.J.; Park, J.-Y.; Kim, Y.M.; Park, S.-J.; Cho, J.K.; Park, K.H.; Ryu, Y.B.; Lee, W.S. Selective and slow-binding inhibition of shikonin derivatives isolated from Lithospermum erythrorhizon on glycosyl hydrolase 33 and 34 sialidases. Bioorg. Med. Chem. 2012, 20, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Chaouche, T.; Haddouchi, F.; Bekkara, F.A. Identification of Shikonin from the roots of Echium pycnanthum Pomel. Asian J. Pharm. Clin. Res. 2012, 5, 30–32. [Google Scholar]
- Sugahara, K.; Fujita, T.; Watanabe, S.; Sakamoto, M.; Sugimoto, K. A convenient lactonization of 2- and 3-cyclopropylalkanoic acids to γ- and δ-lactones. Synthesis 1990, 783–784. [Google Scholar] [CrossRef]
- Kantorowski, E.J.; Eisenberg, S.W.E.; Fink, W.H.; Kurth, M.J. Six- vs Seven-Membered Ring Formation from the 1-Bicyclo[4.1.0]heptanylmethyl Radical: Synthetic and ab Initio Studies. J. Org. Chem. 1999, 64, 570–580. [Google Scholar] [CrossRef]
- Maercker, A.; Theysohn, W. Versuche zur Umlagerung von 2-Cyclopropyl-äthyl-Anionen. Liebigs Ann. Chem. 1972, 759, 132–157. [Google Scholar] [CrossRef]
- Muehling, O.; Wessig, P. Photochemical synthesis of benzoyl spiro[2.2]pentanes; Photochem. Photobiol. Sci. 2006, 5, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.H.; Wallis, E.S. Kinetics of the addition of bromine to some cyclopropanes in aqueous acetic acid. J. Org. Chem. 1956, 21, 663–6966. [Google Scholar] [CrossRef]
- Henrick, C.A.; Willy, W.E.; Staal, G.B.; Ludvik, G.F. Ovicidal activity and its relation to chemical structure for the two-spotted spider mite (Tetranychus urticae Koch) in a new class of miticides containing the cyclopropyl group. J. Agric. Food Chem. 1976, 24, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Zbiral, E.; Werner, E. Reaktionen mit phosphororganischen Verbindungen, 10. Mitt.: [Oxydation mit Pb(OAc)4, (C6H5COO)2, C6H5J(OAc)2 und PbO2]. Zur Darstellung von α-Ketosäuremethylestern und α-Ketosäurethiophenylestern. Monatsh. Chem. 1966, 97, 1797–1820. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | Compound | R |
|---|---|---|---|
| 1 | H | 2 |  |
| 3 |  | 4 |  |
| 5 |  | 6 |  |
| 7 |  | 8 |  |
| 9 |  | 10 |  |
| 11 |  | 12 |  |
| 13 |  | 14 |  |
| 15 |  | 16 |  |
| 17 |  | 18 |  |
| 19 |  | 20 |  |
| 21 |  | 22 |  |
| 23 |  | 24 |  |
| 25 |  | 26 |  |
| 27 |  | 28 |  |
| 29 |  | 30 |  |
| 31 |  | 32 |  |
| 33 |  | 34 |  |
| Cell Line | IC50 Values of 5 (µM) | IC50 Values of 3 [17] (µM) | IC50 Values of 2 [14] (µM) |
|---|---|---|---|
| WM9 | 1.5 ± 0.1 | 3.2 ± 0.8 | 2.7 ± 0.3 |
| WM164 | 4.5 ± 0.5 | 4.9 ± 1.7 | 8.3 ± 0.3 |
| MUG-Mel2 | 2.4 ± 0.2 | 3.2 ± 0.3 | 7.2 ± 0.5 |
| HEK293 | 3.4 ± 0.2 | 5.4 ± 0.7 | n.d. |
| Cell Line | 10 µM (24 h) | 20 µM (24 h) | 10 µM (48 h) | 20 µM (48 h) |
|---|---|---|---|---|
| WM9 | 0.0311 | 0.0014 | 0.1467 | 0.0515 |
| WM164 | 0.3313 | 0.0153 | 0.0553 | 0.0065 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretschmer, N.; Hufner, A.; Durchschein, C.; Popodi, K.; Rinner, B.; Lohberger, B.; Bauer, R. Synthesis and Pharmacological In Vitro Investigations of Novel Shikonin Derivatives with a Special Focus on Cyclopropane Bearing Derivatives. Int. J. Mol. Sci. 2021, 22, 2774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052774
Kretschmer N, Hufner A, Durchschein C, Popodi K, Rinner B, Lohberger B, Bauer R. Synthesis and Pharmacological In Vitro Investigations of Novel Shikonin Derivatives with a Special Focus on Cyclopropane Bearing Derivatives. International Journal of Molecular Sciences. 2021; 22(5):2774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052774
Chicago/Turabian StyleKretschmer, Nadine, Antje Hufner, Christin Durchschein, Katrin Popodi, Beate Rinner, Birgit Lohberger, and Rudolf Bauer. 2021. "Synthesis and Pharmacological In Vitro Investigations of Novel Shikonin Derivatives with a Special Focus on Cyclopropane Bearing Derivatives" International Journal of Molecular Sciences 22, no. 5: 2774. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052774