
Microglial Heterogeneity and Its Potential Role in Driving Phenotypic Diversity of Alzheimer’s Disease

 , , , ,
, , , , 
Abstract
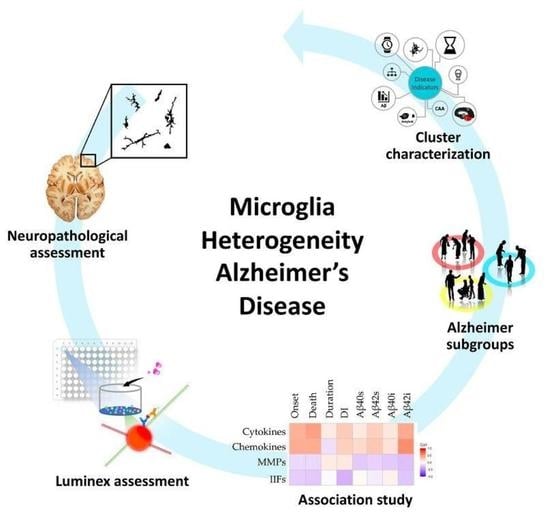
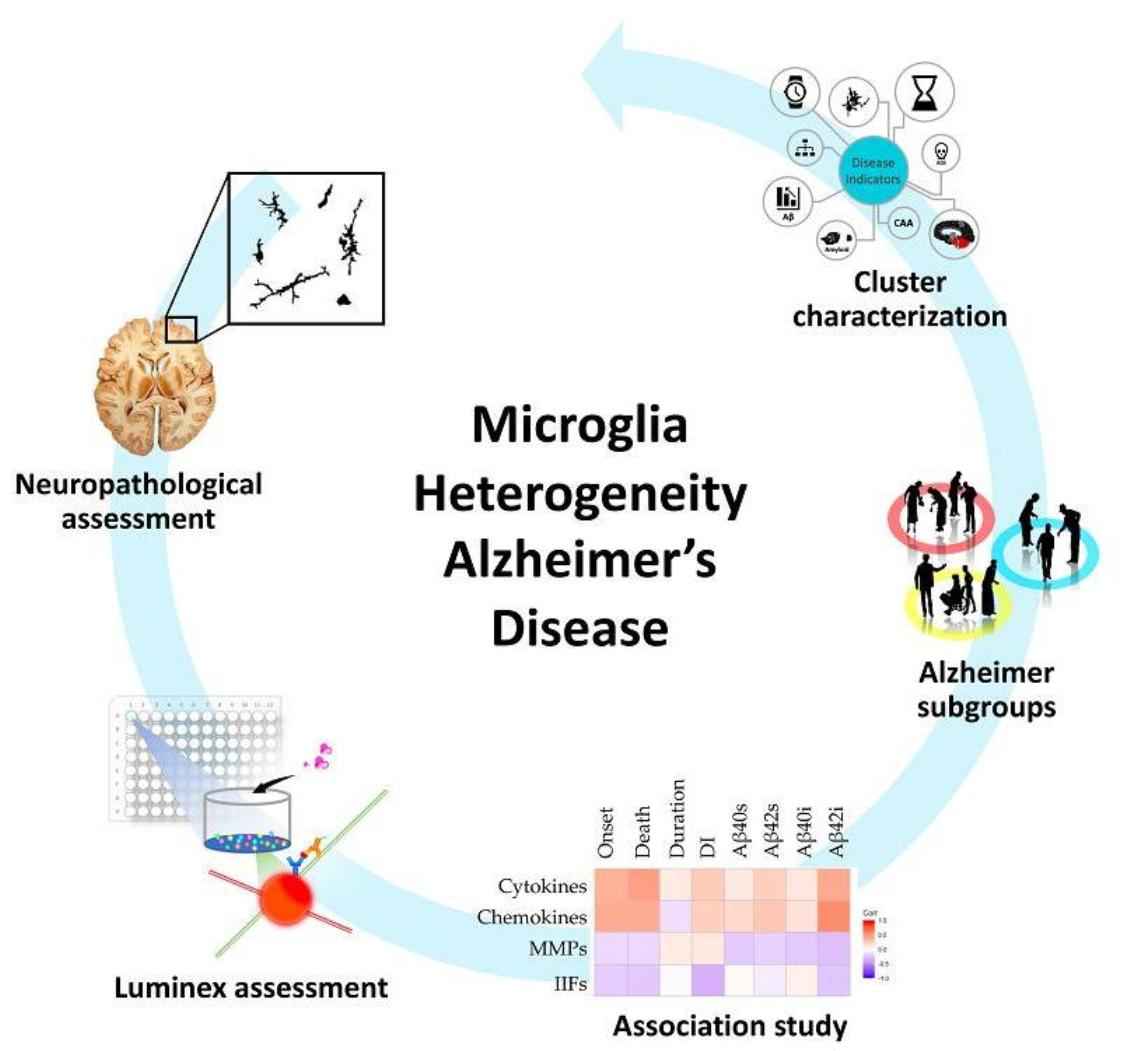
:
1. Introduction
2. Results
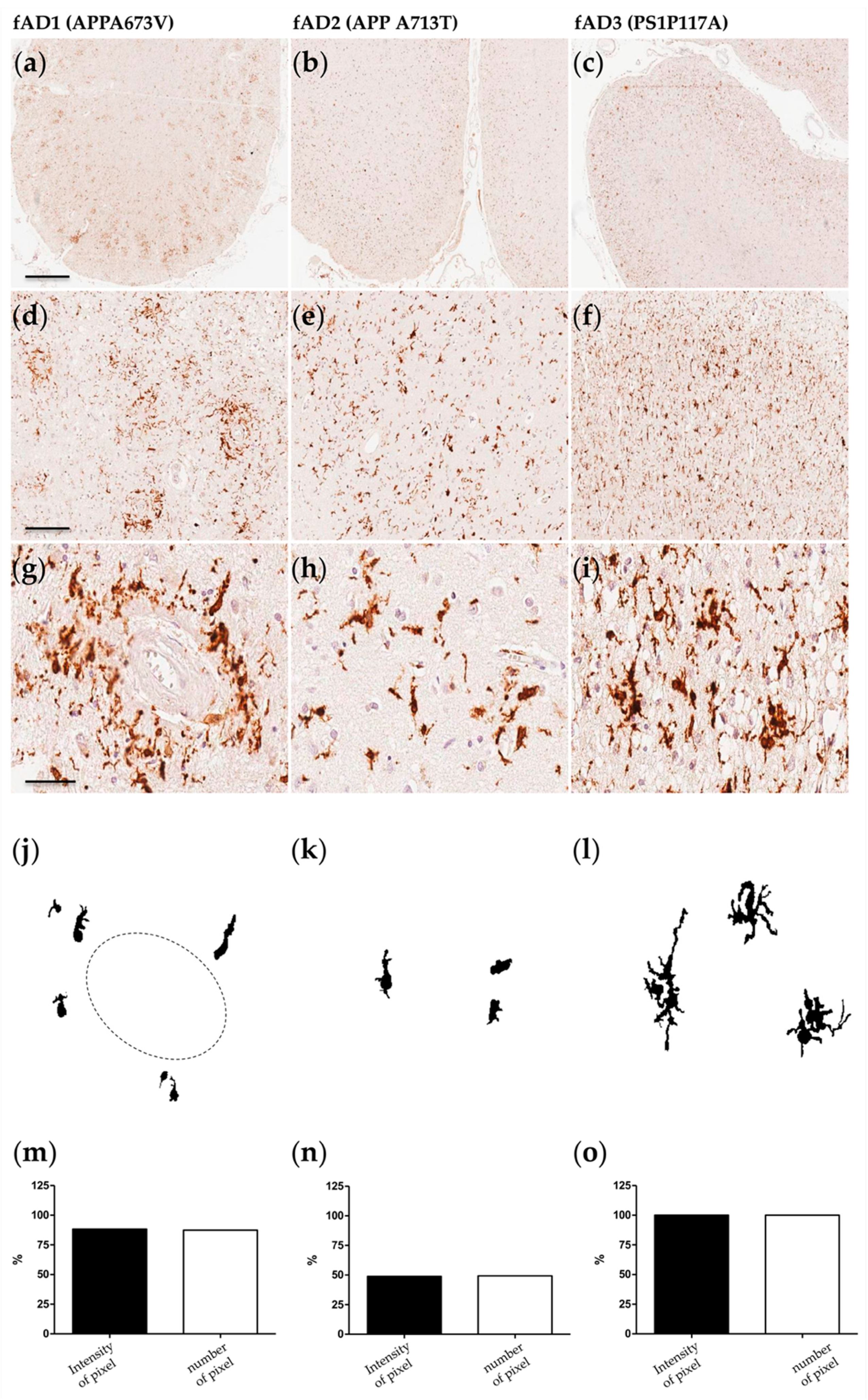
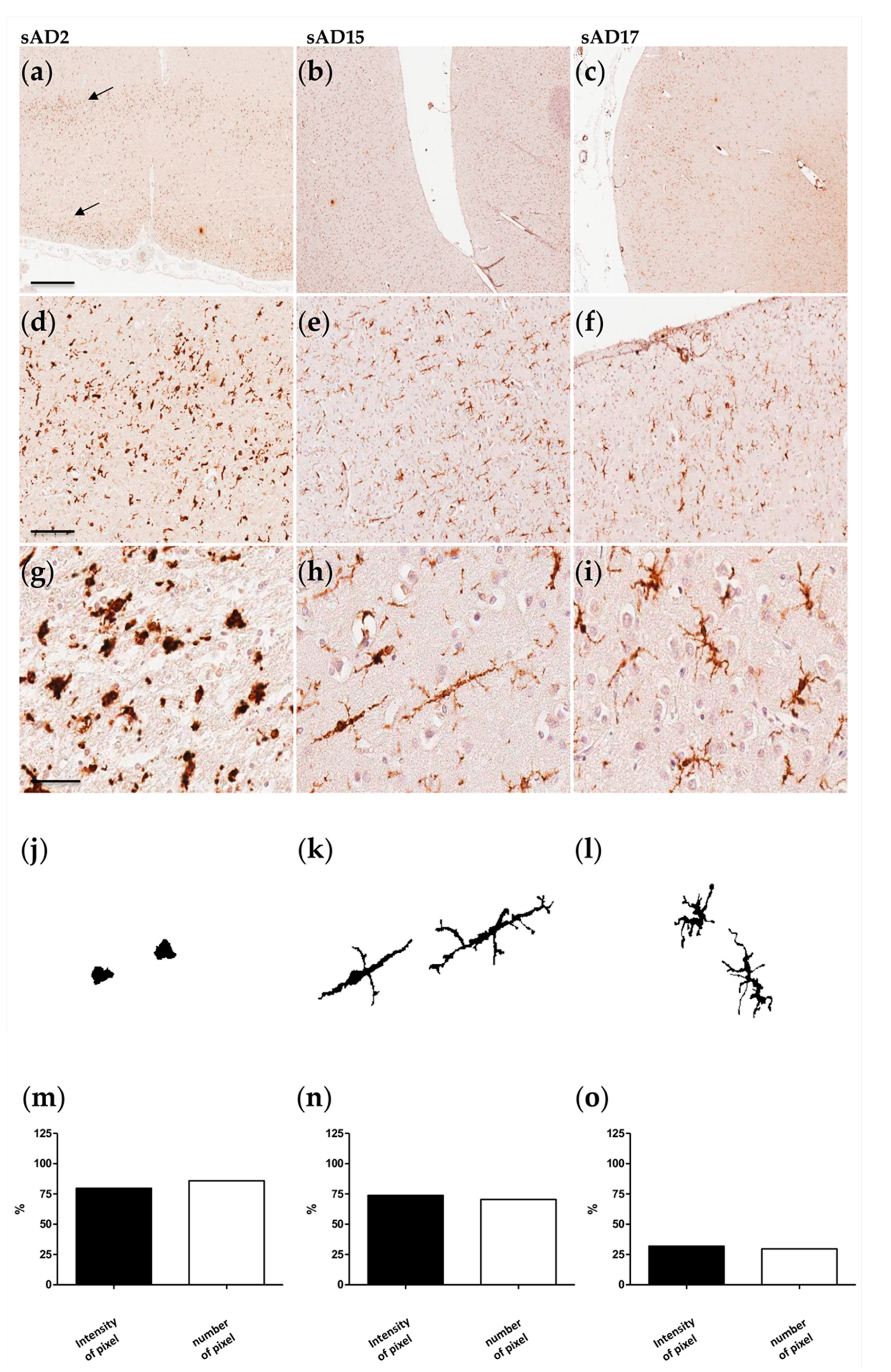
2.1. Morphological/Functional Profiling of Microglia in AD Brain Samples
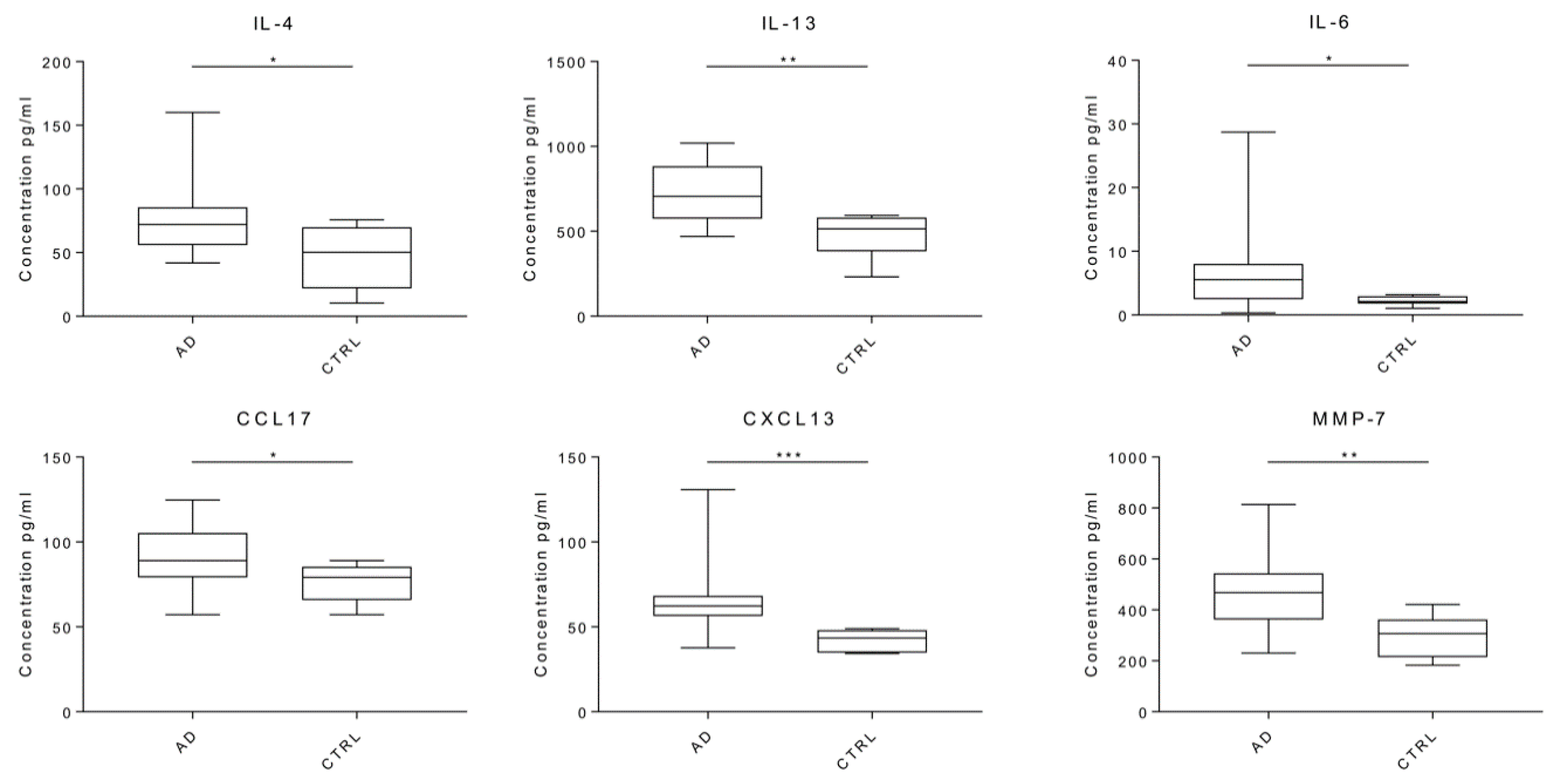
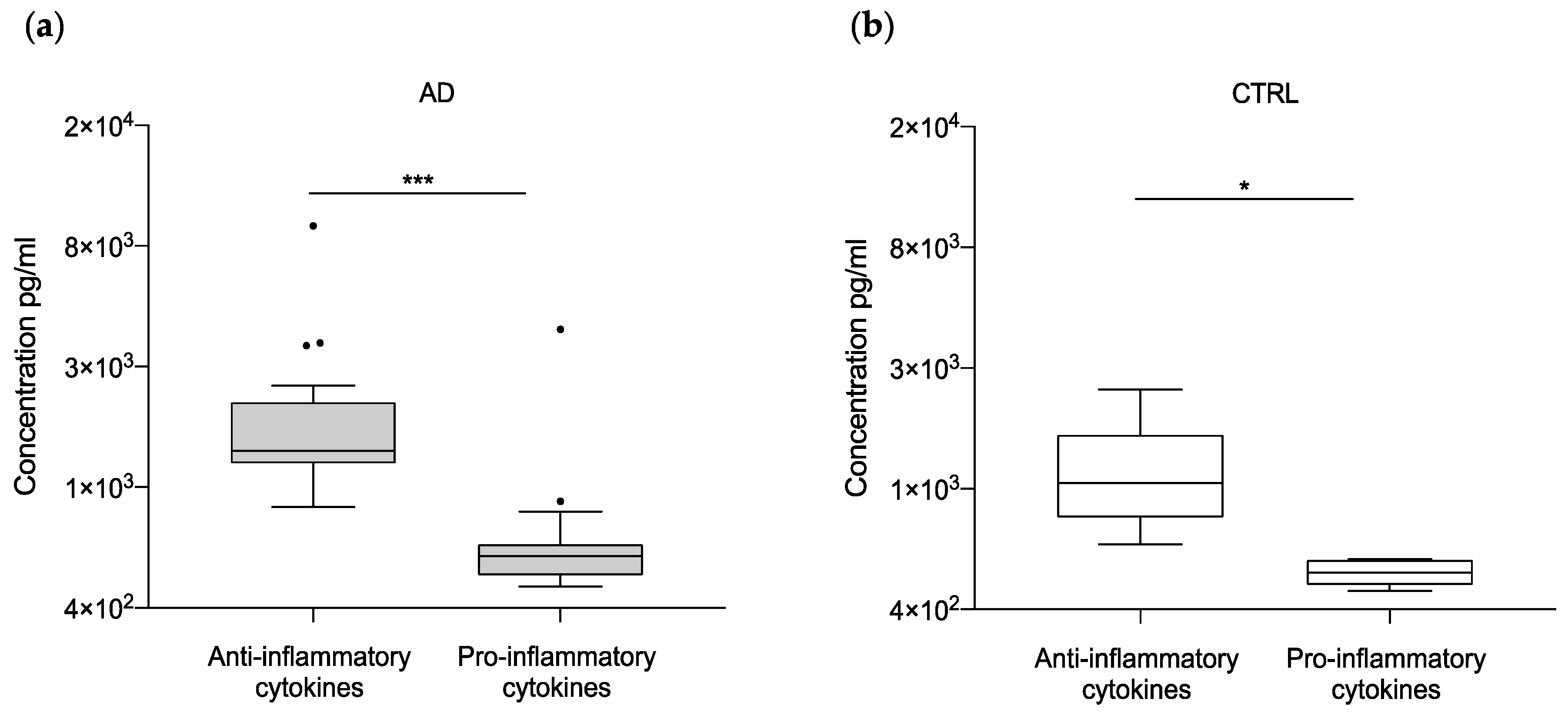
2.2. Neuroinflammatory Cytokines in AD Patients
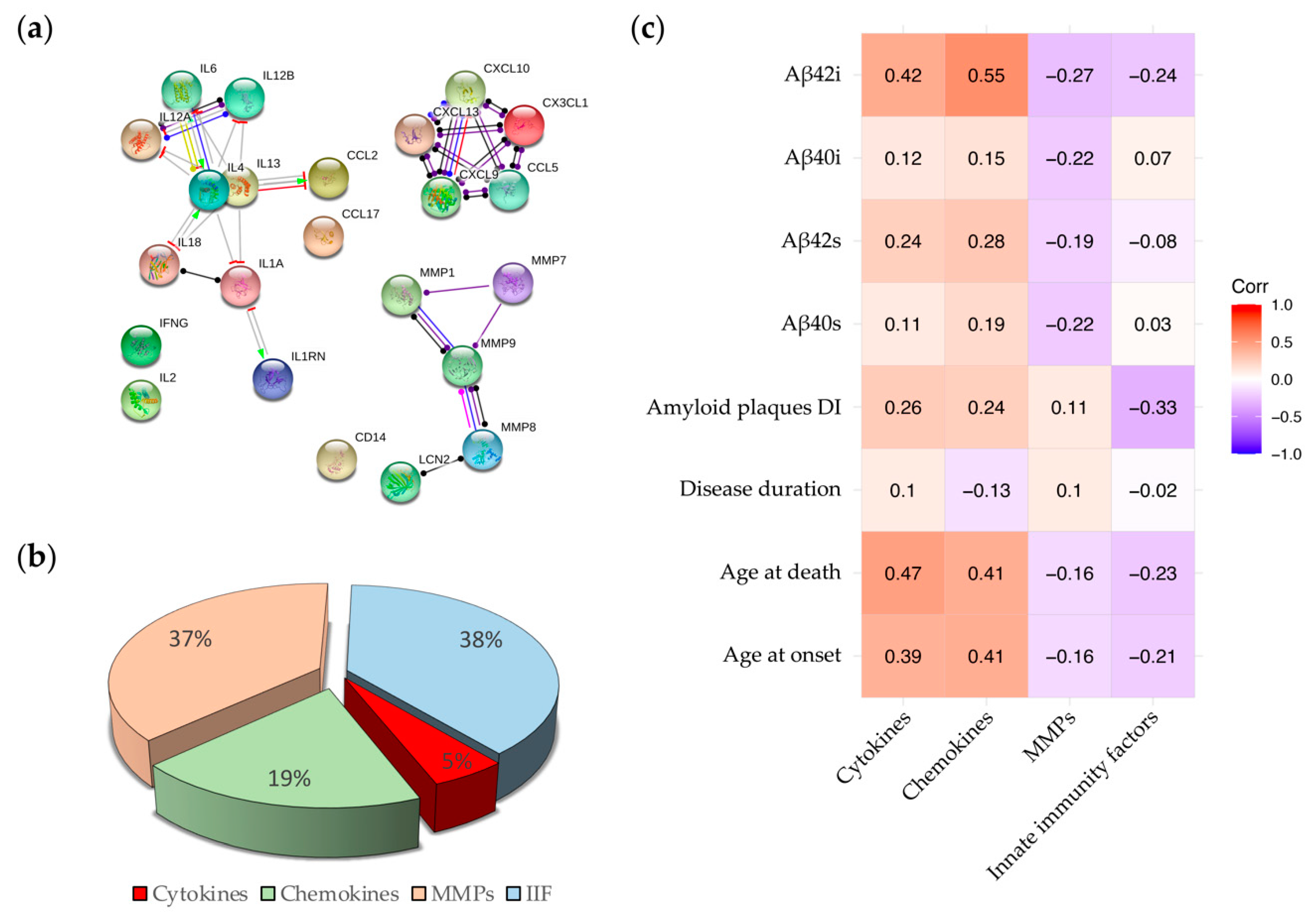
2.3. Correlations between Inflammatory Molecules and Clinical, Neuropathological and Biochemical Features of AD Cases
- the levels of Aβ in the fractionated brain homogenates are positively correlated with cytokines and chemokines and negatively correlated with MMPs and IIFs (Figure 5c). These data are in compliance with the hypothesis that the release of cytokines/chemokines is induced by the increasing production and accumulation of Aβ in AD brains [44,45,46] and suggest a possible protective role of LNC2 and CD14, composing the IIFs family, which are released by activated microglia and are associated with lower levels of Aβ in the brain.
- the DI was positively correlated with chemokines and cytokines and inversely correlated with LNC2 and CD14 (Figure 5c). So, the higher is the concentration of LNC2 and CD14 in the brain, the lower is the DI value, which implicates the presence of a lower number of amyloid deposits with a trend to form larger plaques. This result may suggest a role of the innate immunity molecules in slowing amyloidosis associated with AD.
- Among clinical parameters, age at onset and age at death are positively correlated with the levels of chemokines and cytokines (Figure 5c) while disease duration shows a very weak correlation with the four subgroups of inflammatory factors.
- the correlation between the MMPs family and all the disease indicators are globally weak (Figure 5c). This weakness results from the opposite effect of MMPs. Individually analyzed, MMP-1 and MMP-8 show a robust association, positive and negative respectively, with the age at onset/death and the Aβ42i levels (Figure S3). MMP-9 shows a negative association with Aβ40 (both soluble and insoluble), and MMP-7 shows a mild/weak positive association with all the disease indicators.
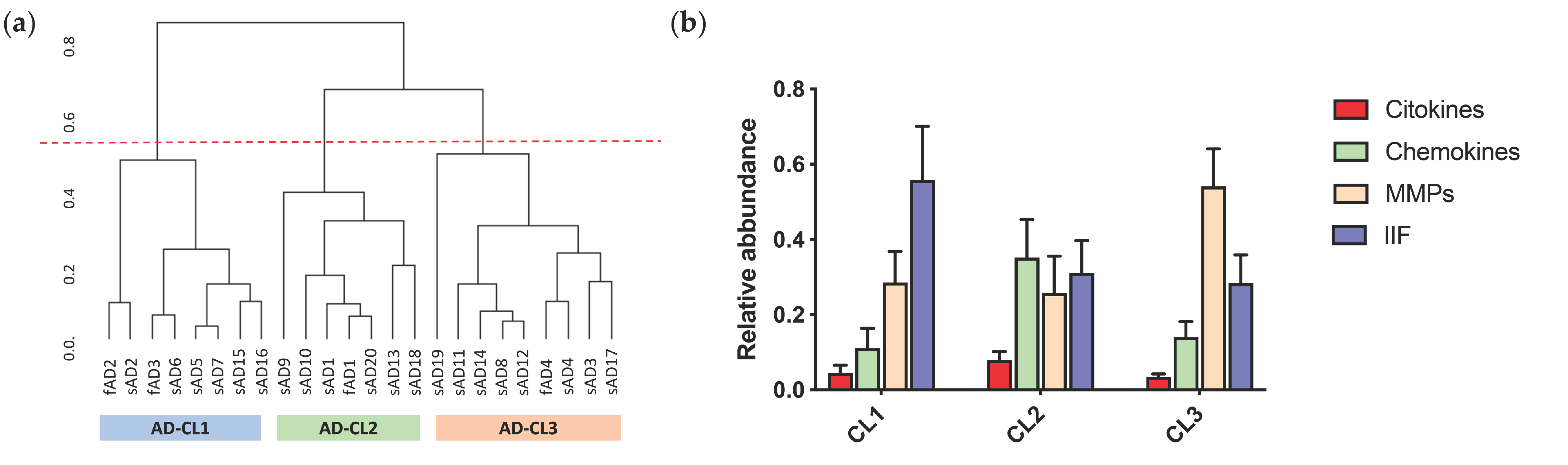
2.4. Patients Stratification and Cluster Composition
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Neuropathological Assessment
4.3. Immunohistochemistry
4.4. Image Analysis
4.5. Preparation of Brain Homogenates and Measurement of the Levels of Neuroinflammatory Cytokines
4.6. Measurement of Aβ Levels in Brain Samples
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AD-CL | Alzheimer’s Disease Cluster |
| fAD | Familiar Alzheimer’s Disease |
| sAD | Sporadic Alzheimer’s Disease |
| Aβ | Amyloid-beta |
| Aβi | Amyloid-beta insoluble |
| Aβs | Amyloid-beta soluble |
| APP | Amyloid Precursor Protein |
| CAA | Cerebral Amyloid Angiopathy |
| CNS | Central Nervous System |
| CD14 | Cluster Differentiation 14 |
| CCL | Chemokine (C-C motif) ligand |
| CXCL | Chemokine (C-X-C motif) ligand |
| CX3CL | Chemokine (C-X3-C motif) ligand |
| DI | Dispersion index |
| HCA | Hierarchical Cluster Analysis |
| IFN-γ | Interferon-gamma |
| IHC | Immunohistochemistry |
| IIF | Innate immunity Factor |
| IL | Interleukin |
| IL-1rn | Interleukin-1 receptor antagonist |
| LCN2 | Lipocalin-2 |
| LPS | Lipopolysaccharides |
| MMP | Matrix Metalloproteinase |
| MS | Multiple Sclerosis |
| OOR | Out of range |
| p-tau | Hyper-phosphorylated tau |
| PS1 | Presenilin-1 |
| PPI | Protein-Protein Interaction |
| TNFα | Tumor necrosis factor-alpha |
References
- Morris, J.C. Early-stage and preclinical Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2005, 19, 163–165. [Google Scholar] [PubMed]
- Khoury, R.; Grossberg, G.T. Deciphering Alzheimer’s disease: Predicting new therapeutic strategies via improved understanding of biology and pathogenesis. Expert Opin. Ther. Targets 2020, 24, 859–868. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Crosswell, A.; van Engen, C.; Limper, M.; Hoozemans, J.J.M.; Veerhuis, R.; van Gool, W.A.; Rozemuller, J.M. Neuroinflammation in Early Stages of Alzheimer’s Disease. In Advances in Alzheimer’s and Parkinson’s Disease; Springer: New York, NY, USA, 2007; pp. 113–121. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Hoozemans, J.J.M.; Veerhuis, R.; Rozemuller, J.M.; Eikelenboom, P. Neuroinflammation and regeneration in the early stages of Alzheimer’s disease pathology. Int. J. Dev. Neurosci. 2006, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Latta, C.H.; Brothers, H.M.; Wilcock, D.M. Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience 2015, 302, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; McGinnis, S.M.; Xia, C.; Price, B.H.; Atri, A.; Murray, M.E.; Mendez, M.F.; Wolk, D.A. Approach to atypical Alzheimer’s disease and case studies of the major subtypes. In Proceedings of the CNS Spectrums; Cambridge University Press: Cambridge, UK, 2017; Volume 22, pp. 439–449. [Google Scholar]
- Mattsson, N.; Schott, J.M.; Hardy, J.; Turner, M.R.; Zetterberg, H. Selective vulnerability in neurodegeneration: Insights from clinical variants of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Lemmin, T.; Stöhr, J.; Nick, M.; Wu, Y.; Maxwell, A.M.; Watts, J.C.; Caro, C.D.; Oehler, A.; Keene, C.D.; et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E782–E791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fede, G.; Catania, M.; Maderna, E.; Ghidoni, R.; Benussi, L.; Tonoli, E.; Giaccone, G.; Moda, F.; Paterlini, A.; Sorrentino, S.; et al. Molecular subtypes of Alzheimer ’ s disease. Sci. Rep. 2018, 8, 3269. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, C.L.; Daugs, I.D.; Spina, S.; Vidal, R.; Kokjohn, T.A.; Patton, R.L.; Kalback, W.M.; Luehrs, D.C.; Walker, D.G.; Castaño, E.M.; et al. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol. Neurodegener. 2008, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.D.; Fletcher, P.D.; Golden, H.L. The paradox of syndromic diversity in Alzheimer disease. Nat. Rev. Neurol. 2012, 8, 451–464. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef] [PubMed]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Villacampa, N.; Heneka, M.T. Microglia in Alzheimer’s disease: Local heroes! J. Exp. Med. 2020, 217, e20192311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2019; Volume 2034, pp. 57–67. [Google Scholar]
- Raivich, G. Like cops on the beat: The active role of resting microglia. Trends Neurosci. 2005, 28, 571–573. [Google Scholar] [CrossRef]
- Abbas, N.; Bednar, I.; Mix, E.; Marie, S.; Paterson, D.; Ljungberg, A.; Morris, C.; Winblad, B.; Nordberg, A.; Zhu, J. Up-regulation of the inflammatory cytokines IFN-γ and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APPSWE transgenic mice. J. Neuroimmunol. 2002, 126, 50–57. [Google Scholar] [CrossRef]
- Asari, Y.; Majima, M.; Sugimoto, K.; Katori, M.; Ohwada, T. Release site of TNFα after intravenous and intraperitoneal injection of LPS from Escherichia coli in rats. Shock 1996, 5, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Wang, D.B.; Dayton, R.D.; Zweig, R.M.; Klein, R.L. Transcriptome analysis of a tau overexpression model in rats implicates an early pro-inflammatory response. Exp. Neurol. 2010, 224, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuena, A.R.; Casolini, P.; Lattanzi, R.; Maftei, D. Chemokines in Alzheimer’s disease: New insights into prokineticins, chemokine-like proteins. Front. Pharmacol. 2019, 10, 622. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-X.; Tan, M.-S.; Yu, J.-T.; Tan, L. Matrix Metalloproteinases and Their Multiple Roles in Alzheimer’s Disease. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Jha, M.K.; Lee, S.; Park, D.H.; Kook, H.; Park, K.G.; Lee, I.K.; Suk, K. Diverse functional roles of lipocalin-2 in the central nervous system. Neurosci. Biobehav. Rev. 2015, 49, 135–156. [Google Scholar] [CrossRef]
- Yu, Y.; Ye, R.D. Microglial Aβ Receptors in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2014, 35, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Paresce, D.M.; Ghosh, R.N.; Maxfield, F.R. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 1996, 17, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Giaccone, G.; Morbin, M.; Moda, F.; Botta, M.; Mazzoleni, G.; Uggetti, A.; Catania, M.; Moro, M.L.; Redaelli, V.; Spagnoli, A.; et al. Neuropathology of the recessive A673V APP mutation: Alzheimer disease with distinctive features. Acta Neuropathol. 2010, 120, 803–812. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [Green Version]
- Levtova, N.; Healy, L.M.; Gonczi, C.M.C.; Stopnicki, B.; Blain, M.; Kennedy, T.E.; Moore, C.S.; Antel, J.P.; Darlington, P.J. Comparative morphology and phagocytic capacity of primary human adult microglia with time-lapse imaging. J. Neuroimmunol. 2017, 310, 143–149. [Google Scholar] [CrossRef]
- Rossi, G.; Giaccone, G.; Maletta, R.; Morbin, M.; Capobianco, R.; Mangieri, M.; Giovagnoli, A.R.; Bizzi, A.; Tomaino, C.; Perri, M.; et al. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar] [CrossRef]
- Spittau, B. Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef]
- Chen, A.; Oakley, A.E.; Monteiro, M.; Tuomela, K.; Allan, L.M.; Mukaetova-Ladinska, E.B.; O’Brien, J.T.; Kalaria, R.N. Multiplex analyte assays to characterize different dementias: Brain inflammatory cytokines in poststroke and other dementias. Neurobiol. Aging 2016, 38, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertig, U.; Hanisch, U.K. Microglial diversity by responses and responders. Front. Cell. Neurosci. 2014, 8, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Bai, F.; Zhang, Z. Inflammatory Cytokines and Alzheimer’s Disease: A Review from the Perspective of Genetic Polymorphisms. Neurosci. Bull. 2016, 32, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflamm. 2005, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Lue, L.F.; Walker, D.G.; Rogers, J. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; et al. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2020, 17, 157–172. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Fernández-Arjona, M.D.M.; Grondona, J.M.; Granados-Durán, P.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglia Morphological Categorization in a Rat Model of Neuroinflammation by Hierarchical Cluster and Principal Components Analysis. Front. Cell. Neurosci. 2017, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, C.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; Knop, A.; Paza, E.; Fidzinski, P.; Kraus, L.; Snijders, G.J.L.; Kahn, R.S.; et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 2019, 22, 78–90. [Google Scholar] [CrossRef]
- Bauer, J.; Strauss, S.; Schreiter-Gasser, U.; Ganter, U.; Schlegel, P.; Witt, I.; Yolk, B.; Berger, M. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 1991, 285, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; O’Banion, M.K. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Carlson, T.J.; Olschowka, J.A.; Kyrkanides, S.; Matousek, S.B.; O’Banion, M.K. Chronic Interleukin-1 Expression in Mouse Brain Leads to Leukocyte Infiltration and Neutrophil-Independent Blood Brain Barrier Permeability without Overt Neurodegeneration. J. Neurosci. 2007, 27, 9301–9309. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Barreto, G.E.; Rashid, M.; Perveen, A.; Ashraf, G.M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int. Immunopharmacol. 2020, 84, 106479. [Google Scholar] [CrossRef]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef]
- Rubenstein, J.L.; Wong, V.S.; Kadoch, C.; Gao, H.-X.; Barajas, R.; Chen, L.; Josephson, S.A.; Scott, B.; Douglas, V.; Maiti, M.; et al. CXCL13 plus interleukin 10 is highly specific for the diagnosis of CNS lymphoma. Blood 2013, 121, 4740–4748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupprecht, T.A.; Plate, A.; Adam, M.; Wick, M.; Kastenbauer, S.; Schmidt, C.; Klein, M.; Pfister, H.-W.; Koedel, U. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J. Neuroinflamm. 2009, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellebjerg, F.; Bornsen, L.; Khademi, M.; Krakauer, M.; Olsson, T.; Frederiksen, J.L.; Sorensen, P.S. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 2009, 73, 2003–2010. [Google Scholar] [CrossRef]
- Huber, A.K.; Irani, D.N. Targeting CXCL13 During Neuroinflammation. Adv. Neuroimmune Biol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.; Piccio, L.; Mikesell, R.J.; Klawiter, E.C.; Parks, B.J.; Naismith, R.T.; Cross, A.H. CXCL13 is a biomarker of inflammation in multiple sclerosis, neuromyelitis optica, and other neurological conditions. Mult. Scler. J. 2013, 19, 1204–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ke, K.-F.; Liu, Z.; Qiu, Y.-H.; Peng, Y.-P. Th17 Cell-Mediated Neuroinflammation Is Involved in Neurodegeneration of Aβ1-42-Induced Alzheimer’s Disease Model Rats. PLoS ONE 2013, 8, e75786. [Google Scholar] [CrossRef]
- Dodel, R.; Balakrishnan, K.; Keyvani, K.; Deuster, O.; Neff, F.; Andrei-Selmer, L.-C.; Roskam, S.; Stuer, C.; Al-Abed, Y.; Noelker, C.; et al. Naturally Occurring Autoantibodies against -Amyloid: Investigating Their Role in Transgenic Animal and In Vitro Models of Alzheimer’s Disease. J. Neurosci. 2011, 31, 5847–5854. [Google Scholar] [CrossRef] [Green Version]
- Szabo, P.; Mujalli, D.M.; Rotondi, M.L.; Sharma, R.; Weber, A.; Schwarz, H.-P.; Weksler, M.E.; Relkin, N. Measurement of anti-beta amyloid antibodies in human blood. J. Neuroimmunol. 2010, 227, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix metalloproteinase-9 degrades amyloid-?? fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miners, J.S.; Baig, S.; Palmer, J.; Palmer, L.E.; Kehoe, P.G.; Love, S. Aβ-degrading enzymes in Alzheimer’s disease. Brain Pathol. 2008, 18, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Ren, Y.; Liu, C.C.; Kurti, A.; Baker, K.E.; Bu, G.; Asmann, Y.; Fryer, J.D. Lipocalin-2 protects the brain during inflammatory conditions. Mol. Psychiatry 2018, 23, 344–350. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef]
- Fassbender, K.; Walter, S.; Kühl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlhauser, F.; Liu, Y.; Ulmer, A.J.; et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2004, 18, 203–205. [Google Scholar] [CrossRef]
- Liu, Y.; Walter, S.; Stagi, M.; Cherny, D.; Letiembre, M.; Schulz-Schaeffer, W.; Heine, H.; Penke, B.; Neumann, H.; Fassbender, K. LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 2005, 128, 1778–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.G.; Ding, J.Q.; Chen, S. Di Microglia in the aging brain: Relevance to neurodegeneration. Mol. Neurodegener. 2010, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Baranello, R.; Bharani, K.; Padmaraju, V.; Chopra, N.; Lahiri, D.; Greig, N.; Pappolla, M.; Sambamurti, K. Amyloid-Beta Protein Clearance and Degradation (ABCD) Pathways and their Role in Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Rosenberg, G.A. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015, 1623, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-J.; Han, J.E.; Woo, M.-S.; Shin, J.A.; Park, E.-M.; Kang, J.L.; Moon, P.G.; Baek, M.-C.; Son, W.-S.; Ko, Y.T.; et al. Matrix Metalloproteinase-8 Plays a Pivotal Role in Neuroinflammation by Modulating TNF-α Activation. J. Immunol. 2014, 193, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.S.; Lim, H.K.; Lee, J.Y.; Kim, D.J.; Park, S.; Lee, C.; Lee, C.U. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2008, 436, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, W.; Guo, Y.; Xu, J.; Yin, M. CX3CL1/CX3CR1 in Alzheimer’s Disease: A Target for Neuroprotection. Biomed. Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Monsell, S.E.; Beach, T.G.; Bigio, E.H.; Bu, Y.; Cairns, N.J.; Frosch, M.; Henriksen, J.; Kofler, J.; Kukull, W.A.; et al. Multisite assessment of NIA-AA guidelines for the neuropathologic evaluation of Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Maderna, E.; Cattaneo, C.; Cacciatore, F.; Catania, M.; Di Fede, G.; Tagliavini, F.; Giaccone, G. Divergent cognitive status with the same braak stage of neurofibrillary pathology: Does the pattern of amyloid-β deposits make the difference? J. Alzheimer’s Dis. 2015, 43, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Linkov, F.; Ferris, R.L.; Yurkovetsky, Z.; Marrangoni, A.; Velikokhatnaya, L.; Gooding, W.; Nolan, B.; Winans, M.; Siegel, E.R.; Lokshin, A.; et al. Multiplex analysis of cytokines as biomarkers that differentiate benign and malignant thyroid diseases. Proteom. Clin. Appl. 2008, 2, 1575–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.r-project.org/ (accessed on 4 July 2020).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitchison, J. The Statistical Analysis of Compositional Data; Blackburn Press: Caldwell, NJ, USA, 2003; ISBN 9781930665781. [Google Scholar]
- Everitt, B.; Hothorn, T. An Introduction to Applied Multivariate Analysis with R: Use R! Springer Science+Business Media, LLC: New York, NY, USA, 2011. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Cases μ1/2 | Controls μ1/2 | Rate | p-Values | |
|---|---|---|---|---|---|
| IL-1rn | 744.120 | 707.42 | 1.051 | 0.4316 | |
| IL-4 | 72.155 | 50.18 | 1.578 | 0.0357 | * |
| IL-13 | 706.18 | 514.71 | 1.372 | 0.0050 | ** |
| IFN-γ | 51.54 | 48.13 | 1.071 | 0.5682 | |
| IL-1ɑ | 9.67 | 8.67 | 1.115 | 0.1261 | |
| IL-2 | 332.09 | 296.64 | 1.119 | 0.0584 | |
| IL-6 | 5.85 | 2.11 | 2.770 | 0.0183 | * |
| IL-12 p70 | 134.48 | 124.08 | 1.083 | 0.4364 | |
| IL-18 | 66.68 | 62.27 | 1.071 | 0.0863 | |
| CCL2/MCP1 | 103.29 | 54.43 | 1.898 | 0.0971 | |
| CCL5/RANTES | 29.19 | 15.27 | 1.912 | 0.0518 | |
| CCL17/TARC | 89.11 | 79.17 | 1.126 | 0.0447 | * |
| CX3CL1/Fractalkine | 6793.00 | 6225.31 | 1.091 | 0.4622 | |
| CXCL9/MIG | 869.11 | 493.92 | 1.760 | 0.1538 | |
| CXCL10/IP-10 | 10.98 | 6.49 | 1.693 | 0.1156 | |
| CXCL13/BLC/BCA-1 | 62.37 | 43.41 | 1.437 | 0.0008 | *** |
| MMP-1 | 226.17 | 181.96 | 1.243 | 0.0584 | |
| MMP-7 | 468.31 | 306.91 | 1.526 | 0.0041 | ** |
| MMP-8 | 5396.34 | 3516.87 | 1.534 | 0.1740 | |
| MMP-9 | 12,305.59 | 11,272.82 | 1.092 | 0.7047 | |
| CD14 | 10,364.04 | 8948.05 | 1.158 | 0.2099 | |
| Lipocalin-2/NGAL | 5585.46 | 4475.46 | 1.248 | 0.1286 |
| AD-CL1 (n = 8) | AD-CL2 (n = 7) | AD-CL3 (n = 9) | p-Value | |
|---|---|---|---|---|
| Age at onset | 56.13 (±12.06) | 67.15 (±18.48) | 60.88 (±9.21) | ns |
| Age at death | 63.38 (±12.95) | 73.86 (±15.23) | 67.3 (±9.06) | ns |
| Disease duration | 7.25 (±3.60) | 6.71 (±3.84) | 13.2 (±0.10) | ns |
| Brak stage | 5.88 (±0.22) | 4.86 (±1.12) | 5.22 (±0.97) | ns |
| DI | 144.01 (±62.67) | 175.82 (±94.83) | 159.76 (±59.72) | ns |
| CAA | 2.64 (±0.87) | 2.86 (±0.83) | 2.00 (±0.92) | ns |
| IBA1 pp intensity | 1.93 × 108 (±1.19 × 108) | 1.59 × 108 (±1.07 × 108) | 1.01 ×108 (±8.27 × 108) | ns |
| IBA1 pp number | 2.66 × 106 (±1.65 × 106) | 2.07 × 106 (±1.45 × 106) | 1.29 × 106 (±1.07 × 106) | ns |
| Aβ40s | 39.191 (±56.72) | 273.26 (±428.71) | 10.92 (±10.26) | ns |
| Aβ42s | 1.76 (±0.99) | 8.22 (±11.00) | 2.90 (±2.02) | * |
| Aβ40i | 3860.80 (±10450.27) | 45,565.88 (±102746.20) | 111.91 (±84.84) | ns |
| Aβ42i | 49.88 (±27.72) | 102.01 (±37.34) | 65.32 (±42.59) | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, S.; Ascari, R.; Maderna, E.; Catania, M.; Ghetti, B.; Tagliavini, F.; Giaccone, G.; Di Fede, G. Microglial Heterogeneity and Its Potential Role in Driving Phenotypic Diversity of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052780
Sorrentino S, Ascari R, Maderna E, Catania M, Ghetti B, Tagliavini F, Giaccone G, Di Fede G. Microglial Heterogeneity and Its Potential Role in Driving Phenotypic Diversity of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(5):2780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052780
Chicago/Turabian StyleSorrentino, Stefano, Roberto Ascari, Emanuela Maderna, Marcella Catania, Bernardino Ghetti, Fabrizio Tagliavini, Giorgio Giaccone, and Giuseppe Di Fede. 2021. "Microglial Heterogeneity and Its Potential Role in Driving Phenotypic Diversity of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 5: 2780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052780