
Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases

1
Protein Engineering, Lilly Biotechnology Center, Eli Lilly and Company, San Diego, CA 92121, USA
2
Professional Scientific Services, Eurofins Lancaster Laboratories, Lancaster, PA 17605, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(6), 2805; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062805
Submission received: 26 January 2021
/
Revised: 2 March 2021
/
Accepted: 6 March 2021
/
Published: 10 March 2021
(This article belongs to the Special Issue Biomarkers in Inflammatory and Chronic Degenerative Diseases)
Abstract
:Since 2015, 170 small molecules, 60 antibody-based entities, 12 peptides, and 15 gene- or cell-therapies have been approved by FDA for diverse disease indications. Recent advancement in medicine is facilitated by identification of new targets and mechanisms of actions, advancement in discovery and development platforms, and the emergence of novel technologies. Early disease detection, precision intervention, and personalized treatments have revolutionized patient care in the last decade. In this review, we provide a comprehensive overview of current and emerging therapeutic modalities developed in the recent years. We focus on nine diseases in three major therapeutics areas, diabetes, autoimmune, and neurological disorders. The pathogenesis of each disease at physiological and molecular levels is discussed and recently approved drugs as well as drugs in the clinic are presented.
1. Introduction
There are over 20,000 drugs in the market for treatment of human diseases [1]. Since only 2% of the available drugs were approved in the last 10 years, the US Food and Drug Administration (FDA) pioneered a few initiatives to expedite drug discovery and approval. These designations include breakthrough, fast track, priority review, and accelerated approval (Table 1). The number of the expediated approvals regulated by the Center for Drug Evaluation and Research (CDER) of FDA has been steadily rising to quickly bring medicine to patients and to encourage investment in breakthrough drugs and modalities. Small molecules and antibodies remain dominant among approvals each year and cell and gene therapies are emerging as promising novel modalities (Table 2).
1.1. Small Molecule and Antibody-Based Therapeutics
Almost 170 novel small molecules, targeting extracellular or intracellular proteins, have been approved since 2015 (Table 3) for cancer and neurological diseases such as pain, schizophrenia, and Parkinson’s disease. Over 80% of the small molecules are formulated for oral delivery for patient convenience. Monoclonal antibodies (mAbs) are the second dominant class of drugs with an investment rate of return (13%) of almost double than small molecule (7%) [2,3]. Since the approval of the first mAb Orthoclone OKT3 in 1986, over 1000 antibodies have entered the clinical trials. Forty-three of the 80 (all IgG-based) approved mAbs were granted approval in the past 5–6 years (Table 3). This remarkable increase in approval was driven by the advancement in protein engineering, development in mAb discovery platforms, and computational predictions to allows the shift from murine or chimeric to humanized/human mAbs with greater potency and reduced immunogenicity. The market value generated by therapeutic antibodies was about $166 billion in 2019. The first approved human antibody was Humira (adalimumab) targeting tumor necrosis factor (TNF) for the treatment of rheumatoid arthritis. Adalimumab was discovered by phage display technology and approved in 2002. Adalimumab remained the best-selling drug from 2015 till 2019 with the global peak sales of $19.7 billion in 2019. Keytruda (pembrolizumab), a humanized mAb approved in 2014, targets PD-1/PD-L1 and is administered to cancer patients based on their specific molecular signatures and immune phenotypes. Its global sales soared from $7 billion in 2018 to $11 billion in 2019.
Bispecific antibodies (bsAbs) engage two targets simultaneously to enhance efficacy. The two approved bsAbs are Blincyto (blinatumomab in 2014) and Hemlibra (emicizumab in 2017). Currently, over 85 bsAbs are being investigated in the clinic [4] and over 50 CD3-specific bsAbs are in early development stages for immuno-oncology [5]. Antibody drug conjugates (ADCs) have become a promising modality among cancer therapies, with eleven ADCs approved thus far. In addition to cytotoxins as payload, immune-stimulating molecules, such as Toll-like receptor (TLR) agonists and light activatable IRDye700DX, are being tested in cancer. ADCs have been gradually transitioned into bifunctional conjugated antibodies to serve beyond cell-killing in cancer. This has been achieved by the use of other payloads, such as steroid for immunological indication. AbbVie’s molecule, ABBV-3373, is leading this effort in the clinic for rheumatoid arthritis. ABBV-3373 is comprised of an anti-TNF antibody conjugated to a glucocorticoid receptor modulator. ADCs have also been utilized as a conditioning approach for patients prior to stem cell transplants or gene therapy. The CD117-directed ADC (MGTA-117) conjugated to a cytotoxin payload, developed by Magenta Therapeutics and currently in preclinical stage, aims to selectively remove disease-causing cells to lower the risk of rejecting the donor stem cells [6]. In addition, a CD45-ADC has shown promising results in achieving immune reset in preclinical models of multiple sclerosis, systemic sclerosis, and inflammatory arthritis [7].
Nanobodies (single-domain antibody fragments) are smaller in size (12–30 kDa), yet maintain target specificity of antibodies [8]. In 2019, the first nanobody-based drug, Cablivi (caplacizumab), was approved for the treatment of acquired thrombotic thrombocytopenic purpura. Caplacizumab is a bivalent von Willebrand factor (vWF)-directed nanobody inhibiting the interaction of vWF with platelets.
1.2. Peptide-Based Therapeutics
Peptide therapeutics (<40 amino acids) have the potential to combine the advantages of small molecules with antibodies/nanobodies. For example, peptides can occupy comparable surface area on the targets as antibodies, hence they can have high potency and less toxicity. At the same time, peptides can have high ligand efficiency like small molecules. Peptide therapeutics initially emerged from natural peptides, such as insulin and adrenocorticotropic hormone (ACTH) and eventually migrated to their synthetic analogs with improved pharmaceutical properties [9]. Peptides are natural ligands of many G-protein-coupled receptors (GPCRs), making the GPCRs the dominant targets for therapeutic peptide discovery. Other molecular targets include cell-surface receptors, such as cytokine receptors, extracellular domains of ion channels, and structural proteins. A small number of intracellular targets has been interrogated by cell penetrating peptides.
Over the past decades, de novo peptide discovery platforms and rational designs have been employed to discover novel peptides. As a result, new generation of peptides with improved specificity, potency, and developability have been developed [9,10]. In addition, new peptide scaffolds, such as macrocyclic, bi-cyclic, stapled helical, and α/β peptides with enhanced physicochemical properties and potential membrane permeability have emerged [10,11]. Peptide-conjugates has been a popular strategy. For example, half-life extension of peptides can be achieved by conjugations to PEG, lipid, albumin, or Fc; intracellular uptake of RNA therapeutics can be made possible by the conjugation to a cell-penetrating peptide; and targeted cytotoxicity can be induced by conjugations of target specific peptides to toxic agents [9,11,12]. As of September 2020, about 67 peptide drugs were approved in the US [9] with 11 of them approved in the past five years. Over 200 peptides are in pre-clinical development and more than 170 peptide drug candidates are in various stages of clinical trials [13,14] for diabetes, oncology, inflammation, and infectious diseases. Peptides are leading the global revenue in diabetes. In 2019, the best-selling peptide drug, Trulicity (dulaglutide) generated $4.4 billion sales globally, followed by Victoza (liraglutide) with $3.3 billion, and Ozempic (semaglutide) with $1.7 billion (Supplementary Materials Figure S1) [15,16].
Most of the approved peptide therapeutics are administrated via parenterally (e.g., injection). However, peptides can be engineered and/or formulated for local or systemic delivery via oral or intranasal routes. Analogs of vasopressin (DDAVP/desmopressin acetate), calcitonin (Ostora), insulin (Oral-lyn), somatostatin (Octreotide), parathyroid hormone (PTH) (Oral PTH (1–34)), thyroid hormone-releasing hormone (Levothyroxine), uroguanylin (Trulance/plecanatide), and glucagon-like peptide 1 (GLP-1) (Rybelsus/semaglutide) are formulated for oral administration [17,18,19]. Linzess (linaclotide) is an example of a peptide engineered for local delivery to the gastrointestinal (GI) tract with minimal systemic exposure. Rybelsus (semaglutide) is the first oral GLP-1 analog for type 2 diabetes approved in 2019. The injectable formulation of semaglutide (Ozempic) was approved in 2017. A common strategy to facilitate oral delivery is the use of excipients such as protease inhibitors and permeation enhancers [17,20]. The use of carriers such as nanoparticles, bioadhesive patches, endogenous transporters, and cell-penetrating peptides were also tried [17,18].
1.3. Cell and Gene Therapies
Personalized treatment and precision medicine are enabled through cell and gene therapies (oligonucleotides, viral vector delivery and gene editing). Currently, four cell-based therapies have been granted regulatory approval. They are PROVENGE (sipuleucel-T, 2010), Kymriah (tisagenlecleucel, 2017), Yescarta (axicabtagene ciloleucel, 2017), and Tecartus (brexucabtagene autoleucel, 2020). PROVENGE is dendritic cell-based immunotherapy for prostate cancer and the later three are CAR T therapies for the treatment of non-solid tumors. Cell-based therapies generally require a substantial number of autologous cells (e.g., stem cells and immune cells) that undergo ex vivo genetic engineering and cell expansion. Most of the active CAR Ts, in phase III, target non-solid tumors including multiple myeloma and lymphoma. Currently, over 1200 active cell-based agents are being investigated for immuno-oncology [5]. While promising, extensive characterization is required to evaluate safety, efficacy, and compatibility of cell-based therapies.
Gene therapies target protein expression at the nucleic acid level by using viral vector- and oligonucleotides-based genetic engineering and gene editing. Currently approved gene therapies are summarized in Table 4. Targeting RNA, ribosome, or translated protein in the cytosol by oligonucleotides (ONs) can modulate protein expression of hard-to-drug targets. ONs are categorized into antisense oligonucleotide (ASO), aptamer, short interfering RNA (siRNA), mRNA, ribozyme, and modified mRNA (modRNA) [11,14].
Gene editing, including zinc finger nuclease (ZFN), transcription activator-like effector nucleases (TALENs) and clustered regularly-interspaced short palindromic repeats (CRISPR) [21,22], and adenosine deaminase acting on RNA (ADAR) [23,24] offers site-specific genetic engineering. ZFN, TALENs, and CRISPR are DNA editing platforms and ADAR is an RNA-directed technology. Since cellular delivery of Cas9 protein or mRNA remains challenging, the CRISPR/Cas9 guide RNA has partnered with CAR T platform to provide a precise ex vivo DNA targeting [25,26] for treatment of blood cancers and disorders [27,28,29]. The earlier generation of gene editing, ZFN and TALENs could correct DNA mutations in mitochondria (mtDNA) by AAV delivery in animal models that CRISPR/Cas9 has not been able to achieve [30,31] due to the challenges associated with the delivery into mitochondria. A recent research study introduced a new CRISPR-free gene editing to enable precise manipulation of mtDNA. The RNA-free DddA-derived cytosine base editors (DdCBEs) utilizes an engineered interbacterial toxin (split-DddA) fused to TALE assay protein to catalyze C-G to T-A conversions in human mtDNA in vitro with high specificity [32]. Innovations in gene editing may offer advantages for base editing in cells and organelles beyond mitochondria.
The proteolysis-targeting chimera (PROTAC) technology utilizes small molecules to degrade intracellular disease-causing proteins by ubiquitin-proteasome system (UPS) machinery. Two PROTACs agents, ARV-110 and ARV-471 are under investigation in clinical studies [33,34]. Lysosomal degradation pathway such as LYTAC (lysosome-targeting chimaera) [35] for extracellular and membrane-bound proteins, AUTAC (autophagy-targeting chimera) [36] for intracellular protein and damaged organelles, and ATTEC (autophagosome-tethering compound) [37,38] for intracellular proteins have recently emerged. LYTAC is an antibody-based mannose-6-phosphate (M6P) to induce lysosomal protein degradation.
Microbiome-based therapies have been an emerging topic [39,40] in autoimmune [41,42], neurodegenerative [43,44], and oncology [45]. Microbiota may provide a unique insight in the mechanism of disease onset and progression, along with a novel therapeutic approach.
Interestingly, drugs that were approved 6–20 years ago make up the main portion of the global revenues (Figures S1–S3). In diabetes, peptide-drugs remain the dominant modality with a clear transition in focus from insulin products to incretin therapies in the recent five years. In immunology, the top selling drugs in the last five years include TNF antibodies, Humira (adalimumab) and Enbrel (etanercept), both approved almost 20 years ago. mAbs are the main molecular modality in immunology. More than half of the top-selling neurology therapeutics are against multiple sclerosis and the rest are against neuropsychiatric diseases and seizures. Ocrevus (orelizumab) is a mAb targeting multiple sclerosis that was approved in 2017 and generated $3.8 billion in 2019. Neurodegenerative diseases remain the most challenging area with very few disease-modifying drugs.
In this review, we provide a comprehensive overview on new therapeutic modalities in diabetes, autoimmune, and neurological diseases. We also outline treatment strategies used in the early 2000 s and the advancement of novel therapeutics to date. The novel entities under development today establish the trends and inspiration for the forthcoming drugs.
2. Diabetes
Diabetes, characterized by insufficient production or usage of insulin, has become a significant public health concern. Currently, 463 million people are suffering from diabetes worldwide and this number is projected to surpass 700 million by 2045 [46], causing a significant increase in annual medical expenses [47]. Diabetes mellitus can be divided into three main categories: type 1 (T1D), type 2 (T2D), and gestational diabetes. Despite different etiologies, all three types of diabetes are associated with elevated blood glucose levels. T1D is an autoimmune disorder caused by genetic, nutritional, and environmental disorders. Disease onset is associated with B-lymphocyte stimulation by autoreactive CD4+ helper cells, activation of CD8+ T cells, and polarization of M1 macrophages in the islet of the pancreas. This results in islet β-cell death, deficiency of insulin production, and lack of glucose sensing [48,49,50,51,52]. T1D is diagnosed at all ages but it mostly occurs in childhood, making it one of the most common chronic disorders in children [46]. T2D is commonly diagnosed in adults and includes about 90% of diabetes cases [46]. It is a lifelong condition caused by insulin resistance, which is insulin inadequacy to evoke the anabolic response to glucose, specifically in skeletal muscle, liver, and white adipocytes. T2D is developed when β-cell can no longer compensate for the peripheral insulin resistance, leading to overt hyperglycemia [53]. Obesity is the primary cause of insulin resistance and hence, T2D. Other risk factors include age, race, hormone, and poor lifestyle [54]. Gestational diabetes occurs during pregnancy due to hormone-change-induced insulin resistance [46,55]. In this section, we will focus on the current and ongoing innovative treatments for T1D and T2D (Figure 1).
2.1. Treatment Strategies
Traditional therapy for T1D is centered around exogenous insulin replacement and close monitoring of blood glucose and hemoglobin A1c (HbA1c) levels. The typical insulin regimens involve multiple daily injections of long-lasting insulin to maintain basal insulin levels, injections or inhalations of rapid-acting insulin to regulate post-meal blood glucose levels, and/or continuous subcutaneous insulin infusions [56,57,58]. The injectable amylin analog, Pramlintide, is the only FDA approved non-insulin treatment for T1D. Amylin is a hormone peptide that stimulates glucagon secretion and limits food intake by delaying gastric emptying [59,60]. Administered in combination with insulin, Pramlintide helps maintain optimal weight and HbA1c levels in T1D patients [61]. Insulin and diet control are also part of treatment plans for T2D and are supplemented with metformin [62], rosiglitazone [63] or rioglitazone [64] to increase insulin sensitivity; with glibenclamide, gliclazide [65], glipizide [66], glimepiride [67], or tolbutamide [68] to enhance insulin production; with α-glucosidase inhibitor acarbose [69] or miglitol [70] to slow down carbohydrate digestion; with sodium-glucose transporter 2 (SGLT2) inhibitors canagliflozin [71,72], dapagliflozin [73] or empagliflozin [74] to block glucose circulation from kidney to blood; with incretin and analogs including exenatide, lixisenatide, liraglutide, albiglutide, semaglutide, dulaglutide [75,76,77]; or with dipeptidyl peptidase-4 (DPP-4) inhibitors alogliptin [78], sitagliptin [79], saxagliptin [80], or linagliptin [81,82] to prevent incretin degradation.
Thus far, insulin and its analogs have demonstrated superior efficacy for the treatment of T1D and T2D [53,56,83,84,85] compared to other drugs [16,86]. In 2018, the insulin sale was worth S21.3 billion globally and accounted for 43.7% of the diabetes drug market [87]. While highly efficacious, the disadvantages are also substantial. Besides weight gain, insulin therapy increases the risk of hypoglycemia, which can be life-threatening [46,88]. Some patients may develop insulin antibodies over time [89]. Insulin therapy also affects the lifestyle of patients and caregivers since the blood glucose level and carbohydrate intake should be monitored closely. Medical training is necessary for daily insulin injections. Patients with low income or from underdeveloped countries and regions may have limited access to insulin [46,90]. These problems were quickly reflected in the drug market. In the U.S., the bestselling medicine for diabetes was insulin glargine (Lantus, Sanofi) with a revenue of over $7 billion in 2015. Lantus sales were dropped to $3 billion in 2019 with the advent of incretin analog Trulicity (dulaglutide, Eli Lilly) and DPP-4 inhibitor Januvia (sitagliptin, Merck) [16,86]. T1D patients still rely on insulin treatment, however, incretin therapy was established as a new trend for T2D treatment. Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the two Incretin hormone family members. Incretin therapy slows down gastric emptying, preserves insulin secretion (insulinotropic effects), suppresses glucagon secretion (glucagonostatic effects) [91], and is associated with a much lower risk of hypoglycemia [76]. The biggest drawback of incretin therapy was the short half-life. The native GLP-1 degrades within 1.5–5 min in plasma [92] and the first incretin drug exenatide with a half-life of 2.4 h requires twice-daily injections. Trulicity overcame this problem by linking two GLP-1 peptides to a human immunoglobulin Fc fragment. The result was superior efficacy and extended half-life (4 days), requiring once-weekly injection [75,76,77]. The next-generation incretin therapies include oral GLP-1R agonist called Rybelsus (semaglutide, Novo Nordisk) [93,94] and the investigational dual GIP-R and GLP-1R receptor agonist tirzepatide (Eli Lilly). Tirzepatide has shown high efficacy in glucose control, and weight loss in clinical studies [95,96,97]. The clinical phase III data, communicated in a press release in December 2020, indicated that 51.7% of the participants can reach normal HbA1c levels with the highest dose of tirzepatide [98]. Tirzepatide was engineered from the native GIP sequence [95] and has a five-fold higher affinity to GIP-R than GLP-1R [99]. A recent publication has suggested that tirzepatide activates GIP and GLP-1 receptors differently. By investigating the downstream signaling molecules, the authors show that tirzepatide resembles GIP at GIP-R but has a biased agonism against GLP-1R in favor of cAMP generation over β-arrestin recruitment. In fact, the observed pharmacology of tirzepatide, such as enhanced insulin secretion, was linked to its imbalanced activity against GIP-R and biased activity towards GLP-1R [99]. Tirzepatide is associated with fewer gastrointestinal adverse effects that are commonly occurring during GLP-1R agonism but has never been reported with GIP-R activity [100]. GIP-R activation may provide additional benefits, including the increased lipid buffering ability governed by the adipocytes in white adipose tissue and the suppressed appetite for bodyweight reduction [101]. Tirzepatide is linked to a C20 unsaturated di-acid acyl chain achieving a weekly injection dosing regimen [99].
The significance of bias agonism was also investigated in preclinical research. A GLP-1R biased agonist, P5 (peptide sequence: ELVDNAVGGDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS), was selected from an autocrine-based library in which random sequences were added to the N-terminus of Exendin 9–39 [102]. P5 triggers the calcium signal transduction similar to GLP-1 and Exendin-4 in cells but recruits β-arrestin 1 much weaker. It is important to note that β-arrestin activation results in receptor internalization and desensitization [103]. Consequently, P5 demonstrated an enhanced GLP-1R-dependent glucose tolerance in mice with just a single dose of treatment. In the chronic setting, P5 treatment also displayed a more efficient blood glucose control in the diabetic mouse model than Exendin-4. Two mechanisms may be involved in such an effect. First, the P5 treatment might have induced upregulation of the insulin sensitivity-related genes, such as proliferator-activated receptor gamma (PPARγ), Glut4, CD36, and tumor necrosis factor-α (TNF-α). Second, PPARγ might have escalated GIP-R expression during adipocyte differentiation [104]. GIP circulation was also improved with the P5 treatment in mice. GIP is known to inspect insulin levels in adipocytes and the GIP-R to GLP-1R ratio has been reported to be related to insulin resistance [105]. Therefore, fine-tuning the agonism between incretin receptor expression as well as their downstream signaling might be a new path forward for diabetes therapy.
Glucagon, a peptide hormone produced by α-cells, plays a key role in diabetes pathology [106,107] by raising glucose concentration in the blood for immediate management of hypoglycemia. The only dry nasal glucagon spray in the market, Baqsimi (glucagon, Eli Lilly), has been approved for treating severe hypoglycemia for four-year-old patients or older [108]. Combination of GLP-1, GIP, and glucagon agonism has shown optimal weight and glycemic control in mice and could cause fewer complications, such as hepatosteatosis and dyslipidemia [109,110,111].
2.2. Disease Modifying Strategies
Thus far, all commercially available treatments provide remedies to compensate for the impaired insulin production or utilization. Next, we summarize the innovative approaches to cure diabetes. These therapeutics can be divided into immune therapy to control autoimmunity in T1D, cell therapy to rescues β-cell destruction, and systemic approaches that merge therapeutics and medical devices.
The goal of immune therapy for T1D is to modulate the unwanted immune response and disrupt the T-cell mediated β-cell death. The universal immunosuppressant cyclosporine was able to restore β-cell function in a sub-population of T1D. However, toxicities and side effects were also significant, preventing the continuous administration of the drug [112,113]. Although not fully successful, this finding highlighted the possibility of immune system modulation as a treatment for T1D. Consequently, the focus was turned to target T1D-related autoantigens, such as glutamic acid decarboxylase (GAD) and hsp60 p277 peptide. This method was thought to cause fewer adverse events than universal immunosuppressants. GAD was expected to be a major autoantigen target for T1D, as the anti-GAD level correlates with β-cell destruction and serves as a biomarker for the disease progression [114,115]. Injecting GAD to the T helper 2 (Th2) deficient non-obese diabetic (NOD) mice induced GAD-specific Th2 immune reaction and halted disease development [116,117,118]. However, treatment with aluminum hydroxide GAD failed to regulate insulin production in two separate clinical phase II trials, although the elevated GAD antibody titers were observed [119,120]. The disconnection between preclinical and human studies might have been due to the lack of continuity among treatment regimens, including timing, route, and dose. In particular for this case, the animal models were treated with GAD before the full development of T1D, while it is hard to do so for humans. Therefore, the efficacy of autoantigen-targeting drugs and the treatment regimen need to be further optimized for higher potency and less adverse events [120,121,122,123,124]. Alternatively, drugs targeting non-autoantigen, such as CD3, have made significant progress in preventing the onset of T1D.
CD3 is a T cell co-receptor that plays a role in antigen recognition. CD3 antibody caps the T-cell receptor (TCR)-CD3 complexes on the regulatory T cells (Tregs), resulting in internalization or shedding of the complex. This process temporarily creates silent T cells temporarily and pauses the immune response. In the activated effector T cells, the antibody-CD3 interaction triggers apoptotic signal cascade instead, resulting in the depletion of about 25% of T cells [125]. Teplizumab, a humanized anti-CD3 mAb, was shown to rescue insulin production and improve HbA1c levels in several clinical studies [126,127,128,129,130]. The commercialization of teplizumab had a setback due to the unfavorable result of a one-year randomized phase III clinical trial [131]. In this study, patients were administered with six or fourteen days of low or full dose teplizumab. A year later, the percentage of patients who required less than 0.5 U/kg insulin per day and had HbA1C under 6.5% was determined. No difference was observed between the treated versus placebo groups [130]. However, in a follow-up phase III study with the same patient population, the patients who were treated with fourteen days of full-dose teplizumab showed improved C-peptide after two years compared to the placebo group [128]. Moreover, in 2019, a phase II clinical study showed that teplizumab treatment slowed T1D progression by two years in the T1D high-risk population [132]. FDA granted Breakthrough therapy designation to teplizumab as the first immune therapy for T1D [133]. Another monoclonal CD3 antibody, otelixizumab, was shown to restore β-cell function and reduce insulin dose during the T1D onset. Unfortunately, adverse events, including Epstein–Barr virus (EBV) infection and acute mononucleosis-like symptoms was observed, possibly caused by cytokine release change and abnormal CD8+ response [134]. Two phase III study was conducted using low doses of otelixizumab and no efficacy was detected [135,136].
Additional drugs for suppressing T-cell immune response were tested for treating T1D. These include CD20 antibody rituximab, CD2 antibody [137], and CD80 and CD86 co-antibody abatacept. All the above drugs showed positive impact on insulin C-peptide stabilization in clinical trials [137,138,139]. A phase II clinical study indicated that rituximab reinstated the β-cell function by depleting B-lymphocytes. However, these drugs are only effective in certain patients (non-progressors), in which the disease might have been caused by heterogeneous T cell populations. This may explain why none of the drugs has moved forward clinically as of today [140,141]. However, preclinical research indicated that CD19 is upregulated in NOD mice and induces invasive insulitis by presenting the membrane associated antigen IGRP, which is critical for autoreactive T cell expansion [142]. Therefore, blocking CD19 signaling may serve as a new direction for treating T1D and its therapeutical application is worth further assessment by scientists and researchers.
Proinsulin vaccines provide a different option for treating T1D. In 2019, ActoBio Therapeutics launched a phase Ib/IIa clinical trial using an oral capsule vaccine, AG019, for the treatment of early-onset T1D. AG019 contains engineered Lactococcus lactis, which secretes human proinsulin and the inhibitory cytokine Interleukin 10 (IL-10). The vaccine demonstrated β-cell protection and enhanced T-cell regulation in preclinical studies. The efficacy and safety of AG019 will be accessed in combination with the CD3 antibody in clinical settings [143,144,145]. C19-A3, an HLA-DR4 specific proinsulin peptide, was also evaluated in a phase I study. Patients with HLA-DRB1*0401 genotype were treated for up to two months and IL-10 levels were measured. The results hinted β-cell restoration but no further investigation has been initiated since then [146]. Overall, none of the current immune therapeutic approaches have been successful in achieving an exogenous insulin-free state for patients.
Pancreas transplantation is practiced for some patients suffering from diabetes mellitus [147,148,149]. However, due to the invasiveness and difficulty in finding an appropriate donor, transplant cases have been declining in the past decade [150]. Rather than replacing the whole organ, pancreatic islet transplantation, which restores the β-cell number and function, was inaugurated decades ago. The process has improved in recent years and is now considered as an effective treatment for T1D [151,152,153]. The method, Edmonton Protocol, was first reported in 2000 when seven patients received islet cells. The patients gained sufficient islet masses and could reach the insulin-independent stage as soon as twenty-nine days post-surgery [152]. Despite the encouraging outcome, Edmonton Protocol faces substantial limitations. A follow-up clinical trial with a larger number of participants showed that only 58% of patients acquired insulin independence. Moreover, 76% of patients who had reached insulin independence required exogenous insulin in two years. Partial (28%) or complete (28%) islet graft loss were also observed in all treated patients [154]. Inclusion of immunosuppressants such as sirolimus, tacrolimus, and IL-2 receptor antibody daclizumab is required in Edmonton Protocol to avoid potential adverse alloimmune and autoimmune responses [152]. Consequently, islet transplantation is only available to patients who are suffering from hypoglycemia unawareness or with serious hypoglycemia condition which cannot be controlled using conventional insulin therapy. Children are also not recommended for this procedure [153]. This emphasizes the importance of optimizing the immunosuppressive regimen for greater safety and prolonged insulin independence for islet transplantation [155,156,157,158,159,160]. Matsumoto and colleagues established a new protocol incorporating the immunosuppressant thymocyte globulin antibody, IL-1β antibody Anakinra, and TNFα antibody etanercept. Mycophenolate mofetil and tacrolimus was also used post-procedure for a lasting immunosuppression effect. Rapamycin (mTOR) inhibitor sirolimus from Edmonton Protocol was excluded for its potential role of limiting β-cell survival [161]. All patients under Matsumoto’s protocol remained insulin independent throughout the whole observation period (almost two years) [157]. In another study, T1D patients were administered exenatide and TNFα inhibitor etanercept after islet transplantation. All the patients in the treated cohort demonstrated durable insulin independence up to eighteen months compared to 20% of the untreated cohort that did not receive exenatide and etanercept [158]. Inducing tolerance for donor cells to reduce the chance of graft rejection was also considered [153,162]. In one example, diabetic cynomolgus macaques were treated with the combination of thymocyte globulin antibody and CD20 antibody (rituximab) after islet transplantation. The untreated animals showed graft rejection 6–35 days following the procedure, while the treated group did not reject the transplant and stayed diabetic free for an extended period (48–1500 days). Liver biopsies of the treated animals revealed the depletion of CD3+ and CD20+ lymphocytes might be the key for tolerance induction [163].
Insulin-secreting stem cells are under investigation for treating diabetes. The commonly used stem cells include hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), and monocyte-derived pluripotent stem cells [164,165]. A preclinical study indicated that the transplantation of allogeneic bone-marrow-derived HSCs prevented the disease progression in T1D mice model [166]. In the phase I/II clinical study, 14 of 15 enrolled T1D patients who received HSC transplantation became insulin-independent for up to 35 months [167]. Further clinical research revealed that co-treatment with immunosuppressants is essential for efficacy [168]. Nonetheless, two lengthy follow-up trials up to four years showed that the insulin-independent duration varied among individuals and the relapse rate was high [169,170]. MSCs are self-renewing stromal stem cells responsible for repairing tissues and are widely used for treating T1D and T2D [171]. In a T1D clinical study, MSCs treatment restored the β-cell function without any adverse events [172]. In a separate study, similar β-cell restoration lasted up to two years after receiving the treatment and no adverse events were reported [173]. Adipose tissue-derived MSCs can be engineered to secrete insulin. T1D patients who received co-transplantation of the insulin-releasing MSCs and HSCs showed significant improvement in glucose control and C-peptide levels up to thirty-two months [174,175,176]. Several clinical studies were performed independently to evaluate the efficacy and safety of allogeneic MSCs for treating T2D. No major adverse event was reported and the MSCs treatment demonstrated optimal levels of blood glucose, HbA1c, and insulin C-peptides [177,178,179,180,181]. Accordingly, stem cells, especially MSCs, offer a promising approach for diabetes treatment. As a result, pharmaceutical giants including Sanofi, Novo Nordisk, and Eli Lilly are investing in stem cell therapies [182]. As of today, 217 stem cell clinical trials for diabetes have been registered with 140 of them still ongoing.
Stem cell therapy requires immunosuppressants to achieve the best efficacy and avoid severe adverse events [168]. There is also no evidence that it can be a “one dose for cure” solution for diabetes. To achieve both immune modulation and β-cell restoration functions, cord blood-derived multipotent stem cell (CB-SCs) was developed. Patients’ lymphocytes were co-cultured with CB-SCs in a closed-loop system and then injected back. Mitochondria released by platelet stimulates β-cell proliferation. Therefore, the “educated” lymphocytes demonstrate continuous immune-suppression, leading to β-cell function restoration [165,183,184]. Clinical phase I/II trial results indicated that the therapy modulated immune response and improved β-cell function and metabolic controls for both T1D and T2D patients [165,185,186,187]. Additionally, CAR T cell technology with great potential in treating non-solid cancers such as B-cell lymphoma provided a treatment option for T1D. In one study, the researchers generated and expressed an insulin-specific CAR in the naïve CD4+ T effector cells. Function and phenotype markers, such as CD25, CD127, CD69 and CD62 were assessed in the induced insulin specific Tregs using flow cytometry to ensure their integrity. The induced Tregs proliferated in response to the insulin treatment in culture. Although cells remained viable for seventeen weeks following the transfer of the induced Tregs to diabetic mice, the disease continued [188]. In a separate study, CD8+ T cells were engineered to express the antigen of the antibody mAb287, which was previously reported to selectively target the pathogenic insulin B chain peptide-MHC complex. A single dose of the mAb287-CAR T cell infusion delayed the onset of diabetes by eighteen weeks in the T1D mouse model but the protection dropped over time and disappeared in thirty weeks [189,190].
With the advance of cell therapy, it is now possible to make the islet-like 3D-cell clusters that release insulin based on glucose levels. A recent preclinical study showed that the glucagon releasing islet α-cells isolated from non-diabetic human donors could be transduced into β-cells by using β-cell-specific transcription factors. The transduced β-cells released insulin in a glucose-dependent fashion and could be reaggregated to form monotypic ‘pseudo-islets’. The transplanted pseudo-islets reversed the disease in the diabetic mouse model and maintained the insulin-releasing function for six months [191]. A separate study showed that the induced pluripotent stem cells could be differentiated and matured into islet-like cells. Islet-like organoids expressed β-cell transcription factors and secreted an array of hormones, including insulin, somatostatin, and pancreatic polypeptide. Insulin secretion corresponded with glucose concentration. The organoids demonstrated glucose control function in the diabetic mouse model [192].
The artificial pancreas is a medical device that functions like a healthy pancreas. It is composed of a glucose monitoring system and an infusion pump. A pre-programmed algorithm allows the release of insulin or insulin plus glucagon based on the glucose level. The artificial pancreas device system (APDS) is expected to provide tighter glucose control and avoid the occurrence of hypoglycemia and hyperglycemia [193,194]. In 2016, the first hybrid closed loop APDS was approved for T1D patients and its use resulted in reduced HbA1c. Patients had to input meal information manually and the device provided basal insulin [193,195]. FDA approved the second-generation artificial pancreas in 2019 with a more accurate algorithm. In a recently conducted clinical study, the new generation of APDS demonstrated better glucose management for T1D patients by keeping their glucose levels within the targeted range (70–180 mg/dL) for a longer time [196], making the device the most advanced technology for T1D treatment thus far [197]. Next generation APDS should include a more precise and real-time insulin-releasing algorithm and insulin-glucagon co-treatment formulation.
Gene therapy, which utilizes the plasma DNA or virus to induce insulin expression, could offer an alternative therapy for diabetes. In one study, intramuscular injection of rat proinsulin plasma DNA in mice induced continuous insulin secretion by the skeleton muscle. Mice treated with the proinsulin plasma DNA showed less fatality comparing to the control DNA group in the β-cell toxin streptozotocin-induced diabetic mouse model [198]. In another study, an insulin-expressing adenovirus was delivered to the streptozotocin-induced mouse model. The insulin expression was regulated by the hepatocyte-specific and glucose-sensing promoter. The treated mice secreted insulin in the liver and maintained normal blood glucose levels for over thirty days [199]. Gene therapy for diabetes is promising but research remains in the preclinical space possibly due to ethical and long-term safety concerns.
The gut microbiome has become a rising trend in preclinical and clinical research for treating diabetes. It was reported that treating the NOD mice with vancomycin not only altered the microbiota composition, but also decreased the number of IL-17- and IFNγ-producing T cells (in male mice); thus increasing the risk for developing T1D [200,201]. In another study, knocking out the myeloid differentiation primary response 88 (MYD88) gene, known as the innate immune response adaptor, in NOD mice prevented T1D development, whereas the same knockout mice raised in a germ-free environment remained diabetic [202]. Taxonomic research also indicated that the microbiota composition was directly related to the T1D risk in children [203,204,205]. Microbiota dysbiosis is also correlated with the onset of T2D [206,207,208]. Several clinical studies revealed that metformin treatment in T2D patients introduced a prompt microbiota composition change and gut bacteria proliferation [209,210,211]. The germ-free mice inoculated with human fecal microbiota after metformin treatment exhibited a reduced level of HbA1c [211]. The microbiota composition was also altered in T2D patients who received the GLP-1R agonist liraglutide and was different from the metformin-treated patients [210,212]. Lastly, it is suggested that high fiber diets may help alleviate insulin resistance, possibly due to alteration in the microbiota population in favor of good bacteria [213]. As a result, microbiota therapy is being considered as a treatment option for diabetes [214,215].
3. Autoimmune Diseases
Autoimmune disease occurs when the immune system does not distinguish between foreign and self, causing an immune-system imbalance. The pathogenesis of autoimmune diseases is usually characterized by the expression of autoantibodies, pro-inflammatory cytokines, and autoreactive T cells [216,217]. Autoimmunity has a global incidence ranging from 5–500 per 100,000 cases per year [218]. Therapeutic mAbs and small molecules have dominated the market. Other novel modalities, such as siRNA- and microbiome-based therapies, might also play major roles in not only alleviating symptoms but potentially providing a cure. In this section, traditional and new targets and modalities will be discussed for rheumatoid arthritis, atopic dermatitis, and Crohn’s disease (Figure 2).
3.1. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a systemic autoimmune disease that causes painful inflammation of the joints, damage of the cartilage and bone, and extra-articular manifestations including heart, lungs, and blood vessels [219]. The articular and systemic comorbidities can lead to a poor quality of life and choric long-term effects such as disability. According to the WHO, the global disease prevalence is between 0.3% and 1% of the population [220]. In the US, RA is the third most common type of arthritis affecting over 1.5 million people [221].
It is known that environmental and genetic factors play significant roles in the pathogenesis of the disease. Environmental factors such as smoking, diet, infectious agents, and perturbed gut microbiome have been shown to trigger the disease development and progression in genetically predisposed individuals [222,223]. Genome-wide association studies (GWAS) have linked 117 loci with RA risk [224] and epigenetic factors, such as DNA methylation and microRNAs, contribute to the pathogenesis [223]. In particular, the HLA-DRB1 locus in the major histocompatibility complex class II gene is considered a dominant contributor to the disease [222,225,226]. RA patients with the shared epitope (QKRAA) in the HLA-DRB1 region have shown a high prevalence of autoantibodies, such as rheumatoid factor (RF) and anti-citrullinated protein antibody (ACPA) [222,227,228,229]. RF are antibodies against the Fc region of the IgG molecules and cause macrophage activation and cytokine induction. ACPAs target citrullinated residues on many self-proteins and can activate macrophages or osteoclasts, resulting in bone loss [228,230]. Detection of circulating ACPAs has been considered a key advance for early diagnosis of RA. The first commercially available ACPA test, CCP2 (cyclic citrullinated peptide 2), was introduced in 2002 [231].
The role of adaptive and innate immune systems in pathogenesis of RA is well established. However, the cause of systemic loss of tolerance and localized inflammation in the joint is not completely understood. Leucocyte infiltration into the synovial joint results in synovial membrane inflammation. The accumulated immune cells include adaptive (e.g., T-cell subsets, B cells, plasmablasts, and plasma cells) and innate immune cells (e.g., monocytes, dendritic cells, mast cells, and innate lymphoid cells). Both T-helper-1 (Th1) and T-helper-17 (Th17) cells, which produce IL6, IL17A, IL17F, IL21, IL22, and tumor necrosis factor α (TNF-α) have been considered as the main drivers of the disease [232]. Elevated levels of cytokines and chemokines activate fibroblasts, macrophages, neutrophils, masts cells, and osteoclasts leading to inflammation and tissue damage. This positive feedback loop continues to mediate the release of TNF-α, granulocyte macrophage-colony stimulating factor (GM-CSF), and IL6; activates endothelial cells; and attract additional immune cells to the synovial membrane, ultimately leading to the damage of the adjacent bone and cartilage [219,233].
Several signaling pathways are involved in RA progressions, including SAPK/MAPK (stress-activated protein kinase/mitogen-activated protein kinases) and JAK/STAT (Janus kinase/signal transducers and activators of transcription pathways). It is known that TNF-α is a key initiator for SAPK/MAPK and JAK/STAT pathways. IL6 can activate SAPK/MAPK and PI3K/Akt/mTOR (phosphatidylinositol-3-kinase/AKT/mechanistic target of rapamycin) pathways [234]. JAK/STAT pathway is considered as the initial driver of the proinflammatory response in RA [235] and it consists of four receptor-associated kinases, JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2), which activate STAT family (STAT1, STAT2, STAT3, STAT5A/B, STAT6). Binding of cytokines to their receptors initiates crosstalk between cytokine receptor with JAK proteins, consequently JAK/STAT pathway is activated. Activated JAKs phosphorylate the tyrosine residues of STAT, leading to the formation of phosphorylated STAT homodimer or heterodimer. The dimers are then translocated to the nucleus to module gene expression of inflammatory molecules. More than forty different cytokines and growth factors activate specific combinations of JAK and STAT [236]. In RA, the dysregulation of JAK/STAT activity via suppressor of cytokine signaling (SOCS) cause continuous activation of JAK/STAT in synovial joints, elevates gene expression of matrix metalloproteinase (MMP) and apoptotic chondrocytes, and results in apoptosis resistance [234].
Traditionally, the first line of treatment for RA involves conventional or targeted disease-modifying antirheumatic drugs (DMARDs), including methotrexate with low doses of glucocorticoids. Other DMARDs include sulfasalazine, leflunomide, hydroxychloroquine, and chloroquine [237]. The current biological DMARDs for RA have four main modes of action: TNF inhibition, IL-6R blockade, B-cell depletion, and T-cell inhibition [219]. TNF antagonists are the first clinically successful biologics to treat RA. These including Enbrel (etanercept), Remicade (infliximab), Humira (adalimumab), CIMZIA (certolizumab pegol), and Simponi (golimumab) (Table 5). TNF plays a role in the activation of transcription factor (e.g., NF-κB), proteases (e.g., caspases), and protein kinases (e.g., JUNK/c-Jun N-terminal kinase, MAPK) through TNF receptors 1 and 2. Most TNF inhibitors either block the soluble and membrane-bound TNF interaction with TNF receptors or initiate a reverse signaling cascade leading to cell apoptosis and cytokine suppression [238]. TNF antagonists are well tolerated in 60–70% of patients experiencing long-term lasting effects, but they also have been linked to severe side effects, including infection. The remaining 30–40% of patients face primary (no response to the anti-TNF drug) or secondary failures (loss of efficacy over time), most possibly due to immunogenicity, non-adherence, and/or disease heterogeneity [239]. Two anti-TNF drug conjugates have been developed by AbbVie. ABBV-154 is an anti-TNF steroid conjugate and ABBV-3373 is an anti-TNF conjugated to glucocorticoid receptor modulator for moderate to severe RA [240].
Like TNF, IL-6 signaling plays a critical role in immune activation in RA pathogenesis. Actemra (tocilizumab) is the first IL-6R inhibitor approved for the treatment of RA in the US in 2010. Combination of tocilizumab with DMARDs provides a better efficacy profile compared to monotherapy. Another human IL-6 mAb is Plivensia (sirukumab) [241]. Sirukumab was withdrawn in 2017 due to high reports of death, infection, and malignancies [242]. Kevzara (sarilumab) is the fully human mAb against IL-6R developed by Regeneron and Sanofi. Sarilumab can block both soluble and membrane-bound IL-6R with a 15–22 fold higher affinity compared to tocilizumab [243,244]. In a meta-analysis study, the relative efficacy of three IL-6 inhibitors were compared in active RA patients who had inadequate response to TNF inhibitors or methotrexate. The study showed that 8 mg of tocilizumab as monotherapy or combined with methotrexate was the most effective treatment in such patient population [245].
B-cell depletion therapy by targeting CD20 on B cell is considered an effective therapy for RA [246]. In 2006, Rituxan (rituximab) was approved for moderate to severe RA in combination with methotrexate. Chimeric nature of rituximab resulted in increased immunogenicity. The second generation of CD20 mAbs are humanized antibodies, including ocrelizumab (phase III terminated), veltuzumab (phase II terminated), and the fully human ofatumumab (phase II terminated) [247]. Alternative targets are explored to block T cell co-stimulation. Orencia (abatacept), approved in 2011, is for the treatment of moderate to severe RA. Abatacept targets CD80/CD86 on the surface of antigen-presenting B-cells and monocytes and blocks the costimulatory signal necessary for T-cell activation [248].
Granulocyte macrophage-colony stimulating factor (GM-CSF) is an extracellular drug target for RA treatment. GM-CSF is a hemopoietic growth factor that contributes to the differentiation of myeloid cells, macrophages, and Th17 cells. It binds and activates the GM-CSF receptor, triggering downstream singling of the JAK-STAT, PI3K, MAPK, and NF-κB pathways. Both GM-CSF and GM-CSF receptor are upregulated in the synovial tissue of RA patients [249]. The first human study against GM-CSF receptor was conducted using the mAb mavrilimumab. The EARTH EXPLORER 2 phase II clinical study compared mavrilimumab and the TNF inhibitor golimumab in 138 RA patients with insufficient response to TNF inhibitors. The study showed that the RA patient with insufficient response to DMARD had a lower response rate to mavrilimumab compared to those treated with golimumab. Interestingly, mavrilimumab showed suppressed serum levels of chemokines CCL22 and CCL17, while golimumab showed suppressed levels of CXCL13 and ICAM1. Furthermore, mavrilimumab was able to induce permanent suppression of inflammatory (e.g., CRP, SAA, MMP1, MMP3, IL6, VEGF, IL2R, and CD163) and extracellular matrix markers (e.g., C1M, C3M, and P4NP7S), whereas golimumab only induced a transient change in the expression of those extracellular matrix markers [250]. Additional GM-CSF mAbs are under clinical development. Human mAb namilumab developed by Takeda, currently in phase II, has shown efficacy and safety in RA patients who had an inadequate response to methotrexate or TNF therapy [251]. GSK has also announced start of phase III trials for its GM-CSF antibody, otilimab, in patients with RA in 2019.
JAK inhibitors are carving their share in the market against TNF inhibitors within the last decade. The abnormal activation of JAK/STAT pathway is linked to the elevated levels of IL-6, IFN-γ, and TNF-α and the induction of autoimmunity. Selective JAK1, JAK2, JAK 3, TYK2, and pan-JAK inhibitors are used to interfere with RA progression. Xeljanz (tofacitinib) was the first JAK inhibitor to be approved by the FDA for the treatment of moderate to severe active RA patients who inadequately responded to methotrexate or other biological DMARDs. Although tofacitinib was designed to be a selective JAK3 inhibitor, it has been shown to also block JAK 1 and JAK2. Since then multiple selective JAK inhibitors have entered the market. In 2018, selective JAK 1/JAK2 inhibitor Olumiant (baricitinib) by Eli Lilly was approved for moderate to severe RA in the United States and Europe. Baricitinib inhibits the phosphorylation of STAT3 (pSTAT3) that is induced by IL-6 mediated signaling through JAK1, JAK2, and TYK2 complexes. Baricitinib offers several advantages over biological DMARDs including the oral dosage and efficacy as a monotherapy [234]. The selective JAK1 inhibitor Rinvoq (upadacitinib) developed by AbbVie was approved in 2019 for moderate to severe RA. In 2020, Galapagos NV/Gilead’s once-daily JAK1 inhibitor filgotinib was granted market authorization in Europe and a delayed FDA approval in the US. Decernotinib, an irreversible selective JAK3 inhibitor has shown clinical efficacy in patients with RA in phase IIb clinical studies [252]. A major side effect associated with JAK inhibitors is the increased risk of infections and recurrence of herpes zoster. Despite their differential selectivity, baricitinib, tofacitinib, decernotinib, and upadacitinib have shown an increased risk of herpes zoster reactivation, which strongly correlate with a decline in cell-mediated immunity [235].
Bruton’s tyrosine kinase (BTK) inhibitors initially were introduced to the market for oncology and now are finding their way into fighting autoimmune diseases. The expression of BTK is limited to B and myeloid cells. The dysregulation of BTK signaling in both cell types is shown to be associated with RA. In normal B cells, BTK regulates cell development. Activation of tyrosine kinase by B cell receptors results in BTK and the downstream NF-κB activity. FcRs expressed on myeloid cells also activate the signaling cascade of BTK. In autoimmune diseases such as RA, the overexpression of pre-B cell receptor and overactivation of FcR signaling pathways have been linked to the induction of autoantibodies, making BTK a potential target for RA [253]. Spebrutinib (CC-292) is the first irreversible covalent oral small molecule that inhibits BTK activity in B and myeloid cells. A phase IIa study with 47 patients over the course of four weeks showed that spebrutinib inhibited cellular responses associated with BTK signaling in primary human immune cells and was well tolerated [254]. Bristol–Myers Squibb developed a reversible inhibitor, BMS-986142, and an irreversible inhibitor, branebrutinib [255,256] of BTK. Both drugs have advanced to phase II clinical trials. Evobrutinib, a potent obligate covalent inhibitor with selectivity for BTK, is ongoing in phase IIb clinical studies by Merck [257]. Takeda recently published phase I positive safety results of its BTK selective covalent inhibitor, TK-020, in healthy volunteers. Fenebrutinib, a selective noncovalent BTK inhibitor from Roche, showed an efficacy comparable to that of adalimumab at week twelve in phase II clinical trials. However, the onset response of fenebrutinib is slower than adalimumab, probably due to delayed effect of BTK inhibition on systemic inflammation [258]. AbbVie’s ongoing phase II clinical drug ABBV-599 is a combination of BTK (ABBV-105) and JAK1 (ABT-494) selective inhibitors. Targeting BTK in RA has its complications, however, the use of reversible inhibitors might offer a better strategy due to their selectively for BTK versus other Tec family kinases [259]. Eli Lilly and Hamni dropped their BTK inhibitor, HM71224, after interim results showed little to no efficacy [260]. The approved and clinical drugs for RA are summarized in Table 4.
Chronic RA induces irreversible tissue damage and cell-based therapy can offer a remedy to regenerate and repair the damage. Mesenchymal stem cells (MSCs) in muscles, synovial tissue, placental tissue, and teeth can reduce cartilage degeneration, osteophyte formation, and synovial inflammation. Human MSCs (hMSCs) can be differentiated into various lineages such as chondrocytes, osteoblasts, or adipocytes [261]. In a three-yearlong study, the efficacy of combination treatment using hMSCs plus DMARCs (leflunomide, hydroxychloroquine sulfate, or methotrexate) was investigated in RA patients. The result showed that the treatment was safe with only 4% of patients showing mild side effects that disappeared within few hours. The treatment showed rapid improvement (as early as twelve hours post treatment) in the diet, sleep, and physical strength. Additionally, rheumatoid factors and CCP2 antibody levels showed a slow decline post treatment [262]. Tolerogenic dendritic cells (tolDCs) is an alternative cell-based immunotherapy to restore the immune tolerance in RA patients. TolDCs induce the priming and differentiation of T cells, leading to autoreactive T cell silencing, and Treg induction [263,264]. TolDCs differentiated from CD14+ monocytes were used in a five-day treatment course in phase I study to assess their safety profile in three RA patients with inflamed knee. Although positive safety result was reported, a larger sample pool and a prolonged treatment course are needed to determine safety and efficacy [265]. The safety of allogeneic expanded adipose-derived stem cell therapy was investigated in 53 patients with refractory RA. A long list of adverse events were reported with no evidence of dose-related toxicity, suggesting that further studies are needed to determine the long term efficacy [266]. Invariant natural killer T cells (iNTC) derived from the thymus were injected at the site of joint inflammation in RA mice model. The data showed iNTC treatment improved joint swelling, resorted Th cell subset imbalance, and reduced TNF-α, IFN-γ, and IL-6 levels in this preclinical study [267].
The pathogenesis of RA is suggested to link to the imbalance of the microbial composition in human gut or microbiota dysbiosis, leading to dysfunction of tight junction, permeability of the intestinal barrier, and induction of immune response causing inflammation. A few studies have highlighted the difference in the diversity of gut microbiota in various subset of RA patients compared to the healthy subjects [268]. The commensal bacteria in the gut play a role in generating short-chain fatty acids (e.g., butyrate), which regulate the differentiation and expansion of peripheral immune cells such as Treg and Th cells [269]. A widely used probiotics, Lactobacillus casei (L. casei) have been shown to significantly decrease the expression of Toll-like receptor 2 and TNF-α in arthritis induced rat model. In this study, L. casei was used to treat adjuvant-induced arthritis in rats by restoring the microbiota balance to a healthy state. The treatment attenuated the disease symptoms such as joint swelling, lowered arthritis scores, and prevented bone destruction [270]. In another study, Prevotella histicola (P. histicola) isolated from the commensal bacteria in the human duodenum reduced the RA severity in collagen-induced arthritis mouse model as a result of the suppression of IL-2, IL-17, and TNF-α. Furthermore, the collagen-induced arthritis mice inoculated with P. histicola showed increased number of Treg cells and a reduced Th17 response [271].
Oligonucleotide-based gene therapy such as siRNA and microRNA has shown a significant promise for the treatment of RA in the pre-clinical setting. Unfortunately, limitations associated with the delivery to cells has hindered progress to clinic. To overcome the challenge, peptides against a few RA targets were explored as delivery vehicles in vivo [272]. An example is the encapsulation of the glucan particles (β-1,3-D-glucan), which are isolated from the yeast species Saccharomyces cerevisiae and have immune-modulation and anti-inflammatory activity. The glucan particles were covalently attached to small-molecule amines (ethylenediamine and histamine) and an amphipathic peptide. The complex interact with siRNA via electrostatic interaction to facilitate cell entry and endosomal escape. Successful silencing of selective macrophage genes in vitro and in vivo (such TNF-α) was observed, indicating productive delivery to cells [273,274]. Another approach is to target microRNA to regulate immune response. MicroRNA-135a promotes apoptosis of synovial fibroblasts in RA by regulating PI3K/AKT signaling via regulation of the negative modulator phosphatidylinositol 3-kinase regulatory subunit 2 (PIK3R2). Expression of micorRNA-135a (miR-135a) is predominately high and the expression of PIK3R2 is low in the synovial tissues of RA patients. Treatment with the inhibitor of miR-135a resulted in decreased synovial fibroblasts proliferation, reduced migration and invasion, and enhanced apoptosis. Down-regulation of miR-135a also restored PIK3R2 expression to normal levels, indicating that miR-135a inhibition has potential for treatment of RA [275].
3.2. Atopic Dermatitis
Atomic Dermatitis (AtD) is a chronic autoimmune disease affecting the protective layer of the skin, stratum corneum. The disease is caused by genetic and environmental factors and is prevalent in 20% of children and 1–3% of adults worldwide [276]. AtD results in itchy, red, swollen, and cracked skin. Over 70 genes are associated with AtD in different populations. The First manifestations of AtD usually appear in early childhood and is progressed to allergic sensitizations commonly referred to as atopic march [277]. The immunoglobulin E (IgE)-mediated allergic reaction is associated with atopic march progression and IgE level is suggested to be used for early detection of AtD [278].
The pathogenesis and progression of AtD have been linked to epidermal barrier dysfunction, immune dysregulation, and microbiota dysbiosis. The epidermis provides an outside/inside barrier that prevents the leakage of body fluids, retains water within the cells, and protects against mechanical, chemical, and microbial assault. Filaggrin, loricrin, and involucrin loss of function is associated with impaired skin barrier in the AtD patients. Filaggrin plays an important role in maintaining cellular homeostasis in the skin and its deficiency results in inflammation and dryness of the stratum corneum [279]. Prevalence of the disease might be attributed to the imbalance of Th1 and Th2 cells systemically and in the epidermis. Infiltration of Th9, Th17, and Th22 cells to the skin is also suggested to damage the skin barrier [279,280]. Enhanced expression of Th2 cytokines such as IL-4, IL5, IL-13, IL-25, IL-31, and IL-33 is detected in AtD lesions [281,282]. IL-4 and IL-13 are reported to promote pathogenesis of AtD through multiple mechanism. IL-4 and IL-13 stimulate keratinocytes to express thymic stromal lymphopoietin (TSLP) and act as a common link between barrier defects and Th2 polarization [283]. IL-4 and IL-13 stimulate IgE production from B cells [279], reduce the expression of filaggrin and loricrin by inhibiting the cytoplasmic-to-nuclear translocation of transcription factor gene OVOL1, activate STAT6 and subsequently enhance IL-24 production, which results in activation of JAK1/Tyk2/STAT3 pathway and further reduction in the expression of filaggrin [284]. Two additional Th2 cytokines, IL-5 and IL-31, have been associated with AtD. Expression of IL-5 is elevated systemically and in the lesioned skin, leading to a poor pathology. IL-5 plays a critical role in the activation of eosinophil and JAK/STAT pathway and has been explored as a therapeutic target for AtD [285]. IL-31 expressed by T cells is also upregulated in AtD. It signals through the hydrodimerization of IL-31 receptor A (IL-31RA) and oncostatin M receptor (OSMR) [286]. Lastly, skin microbiota dysbiosis presents an independent risk factor in the development of AtD. Increased colonization of Gram-positive bacterium, Staphylococcus aureus (S. aureus), is found in 90% of AtD patients and has been linked to skin barrier dysfunction and inflammation. S. aureus induces Th1/Th2 immune response, resulting in increased expression of IL-4, IL-13, and IL-22. It can also impair skin barrier function by compromising the expression of filaggrin [287].
Mild-to-moderate AtD is traditionally treated by topical emollients, corticosteroids, and calcineurin inhibitors. Phototherapy and systemic immunosuppressants such as cyclosporine, methotrexate, mycophenolate mofetil, and systemic corticosteroids are offered for moderate to severe AtD. These off brand treatment modalities can be inconvenient, intolerable, and short lived solutions to patients with systemic immune response [288]. Therefore, inhibiting disease-specific targets using biological therapeutics can be much effective approach than surface level topical ointments [289].
A novel approach to treat AtD is by targeting the factors that cause immune imbalance. Phosphodiesterase 4 (PDE4) level is increased in the dermal fibroblasts of skin in AtD patients, resulting in elevated levels of IL-6 and IL-10 [290]. PDE4, abundantly expressed in T cells, causes proinflammatory response through conversion of cyclic adenosine monophosphate (cAMP) to 50–adenosine monophosphate. High levels of cAMP is associated with the suppression of T cells, monocytes, and pro-inflammatory molecules such as IL-4, IL-13, and IL-31 [290,291]. Enhanced PDE4 activity in AtD leads to reduced cAMP levels and inflammation [291]. Mild to moderate AtD has been treated using Pfizer’s PDE4 inhibitor, Eucrisa (crisaborole). A meta-analysis study of five PDE4 inhibitors showed carisaborole is significantly more effective in clearing the skin [291]. Otezla (apremilast), another PDE4 inhibitor is being considered for the treatment of moderate to severe AtD. In phase II, double-blind, placebo-controlled trial, apremilast at 40 mg per dose improved the Eczema Area and Severity Index (EASI) and decreased atopic dermatitis-related biomarkers over twelve weeks [292]. ADCs are employed to improve therapeutic index of PDE4 inhibition. Selective targeting of immune cells was achieved by conjugating the PDE4 inhibitor to a chimeric CD11a antibody. Treatment with the ADC reduced inflammatory response in human monocytes and mouse peritoneal cells [293]. Two additional PED4 inhibitors, E6005 and DRM02, are undergoing clinical evaluation in AtD patients.
IL-13 and IL-4 levels are increased in AtD patients and play an important role in the pathogenesis of the disease. Both cytokines exert their effects by binding to the shared IL-4Rα expressed on T cells, B cells, and macrophages [288]. IL13 engages a heterodimeric receptor composed of IL-4Rα and IL-13Rα1. It also binds to IL-13Rα2 with high affinity. Heterodimerization of IL-4Rα and IL13Rα1 activates JAK2 and TYK2 [294]. Dupixent (dupilumab), an IL4Rα antagonist, was the first fully human IgG4κ mAb approved to treat moderate to severe AtD. Dupilumab sales was grown to $2 billion in 2019 with an expected growth peak of $10 billion in the foreseeable future. Dupilumab inhibits IL-4 and IL-13 signaling by binding to the shared IL-4Rα subunit. As a result, dupilumab inhibits the release of pro-inflammatory cytokines, chemokines, and IgE [295]. Tralokinumab, IL-13 humanized mAb from LEO Pharma, binds to the soluble IL-13 and prevents its interaction with the two IL-13 receptors, IL-13Rα1 and IL-13Rα2. This blocks the heterodimerization of the IL-4Rα and IL-13Rα1 but does not affect binding of the cytokine to IL-13Rα2 subunit [289]. A phase IIb clinical study showed that treatment with 300 mg of tralokinumab for twelve weeks significantly improves AtD lesions within seven days [296]. The result of an ongoing phase III study has suggested 75% improvement in the EASI score at week sixteen, which was maintained for 52 weeks in 50% of the patients. Eli Lilly has acquired Dermira (lebrikizumab), a humanized mAb with high affinity to IL-13. Lebrikizumab is currently in phase III clinical trials. It had a positive safety profile and has demonstrated dose-dependent efficacy across patients after sixteen weeks of treatment [297]. Inhibition of two additional Th2 cytokines, IL-5 and IL-31, are also considered for treatment of AtD. Mepolizumab, a humanized IgG1κ mAb against IL-5 has shown efficacy and safety at 100 mg subcutaneous dose in phase II [298]. An additional IL-5 mAb, benralizumab, is being evaluated in phase II for its ability to prevent eosinophils recruitment to the skin. Nemolizumab, a first-in-class humanized mAb, blocks IL-31 signaling by binding to its receptor IL-31RA. Phase IIb trial with 30 mg of nemolizumab has resulted in a rapid and sustained improvement in cutaneous inflammation and pruritus in AtD patients [299].
AtD has been associated with activation of Th17/IL-23 [281]. AbbVie’s IL-23 antibody, risankizumab, is being evaluated in phase II clinical trials in a subset of AtD patients [300]. Fezakinumab, an IL-22 inhibitor, has entered phase III clinical trials. A randomized, double-blind, phase IIa trial of 60 moderate to severe AtD patients showed fezakinumab is efficacious and well tolerated [301].
The JAK/STAT pathway has been explored for treatment of AtD. Several FDA approved drugs for other indications such as RA and psoriasis are currently in clinical trials for AtD. Eli Lilly drug Olumiant (baricitinib) is an orally approved small molecule antagonist of JAK1 and JAK2 for treatment of RA and is currently in phase III clinical trials for AtD. Phase II results showed that baricitinib in combination with a once daily topical corticosteroids reduced EASI by 65% in 16 weeks [302]. Ruxolitinib is a topical JAK1/JAK2 inhibitor approved for oncology, myelofibrosis, and polycythemia vera. Ruxolitinib was well tolerated and provided rapid and sustained improvements in AtD symptoms through the course of twelve-week treatment during phase II in 2020 [303]. RINVOQ (upadacitinib) is AbbVie’s JAK 1 inhibitor approved for RA. Phase III clinical studies (2020) in AtD showed that 80% of patients receiving 30 mg of upadacitinib achieved EASI 75 after sixteen weeks of treatment compared to 16% in the placebo group [304]. Approved dosage of upadacitinib is 15 mg in RA patients but 30 mg seems to be the optimal dosage for AtD. Long-term studies are still needed to determine the safety and efficacy of the drug at 15 and 30 mg. Pfizer’s first in class JAK1 inhibitor, abrocitinib, was tested at 100 and 200 mg in 391 patients ages 12–18 with moderate to severe AtD in 2020. The result showed that 61% of patient treated with 200 mg and 44.5% treated with 100 mg had an EASI 75 [305]. Gusacitinib (ASN002), a dual JAK/SYK inhibitor, developed by Asana Biosciences showed great safety and efficacy in phase IIb clinicals. However, subsequent studies were terminated in May 2020 for undisclosed reasons [306]. Other drugs including the dual JAK and SYK inhibitor Cerdulatinib (RVT-502), pan-JAK (JAK1, JAK2, JAK3, TYK2) inhibitor Delgocitinib (JTE-052), and JAK3 and tropomyosin receptor kinase A (TrkA) inhibitor SNA-125 are in the early stages of clinical trials [307].
Histamine H4 receptor (H4R) is a novel target for AtD. H4R is expressed in sensory neurons and induces scratching behavior that contributes to skin lesions in AtD patients [308]. Histamine H4R antagonist, ZPL-3893787, was investigated in phase II clinical trials in patients with moderate to severe AtD. ZPL-3893787 had a 50% reduction in EASI score and was well-tolerated with no major side effects [309]. An earlier H4R antagonist, JNJ39758979 developed by Janssen Pharmaceutical, was terminated after reports of agranulocytosis in two cases. Another target that has been shown to induce an itching response is TSLP. TSLP is an epithelial derived cytokine, and its expression is elevated in lesioned skin of AtD patients, leading to T-cell response and IL-5 induction. Two TSLP inhibitors, Tezepelumab (phase IIa) and MK8226 (terminated), have been evaluated in patients with AtD [285,310]. TSLP activates DC cells via TSLPR/IL-7R complex, promotes immature DCs maturation, and the production of OX40 ligand on the cell surface [311]. Elevated OX40 ligand interacts with OX40 on naïve CD4+ T cells in the lymph nodes and induce an inflammatory Th2-type response [283]. A first in class humanized IgG1 OX-40 receptor mAb, GBR 830, is in phase II for moderate to severe AtD. OX40 expression is upregulated in AtD patients, in particular in the lesion sites and it is believed to play a critical role in the disruption of T-cell tolerance. Administration of GBR 830 over four weeks was well tolerated with significant progressive clinical effects [312]. KHK4083 is a fully human OX40 mAb that is effective in depleting activated T-cells and suppressing clonal T-cell expansion. Clinical trials conducted on 22 patients with moderate to severe AD had an acceptable safety profile and sustained symptoms improvement over 155 days of treatment [313].
Another approach has been to target cytokines that are overexpressed in epidermal such as IL-17C and IL-17A. Both cytokines are part of a “feed-forward” process. IL-17C, produced by epidermal keratinocytes and other non-immune cells, induces IL-17A release from T lymphocytes. IL-17A in turn induces expression of IL-17C, hence its naturalization can reduce skin inflammation [288]. However, IL-17C antagonist (MOR106) did not meet expected outcome. Fully human IgG1 IL-17C mAb developed by MorphoSys and Galapagos also failed to meet the expected percentage change in the EASI score during phase II trials [314]. Secukinumab, an approved IL-17A mAb for plaque psoriasis, showed no improvement in the EASI in AtD patients at 300 mg [315]. Targeting IL-33 in AtD patients also failed to meet productive clinical outcomes. IL-33 is significantly up-regulated in keratinocytes in patients with AtD. It stimulates production of IL-5 and IL-13 and compromises the barrier function by reducing the expression of filaggrin and claudin-1 [316]. Sixteen weeks of treatment with IL-33 mAb Etokinumab, developed by AnaptysBio, did not improve EASI [317]. Two additional anti-IL-33 mAbs, PF-06817024 and REGN3500, have shown some potential in treating AtD. The list of approved and clinical drugs for AtD is summarized in Table 6.
The skin microbiome has been considered for treatment of AtD. Higher colonization of S. aureus and reduced density of S. epidermidis and S. hominis (strains that produce antimicrobial peptides cathelicidins and β-defensins) were observed in AtD patients. This imbalance in the skin microbiota and high abundance of S. aureus are associated with disease symptoms. In one study, five AtD patients were given S. hominis or S. epidermidis strains that were isolated from healthy subjects. This resulted in reduction in the density of S. aureus after a single application [318]. In another study, application of an autoinducing peptide (SYNVCGGYF) isolated from S. hominis resulted in prevention of S. aureus mediated epithelial damage and inflammation in murine skin [319]. In 2020, Bayer announced a partnership with Azitra to develop therapeutic products with S. epidermidis strains for eczema-prone skins and AtD. Additionally, transfer of Gram-negative skin bacteria, Roseomonas mucosa, collected from human skin of healthy individuals has shown to reduce S. aureus growth in mouse model of AtD [320]. Safety and activity of transplant with commensal bacterium R.mucosa were evaluated in ten adult and five pediatric patients with AtD in phase I/II. The study showed that the transplantation significantly decreased disease severity, topical steroid requirement, and S. aureus burden [321]. Evelo Biosciences is developing the strategy with an oral single strain of microbes called monoclonal microbial for regulating innate and additive immune systems. Administration of EDP1815, a single strain of Prevotella histicola isolated from healthy human duodenum resulted in systemic anti-inflammatory response in mice. While the mechanism of action is not completely understood, EDP1815 inhibited T cell-mediated inflammation in the small intestine [322].
Notably, siRNA treatment might be an effective approach for AtD [323]. In a study conducted by Kanazawa and colleagues, lipid vesicles coated with a cell-penetrating peptide was used to deliver an siRNA targeting the RelA subunit of the cytokine transcription factor NF-κB. Combination of the liposome (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine/cholesteryl hemisuccinate) with high affinity to cell membrane and the cell penetrating peptide (AT1002: FCIGRL) improved siRNA delivery, resulting in a significant improvement in the lesion sites in mouse models [324,325,326]. Modulation of miRNA expression is used to repair barrier function in AtD [280]. miR-335, an inducer of keratinocyte differentiation is downregulated in AtD. Belinostat, an inhibitor of histone deacetylase approved in hematological malignancies and solid tumors recused the skin barrier by restoring the expression of miR-335 in vitro [327].
Corticosteroids are the first line of therapy in AtD with side effects such as nephrotoxicity and carcinogenesis. Small molecules and biologics are growing at a fast pace and are expected to continue to dominate the market. PDE4 inhibitors are also expected to gain tremendous popularity. Eucrisa (Pfizer; crisaborole ointment, 2.0%) is the only PDE4 inhibitor approved for the treatment of mild-to moderate AtD for adult and pediatric use. Biologics are prescribed as second or third line of treatment. However, the efficacy observed with Dupixdent might shift the order prescribed therapeutics.
3.3. Crohn’s Disease
Crohn’s disease (CD) is an inflammatory bowel disease (IBD) that affects the gastrointestinal tract. Genetic factors have been linked to the disease, but gene polymorphism is still unknown. In patients with CD, the expression of MUC1 gene is downregulate, resulting in reduced levels of mucin in the mucosal layer that provides the first line of immune defense. The gut barrier becomes leaky during the pathogenesis of the disease, allowing antigens to permeate to lamina propria, where immune cells are densely populated. The increased risk of disease has been associated with imbalance of symbiotic commensal microbiota [328]. In the US, IBD affects 3 million individuals at an annual incident rate of 3–20 cases per 100,000 [329]. There is no cure for CD, just fast relief of symptoms by steroids-sparing agents (e.g., thiopurines) and methotrexate in combination with TNF mAbs (e.g., infliximab, adalimumab, and certolizumab pegol) for long-term maintenance [329]. TNF inhibitors have shown to be effective and safe for treating CD. However, 20–40% of patients do not respond to this treatment and 30–40% become non-responsive over time [330].
A prominent feature of CD is the infiltration of T cells to the inflamed gut. Anti-adhesion therapies have been exploited to modulate T cell trafficking to the gut. Integrins on the T-cells interact with mucosal addressin cell adhesion molecule (MAdCAM) on endothelial venules [331] to facilitate T-cell trafficking. Blocking heterodimeric α4β1 and α4β7 integrins prevents transmigration of leukocytes across the endothelium and in turn halts the chronic inflammation of the tissue [332]. α4β7 is highly expressed on intestinal-homing T lymphocytes and α4β1 is involved in T lymphocytes homing to the intestinal and non-intestinal tissues, such as the lung and central nervous system [333]. Natalizumab, a humanized IgG4κ α4β1 mAb, was the first selective adhesion molecule blocker approved for moderate to severe CD in 2008. Blocking α4β1 introduced the risk of off-target events leading to progressive multifocal leukoencephalopathy, a fatal brain infection [332]. Entyvio (vedolizumab), targets the gut-specific α4β7 with no reports of multifocal leukoencephalopathy [329]. Another novel approach is the dual targeting of α4β7 and αEβ7 to improve the therapeutic effect and prolong the remission. The integrin αEβ7 expression is upregulated in the lymphocytes that are infiltrated to the mucosa to mediate lymphocyte retention [333]. Etrolizumab, anti-β7 mAb developed by Roche and Genetech to target both α4β7 and αEβ7, is in phase III clinical for CD. Although efficacy studies are yet to be published, β7 blockade might have caused off-target effects since αEβ7 is expressed in T-cells that are responsible for controlling local infections [331]. PN-943 developed by Protagonist Therapeutics is an oral gut-restricted α4β7-specific antagonistic peptide for the treatment of IBD. The phase I study showed that PN-943 was safe and well tolerated [334]. The oral peptide is currently in a phase II study for ulcerative colitis. Targeting MAdCAM is investigated as a novel treatment option due to its role in immune surveillance. PF-00547659, a fully human mAb developed by Pfizer, is a selective blocker of MAdCAM. In phase II clinical trials, PF-00547659 did not improve the disease with statistical significance in patients with moderate to severe CD who had failed to respond to anti-TNF treatment [335].
A growing interest has shifted to novel small molecules that bind to pro-inflammatory targets in the lymph node [336]. An example is sphingosine 1-phosphate (S1P) with potential in treating CD. S1P mediators interact with G-coupled protein receptors and play an important role in the regulation of immune cell circulation. S1P levels are increased at the site of inflammation, leading to immune cell infiltration and exacerbation of the inflammatory process [336]. Except for S1P4 and S1P5 that are expressed in the lymph node, spleen, lung, and thymus, S1P mediators are ubiquitously expressed [337,338]. Bristol-Myers Squibb’s ZEPOSIA (ozanimod) is an oral modulator of S1P1 and S1P4 approved in 2020 for multiple sclerosis. The drug is currently in phase III for treatment of CD [339]. Biogen terminated its Amiselim (MT-1303), an oral drug against S1P1 after randomized placebo-controlled Phase II studies in CD. It is still early to determine whether S1P modulators can impact the treatment of IBD, however, positive safety and efficacy profiles of oral S1P modulators has placed them under the spotlight [337].
New class of drugs targeting the JAK family known as Tyk2 inhibitors might compensate the poor safety profile of JAK inhibitors. BMS-986165 currently in phase III for psoriasis is also under investigations in patients with moderate to severe CD with the expected conclusion in 2022. Pfizer has developed three drugs targeting TYK2 for different autoimmune indications. These include the first-in-class inhibitor for JAK 1–3/TYK2 (PF-06263276) in chronic plaque psoriasis (NCT01981681), dual TYK2/JAK1 inhibitor (PF-06673518, pre-clinical) and Tyk2/Jak1 (PF-06700841) for ulcerative colitis, CD, and psoriasis in phase II [340]. Johnson and Johnson in partnership with Theravance have developed Pan-JAK/TYK2 inhibitor, TD-1473, currently in phase II clinical trials for CD.
Ustekinumab, initially approved for psoriasis, was approved in 2016 for CD. Ustekinumab binds to the p40 subunit of IL-23 and IL-12 to block interaction with their receptors on T-cell [341]. Another IL-23 mAb approved for psoriasis, Risankizumab, is being investigated for the treatment of CD. An open-label study extension showed that the antibody is effective and the treatment results in increased clinical response and remission rates after twenty-six weeks [342]. Risankiumab is in phase III with a completion date of 2027. In addition, PTG-200 is an oral IL-23R antagonist peptide developed by Protagonist Therapeutics and Janssen Pharmaceuticals. The peptide currently is in Phase II study for CD. The list of approved and clinical drugs for CD is summarized in Table 7.
A novel approach has focused in targeting the gut lumina to promote intestinal healing in CD. Expression level of glucagon-like peptide 2 (GLP-2) is decreased at the sites of inflammation in the colon, while it’s levels are increased in the serum of CD patients [343], indicating that GLP-2 might play a role maintenance of gut barrier. GLP-2 attenuates expression of TNF-α by macrophages in the lamina propria. Native GLP-2 and teduglutide, a synthetic analog of GLP-2, are shown to enhance intestinal barrier function in vivo [344]. A proof of concept pilot study in 71 CD patients receiving a daily injection of teduglutide over twelve weeks showed improved Crohn’s Disease Activity Index compared to the placebo group. Further assessment is needed to understand the effect of teduglutide as a monotherapy or in combination with other therapeutics on systemic inflammation and barrier function [345].
Recommended treatments for CD are corticosteroids, immunosuppressive agents, and biological agents. They relieve short term symptomatic complications and cause severe side effects. Alternatively, several strategies including antibiotic treatment, fecal transplantation, and administration of pre/probiotics are considered for the remission of active disease or preventing relapse [346]. Gut microbiota dysbiosis plays a significant role in CD pathogenesis [347,348]. Adherent-invasive E. coli (AIEC) is a strain of Escherichia coli with adherent and invasive properties that is present in the gut of CD patients. AIEC bacteria adheres to intestinal epithelial cells and induces inflammatory cytokine secretion and disruption of epithelial barrier function [349]. Three anti-adhesive strategies have been employed to reduce AIEC colonization. They include the use of bacteriophage to kill AIEC, anti-adhesive molecules to block adhesion, or bacteriocins [350,351]. Bacteriophage therapy utilizes an engineered or naturally-occurring bacterial viruses (phage) to infect and lyse bacteria [352]. In an in vivo study, administration of three bacteriophage targeting AIEC strain, LF82, resulted in decreased intestinal colonization of LF82 [353]. Another approach to block adherence of LF82 to the epithelial cells is by targeting the FimH receptors at the tip of type 1 pili of AIEC. In pre-clinical studies, EB8018, an oral gut-restricted small molecule against FimH demonstrated a dose-dependent reduction in the density of LF82 and profoundly reduced the levels of TNFα, IL-6, and IL-8. EB8018 is currently being investigated in phase I clinical trials [354]. The third strategy is to use bacteriocins, ribosomally synthesized bactericidal peptides with inhibitory activity against microorganisms [351]. Species-specific antibiotics, Colicins, showed potent activity against biofilm-associated cells in established AIEC biofilms. In addition, colicins were able to effectively kill drug-resistant AIEC biofilms, AIEC bacteria associated with intestinal epithelial cells, and those growing intracellularly within macrophages [355].
Personalized microbial-based therapies might be an alternative treatment option or even a cure for CD patients. A new class of live therapeutics products comprised of synthetically engineered or genetically-modified microbes are considered. There is no approved live therapeutic products but the FDA has issued guidelines for this treatment and clinical trials are to be conducted under an investigational new drug application [45]. Result of a preclinical study showed that an engineered commensal E. coli strain can selectively synthesize and secrete GM-CSF in the presence of nitric oxide, a CD biomarker. The bacteria accumulate at the sites where nitric oxide levels are elevated to secret GM-CSF and turns to an “off” state when nitric oxide levels are low. This approach provides localized delivery of GM-CSF based on levels of nitric oxide. GM-CSF restores mucosal barrier functions and promotes mucosal healing. The efficacy of this approach is yet to be evaluated in the in vivo studies of CD models [356]. Assembly Biosciences in partnership with Allergan has developed capsules containing synthetic live biotherapeutics for treatment of ulcerative colitis, irritable bowel syndromes, and CD.
Stem-cell-based therapy using hematopoietic or mesenchymal stem cells are being considered for mucosal healing in refractory CD patients [357]. A study published in 2020 provided the result of a four-year clinical evaluation of allogeneic bone marrow-derived mesenchymal stromal therapy in twenty-one refractory CD patients. There were no serious adverse events and the fistula size was either significantly reduced or closed after four years post-therapy. In addition, HLA antibodies could not be detected at 24 weeks and four years post-therapy [358].
Celsius Therapeutics is leading the effort to identify novel drug targets for IBD by evaluating longitudinal samples using single-cell genomics and machine learning capabilities. Single-cell genomics allows profiling of a large number of cells in healthy and diseased individuals [359] and can provide an understanding as to why CD patients do not respond to certain therapeutics, such as anti-TNFs. Additionally, novel pathogenic cells or targets can be identified by single-cell genomics. In a study of four independent CD cohorts (n = 441), CD lesions were analyzed with single-cell technology to identify non-TNF responders that express cellular modules such as IgG plasma cells, inflammatory mononuclear phagocytes, activated T cells, and stromal cells or GIMATS in short. It was suggested that GIMATS correlates with the non-responsiveness to TNF therapy, suggesting that single-cell mapping tools can be used to tailor therapeutics based on novel biomarkers in CD patients [360].
The market size of CD therapeutics is expected to grow to $4.7 billion by 2025 according to Grand View Research. TNF inhibitors constitute the largest prescribed therapeutics to CD patients and are expected to remain so in the future. Oral JAK inhibitors are the fastest growing product for treatment of CD [361]. Many adhesive antibody therapeutics for CD are entering the market. Entyvio developed by Takeda is forecasted to be the market leader in in 2026 [362]. While microbiome-based therapy and stem cell therapy have attracted some attention, their overall potential for treatment of CD is yet to be determined.
4. Neurological Diseases
4.1. Chronic Pain
More than 20% of Americans are living with some forms of chronic or severe pain [363]. In early 2020, the International Association of the Study of Pain (IASP) revised the definition of pain that was first developed in 1979. Pain is described as “an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage” [364]. The new definition reinforces the fact that feeling of pain is personal with both sensory and emotional experience and it can be influenced by biological, psychological, and social factors. Chronic pain is related to the persistent stimulation of the nociceptors due to ongoing tissue injury or can result from ongoing damage and dysfunction of the peripheral or central nervous system (CNS), causing neuropathic pain. Chronic pain may last many months or even years causing severe problems [365]. Pain pathways are composed of a complex sensory system and the mechanisms at the induction and maintenance of chronic pain are still poorly understood. Many chronic pains are developed after a primary injury has healed or do not have apparent underlying physical injury. Non-opioid and opioid analgesics are the most common drugs to treat chronic pain. Antidepressants, anticonvulsants, and other CNS-active drugs are also used to treat chronic neuropathic pain [366,367,368]. In this section, we will focus on chronic pain caused by osteoarthritis in the joints and migraine in the CNS. Current and emerging therapies and treatments for each disease are discussed.
4.1.1. Osteoarthritis
Osteoarthritis (OA), a highly prevalent and most common form of arthritis, has touched over 303 million people globally [369] by affecting the joints of knee, hands, hip, and spine [370]. OA is a complex and heterogeneous condition with multifactorial origins such as joint injury, age, and obesity [371,372]. More than 80% of the population shows radiological evidence of OA by the age of 65 [373]. OA joints show features of inflammatory and degenerative diseases with chronic joint pain being the most dominant symptom in OA patients [374]. The chronic OA-related joint pain causes substantial functional limitations, such as poor sleep, disability, and depression. In this section, we provide an overview of the pathogenesis of OA pain, discuss the available therapeutics for pain management, and present the potential disease-modifying strategies for OA.
OA patients experience pain primarily in the periphery (nociception) and can have different levels of central (neuropathy) sensitization [371]. Nerve damage can occur in the injured joint, dorsal root ganglia, and the spinal cord leading to neuropathic pain during the development of OA [375,376]. The inflammation that develops post joint damage is the major contributor to the chronic OA pain. During inflammation, a state of hyperexcitability of nociception is developed in the OA joints and the sensation is relayed to the spinal cord and cortex and then amplified in the central nervous system causing central sensitization. In the healthy state, nociceptive signals are modulated by cortical and brain-stem pathways within the CNS. In OA patients, the dysfunction of both nociceptive ascending and inhibitory descending pain signals results in higher pain intensity and longer duration [371,377,378]. A substantial variety of neurotrophins, cytokines, proteases, neuropeptides, chemokines, and prostaglandins are released at different levels of pain sensitization [371]. In the synovium, the upregulation of nerve growth factor (NGF), TNF-α, IL1β, IL-6, IL-8, and C-reactive protein lead to the increased pain perception [374,378,379,380]. Locally, all of these components can induce matrix metalloproteinases (MMP) and other hydrolytic enzymes, leading to cartilage damage [381]. At the dorsal root ganglia and spinal cord level, NGF, calcitonin gene-related peptide (CGRP), brain-derived neurotrophic factor (BDNF), substance P, vasoactive intestinal peptide (VIP), opioid receptors, and CC chemokine ligand 2 (CCL-2) have been identified as mediators of pain [371,379]. Substance P, serotonin, and glutamate are pain mediators at the brain level [371,382].
As of today, there is no known cure for OA, no intervention against disease progression, and no efficient pain management with acceptable adverse effects [383]. Standard approaches that have been used for the past decades for OA associated nociceptive pain are painkillers and anti-inflammatory drugs for symptom alleviation, including nonsteroidal anti-inflammatory drugs (NSAIDS), acetaminophen, duloxetine, corticosteroid, and opioid receptor agonists [381,384,385] (Table 8). The conventional medications are associated with limited efficacy in pain and function improvement [386]. Significant adverse events are observed with prolonged use, such as gastrointestinal issues, high blood pressure, kidney damage, and increased cardiovascular risk [384,385,387]. The chronic long-term use of opioids is strongly discouraged due to serious side effect and risk of addiction. The newer cyclooxygenase COX-2 selective inhibitors (etoricoxib and rofecoxib) provide comparable efficacy and lower gastrointestinal issues compared to NSAIDS that block both COX-1 and -2 [388]. Alternative medicines to alleviate joint inflammation and pain include intra-articular corticoid injection, chondroitin and glucosamine supplement, and hyaluronic acid injection. However, their efficacies have been controversial. For the management of neuropathic pain, drugs such as duloxetine that inhibit serotonin and norepinephrine reuptake in the brain are recommended [389]. Joint replacement and surgical intervention are effective treatment options in patients with late stage OA. In some cases, a revision surgery may be needed after the joint replacement. Mechanism-based analgesic treatments for the early stage of OA with high efficacy and low side effects or abuse liability are an unmet medical need [390].
In the past five years, biologics and small molecules have been used for treatment of OA associated pain [391]. Three NGF mAbs, tanezumab (Pfizer and Eli Lilly), fulranumab (Janssen and Amgen-development was discontinued in 2016), and fasinumab (Regeneron and Teva) are currently under clinical development. Tanezumab is the most advanced and extensively studied mAb under regulatory review. In two meta-analysis studies, tanezumab showed similar serious adverse events but greater efficacy for pain compared to the placebo group [392,393]. Tanezumab administration also alleviated pain and improved physical function compared to NSAIDS in patients with knee and hip OA [394]. It is suggested that anti-NGF therapy is associated with accelerated joint damage, therefore, exclusion of individuals with preexisting joint abnormalities is highly recommended from these studies [395]. One hypothesis that has not been conclusively proven suggests that alterations in the expression of NGF receptor, TrkA (tropomyosin-related kinase A), in arthritis might disrupt endogenous anti-inflammatory mechanisms, leading to increased joint destruction [396]. AstraZeneca bispecific antibody (MEDI7352) targeting NGF and TNF is currently under investigation in phase I study. LEVI-04 is a p75 neurotrophin receptor fusion protein in phase I study. Small molecules targeting NGF signaling include TrkA inhibitor GZ389988 and pan-Trk inhibitor ONO-4474. Phase II study of GZ389988 was completed with no available results and ONO-4474 study was terminated in 2018.
A few existing drugs were repurposed for treatment of OA [384], including lutikizumab (IL-1α/β mAb) [397,398], adalimumab (TNF mAb) [399,400], GSK3196165 (GM-CSF antibody) [401] and galcanezumab (CGRP mAb) [384,402]. They all showed minimal to no efficacy in reducing pain or improving function for OA, suggesting that the inhibition or activation of the indicated targets might not be relevant in symptomatic OA treatment. An antibody against IL-6 receptor, tocilizumab, has completed the phase III in February 2019 for patients with hand OA but results are not yet available.
Small molecules against opioid receptors (δ, κ, or µ isoforms), cannabinoid receptors, or ion channels are developed for OA pain [391,403]. CR845, a peripherally selective κ opioid receptor agonist, is the most advanced molecule in the clinic. A phase IIa study showed a significant pain reduction with 5 mg dose of CR845 compared to placebo in patients with OA of the hip [404]. A few drugs targeted δ/µ opioid receptors or bradykinin (BK) B2 receptor did not show efficacy [391]. Promising result was reported with drugs targeting sodium channels Nav 1.7 and Nav 1.8. Changes in the expression, trafficking, and redistribution of NaVs following inflammation are attributed to the abnormal firing in different neurons. Phase II study with VX-150, a first-in-class oral inhibitor of Nav 1.8, showed significant pain relief in patients with knee OA [405]. The topically applied Nav 1.7 inhibitor (TV-45070) from Xenon Pharmaceuticals did not show efficacy in OA knee pain over placebo. Transient receptor potential cation channel subfamily V membrane 1 (TRPV1) is involved in the detection of heat, pain, and low pH. Capsaicin found in chili peppers is a selective agonist of TRPV1 for pain. Prolonged exposure to capsaicin desensitizes the receptor, and subsequently inactivates the local pain transmission. A phase II study of CNTX-4975, a synthetic trans-capsaicin, showed that 1 mg of CNTX-4975 via local injection reduced pain associated with walking and improved knee stiffness and physical function in patients with OA knee pain [406]. An oral TRPV1 antagonist, NEO6860, did not show significant improvement compared to placebo in phase II study [407].
AXS-02 (disodiumzoledronate tetrahydrate) is an oral formulation of farnesyl pyrophosphate synthase inhibitor of osteoclasts. Result of phase III study of AXS-02 in patients with knee OA with bone marrow lesions has not become available since 2016. Increased activation of Wnt signaling is shown to contribute to the inflammation and progressive joint destruction in OA. Single intra-articular injection of Lorecivivint (SM04690), a Wnt pathway inhibitor, has reduced both symptoms and structural damage in patients with knee OA [408]. Lorecivivnt is currently in phase III clinical investigation. Preclinical studies with MMPs and aggrecanases (ADAMTs) inhibitors have preventing extracellular matrix degradation. Highly selective MMP-13 inhibitors (ALS 1-0635, PF152, and CL82198) and ADAMTs inhibitors (114810 and nanobody M6495) have shown promising results in slowing disease progression in preclinical studies [409,410].
Cell-based therapies are being considered to restore the balance between anabolic and catabolic activity in OA joint tissue. An example is delivering of allogeneic human chondrocytes overexpressing transforming growth factor-β1 (TGF-β1) to joints by intra-articular injection (Invossa: TissueGene-C or TG-C) [411]. TGF-β proteins play a critical role in regulating osteogenesis and chondrogenesis, stimulate proteoglycan synthesis, and chondrocyte proliferation [412]. The phase III results of TG-C showed improvement in pain, structure, and function in patients with knee OA over placebo. TG-C is currently under FDA review with the potential to become the first cell and gene therapy in OA. Injectable mesenchymal stem cells (MSCs) have also drawn much attention, however the outcomes are controversial in OA [384,409,410,413]. This could possibly be due to the heterogeneity in MSC preparation and the subsets of OA patient included in the study (e.g., only older patients with knee OA). Umbilical cord stem cells have also been investigated for cartilage regeneration. In four clinical studies, delivery of umbilical cord stem cells by intra-articular injection or arthroscopic implantation of collagen scaffolds improved OA symptoms and resulted in tissue regeneration [414]. Extracellular vehicles, such as exosomes, are used to deliver stem cells. Exosomes have shown great potentials in pre-clinical studies for bone and cartilage remodeling in OA [415,416].
Gene therapy for OA has emerged over two decades ago [417]. However, no such entity has yet gained regulatory approval. CRISPR/Cas9-based gene editing has been explored to target genes encoding MMP13, IL-1β, and NGF by intra-articular injection of adeno-associated virus in mouse model [418]. CRISPR-mediated ablation of NGF alleviated OA pain, but worsen the joint damage in surgically induced mouse OA model. Deletion of MMP13 or IL-1β attenuated structural damage but did not result in pain reduction compared to NGF blockade. Ablations of all three genes mitigated OA pain and structural damage in mice, suggesting that combination therapy is advantageous and IL-1β and MMP13 antagonism can be a supplementation to NGF inhibition.
Developing pain medications with high efficacy and low adverse effects is challenging due to the heterogeneity of clinical pain conditions and the complexity of the pathophysiological mechanisms [403]. Lack of translation between pre-clinical studies and clinical trials in human attributes to the challenge. Limitations in the animal model of chronic pain include difficulty in pain measurement in animals, poor correlation between rodent models and human patients, and translation of the analgesic effects from rodents to human. In addition, the placebo effect in OA is significant and has attributed to an average of 75% pain reduction and 71% functional improvement [419]. Up to date, there are no approved drugs that cure OA or slow the progression of the disease. Promisingly, a substantial amount of new pharmacological entities are being developed with new targeting mechanisms and modalities. Moreover, epigenome-based therapeutics provide a new mechanistic approach to target the dysregulation of molecular pathways in OA pathogenesis in preclinical studies [420]. The strategies to optimize tailored analgesia with sustained efficacy will be the next challenging task.
4.1.2. Migraine
Migraine is a common neurological disease that affects billions worldwide [421]. It is a complex disorder that can cause recurrent episodic headaches with no protective purpose. In addition, the head pain is accompanied by a variety of symptoms including nausea, vomiting, light/sound/odor sensitivity, visual effects, and aura [422]. The frequency of the migraine attacks varies, from once in a lifetime to almost daily. Vulnerability to migraine has been associated with several susceptibility loci by GWAS [423]. Migraine involves activation of peripheral trigeminal nociceptive pathways, brain stem, diencephalic nuclei, and the cortex [423]. The mechanisms as to how migraines occur is not completely understood but studies suggest that the interaction among neurons, trigeminovascular system, and neurogenic inflammation play important roles in the pathogenesis [424]. Due to the poorly understood pathophysiology, the therapeutic landscape for migraines is diverse and can be classified into general anti-inflammatories and painkillers, serotonin (5-HT) receptor targeting, dopamine and dopamine receptor blockade, GABA activity enhancement, and CGRP inhibition (Table 9). Small molecules and mAbs are the dominant modalities in treating migraine.
NSAIDs and acetaminophen have a proven track record for acute migraine relief [425] but with considerable side effects for long-term use. They block COX-1 and 2 to inhibit prostaglandin synthesis systemically, therefore preventing inflammation. Migraine is suggested to be a neurovascular disease and is dependent on the activation and sensitization of trigeminal afferents from meninges and associated blood vessels [426]. Many scientific studies have focused on the role of 5-HT receptors in migraine. A significant decrease of serotonin levels in periphery and brain were observed during a migraine attack [427,428]. Triptans, developed in 1990s, are selective and specific serotonin receptor agonist (5-HT1B/1D) acting on the smooth muscle cells of blood vessels. Serotonin receptor agonism by Triptans results in narrowing blood vessels in the brainstem and inhibits release of neurogenic inflammatory mediators like CGRP [429,430]. Triptans are ineffective in about 35% of patients with migraine and induce adverse side effects [428]. Ergotamines work similarly to triptans by activating the 5-HT1D receptors located in the intracranial arteries to constrict blood vessels, slow blood flow around the brain, and inhibit release of vasoactive peptides from the trigeminal nerve terminals [431,432]. β-blockers relieve migraine symptoms through a mechanism not entirely understood, although it may involve modulation of the adrenergic system and/or influence on cranial blood vessels [433,434]. Anti-depressants such as tricyclic anti-depressants and selective serotonin uptake inhibitors increase extracellular levels of serotonin and norepinephrine [427,435,436]. Excess dopamine causes nausea and vomiting, and D2 receptor antagonist are used to alleviate this sensation [425]. Calcium channel blockers inhibit intracellular influx of calcium ions, preventing the constriction of blood vessels involved in migraine attacks [437]. Botox is a Botulinum toxin A, and it is thought to interfere with peripheral and central sensitization and block vasoactive peptides that are released from trigeminovascular endings [438]. Anti-epileptics are used as prevention drugs [439]. It is postulated that anti-epileptics inhibit glutamate-mediated excitation, enhance GABA activity, and reduce CGRP. Lasmiditan is a new class of acute treatment for migraine approved in 2019 [372]. Unlike triptans, lasmiditan is a serotonin receptor agonist selective for 5-HT1F receptors, which are expressed on the nerves responsible for transmission of pain signals. Activation of 5-HT1F potentially inhibits release of CGRP and the neurotransmitter glutamate, hence targeting the trigeminal pathways involved in migraine [440,441].
Anti-CGRP treatments are a new class of drugs developed for preventive treatment of migraine. CGRP is believed to mediate the vasodilator component of neurogenic inflammation, and its expression is elevated during migraine attacks [442]. CGRP’s causative role in migraine has been shown in animal and human studies, when intravenous administration of CGRP induces pain and headaches [443,444]. Blocking CGRP from binding to its receptor reduces neurogenic inflammation, hence preventing migraines. Since 2018, one CGRP receptor mAbs (erenumab) and three CGRP mAbs (fremanezumab, galcanezumab, eptinezumab) have entered the market. Two other CGRP small molecules, atogepant (NCT03939312) and vazegepant (NCT04408794), are in phase III clinical trials. Vazegepant is administered intranasally [445,446].
In addition, several novel entities for pain management are in clinical development. KarXT is combination of xanomeline, a M1/M4 muscarinic acetylcholine receptor (mACR) agonist, and trospium, an approved muscarinic receptor antagonist. KarXT has been tested in early-stage clinical trials for various indications including pain, schizophrenia, and dementia related psychosis with promising results [447]. LX9211, a small molecule inhibitor of adaptor associated kinase 1 (AAK1), has been developed for the treatment of neuropathic pain. A phase I study demonstrated that it is well-tolerance and has no drug-related serious adverse events [448].
4.2. Neurodegenerative Disease
Neurodegenerative disease is the progressive loss of nerve cells within the nervous system. It impairs a person’s movement, speech, memory, or intelligence. Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the most common neurodegenerative diseases affecting 50 million and 10 million people, respectively worldwide [449,450]. In 2018, the economic burden of AD and PD were estimated at $1 trillion USD globally [451] and $52 billion USD in the US, respectively [450]. Direct and indirect costs include medical, social and non-medical care, drug and non-drug treatments, loss of income due to disability, and unpaid informal care by family members. AD and PD are mechanistically and clinically different, but share common features such as progressive nature, increased prevalence later in life, and destruction and irreversible loss of neurons [452]. Protein misfolding, aggregation, and accumulation play critical role in the disease pathogenesis and progression, eventually leading to cell degeneration and dysfunction [453,454,455]. In this section we will focus on the AD and PD and discuss the evolution of therapeutic modalities against each one since the first diagnosis of the disease.
4.2.1. Alzheimer’s Disease
AD is a progressive neurodegenerative disease that accounts for 80% of dementia. It is characterized by the loss of memory and other cognitive functions that interfere with daily life. Such symptoms appear due to decline, destruction, and death of nerve cells in parts of the brain that are involved in memory, language, and social behavior [456,457]. The diagnosis and prediction for AD is difficult as the root cause of AD still remains largely unclear and the non-genetic cases account for 97% of AD [458]. Clinically, AD progression can be divided into five stages: preclinical, mild cognitive impairment (MCI), mild, moderate, and severe dementia [459,460]. Autopsy studies suggest that the pathological hallmarks of AD are the extracellular senile amyloid plaques formed by the fibrillar β-amyloid (Aβ); intracellular neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau; and spreading neuron and synapse damages in the memory related brain regions, such as hippocampus and cerebral cortex [461,462,463,464]. The abnormal Aβ (1–42) level is the earliest pathological sign of the disease, which can be detected in the cerebrospinal fluid (CSF) or by PET imaging. The Aβ (1-42) level plateaus before cognitive impairment and has been widely used as a dynamic biomarker to show β-amyloid deposition. Tau level in CSF is used to determine NFT accumulation and can be measured by fluorodeoxyglucose-PET imaging. Interestingly, Tau levels keep rising and does not reach a plateau until dementia has developed [465]. A Braak staging system is used to evaluate the disease stages based on the distribution of the markers in different brain regions. Disease progression is divided into three stages: transentorhinal (Braak I-II, preclinical), limbic stages (Braak III-IV, early AD), and neocortical stages (Braak V-VI, AD) [466]. Clinical studies showed a strong correlation between Braak staging and cognitive impairment [467,468,469], suggesting that Aβ and NTF can be exploited as therapeutic targets of AD.
Only five approved drugs are in the market for AD treatment since the first patient was diagnose over 100 years ago (Table 10). These small molecule drugs are divided into two categories: acetylcholinesterase inhibitors (AChEIs) and N-methyl-D-aspartic acid (NMDA) receptor agonist. Acetylcholine (ACh) is an important neurotransmitter involved in learning and memory [470]. Synthesis of ACh is greatly reduced in AD patients. AChEIs can alleviate the cognitive impairment by preventing the breakdown of neurotransmitters [471,472]. Tacrine was the first AChEI drug available in the market, however, it was withdrawn from the market due to its liver toxicity and availability of newer AChEIs [473]. Second generation AChEIs, donepezil, rivastigmine, and galantamine are widely used with similar efficacy, fewer side effects, and preferred pharmacokinetics [474,475]. Memantine is a NMDA receptor antagonist that reduces the continuous activation of NMDA receptor and excitotoxic effects of up-regulated glutamate [476]. The use of these neurotransmitter regulating drugs as monotherapy or in combination provides temporary relief but does not delay the progression of AD. Thus, there is a large unmet need for disease modifying treatments (DMTs) for AD. Previous and current drugs were developed to target specific molecular/cellular markers that appear throughout the AD progression process. However, recent failures in clinical trials suggest earlier targeting with both treatment and diagnosing strategies is needed [477].

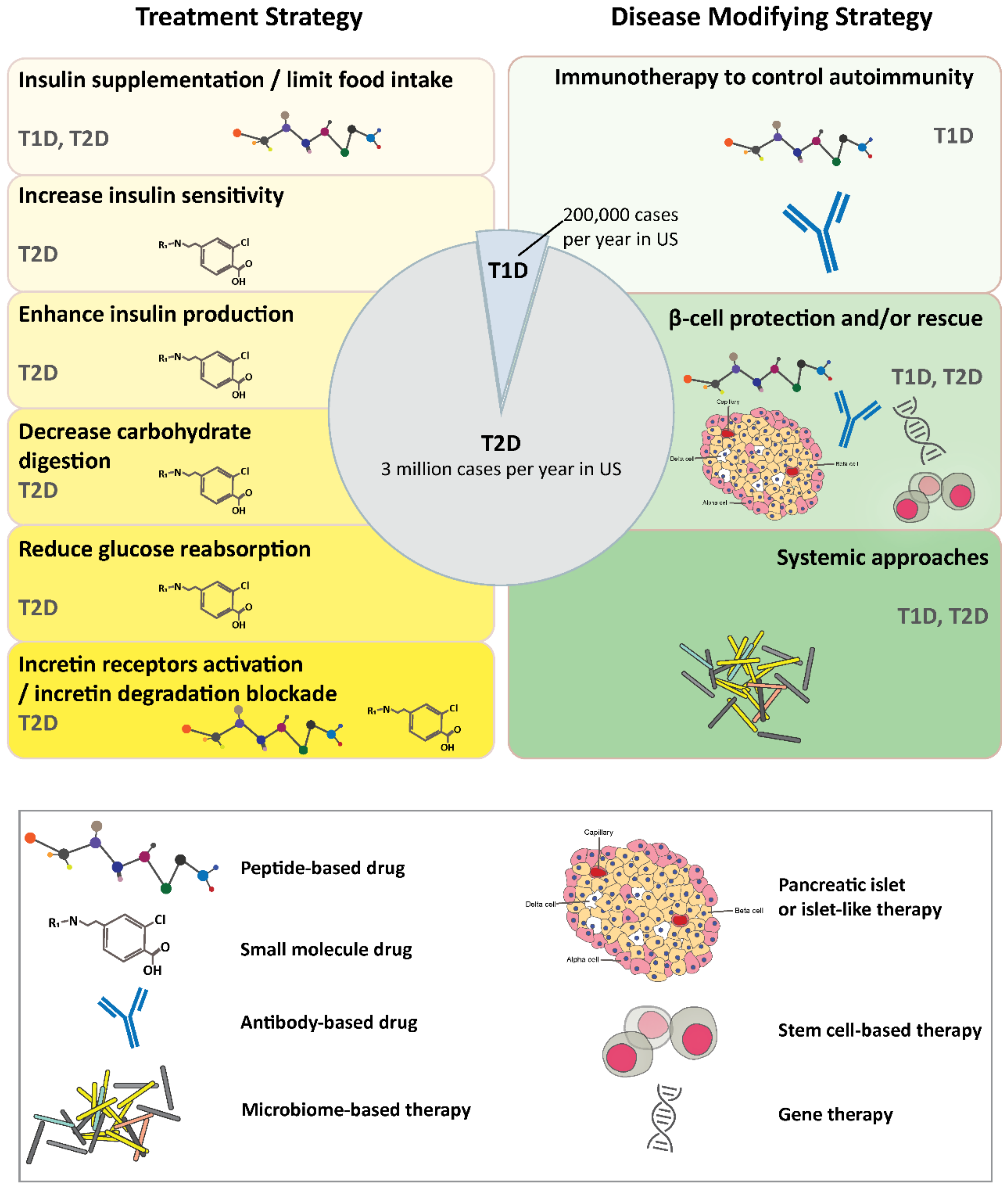{kind=link}
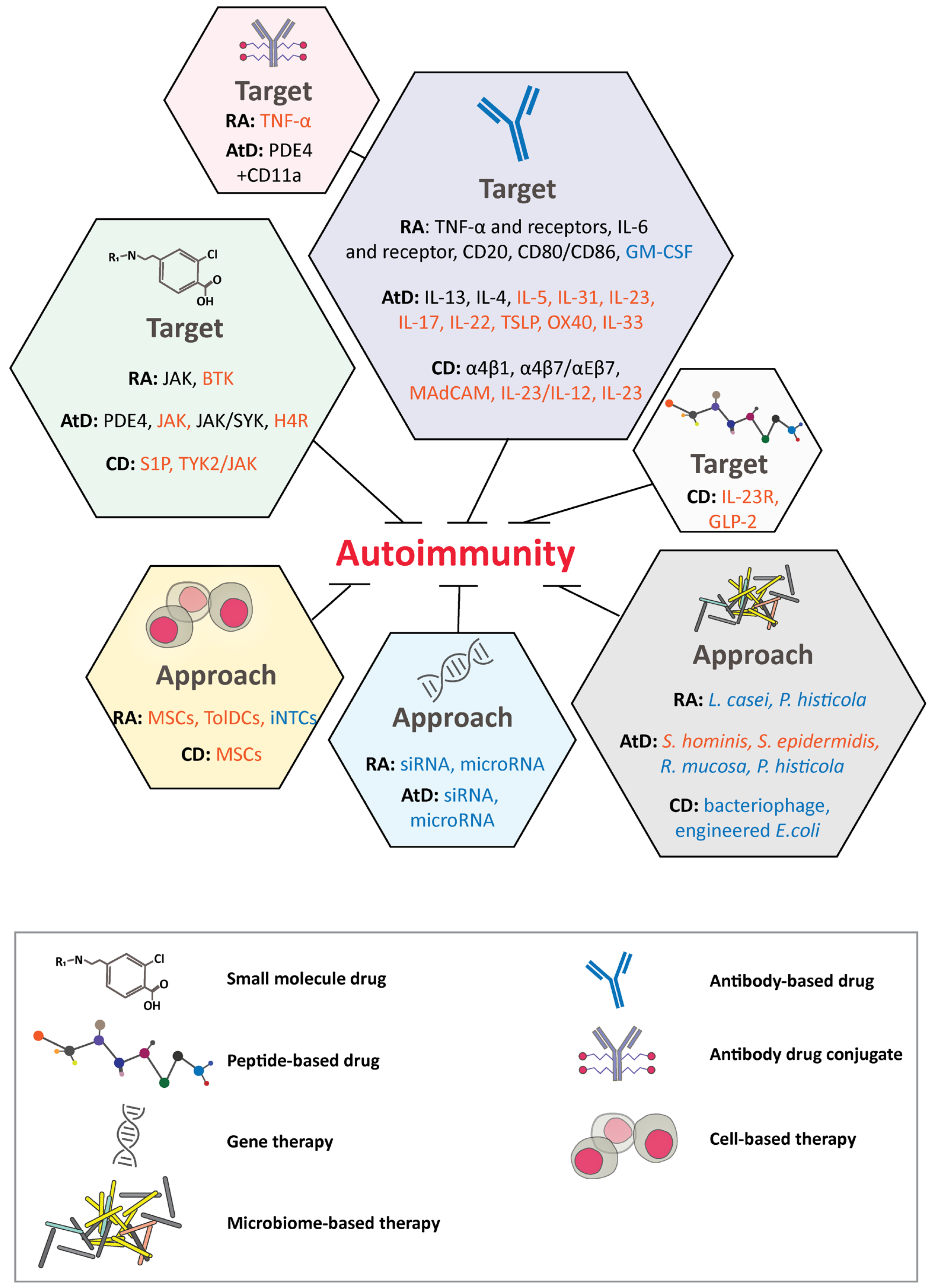{kind=link}
Table 10.
Approved therapeutics for AD.
| Drug | Approval Date | Mechanism of Action | Indication | Status | Reference |
|---|---|---|---|---|---|
| Tacrine | 1995 | AChEI | mild to moderate AD | Discontinued | [478] |
| Donepezil | 1996 | AChEI | mild to moderate AD | Approved | [479] |
| Rivastigmine | 1997 | AChEI | mild to moderate AD | Approved | [480] |
| Galantamine | 2001 | AChEI | mild to moderate AD | Approved | [481] |
| Memantine | 2003 | NMDA receptor agonist | Moderate to severe AD | Approved | [482] |
Amyloid hypothesis [463,483,484] has fueled the development of the majority of the current DMTs. It suggests that formation of Aβ and senile amyloid plaques (insoluble fibrils in the brain) initiates a cascade of events leading to AD. Aβ is generated through the abnormal processing of the 695-residue isoform of amyloid precursor protein (APP). APP is sequentially cleaved by β-secretase (BACE1) and γ-secretase to form Aβ. The aggregation-prone Aβ proteins (Aβ40 and Aβ42) oligomerize and eventually form amyloid plaques, causing neurotoxicity [485]. Aβ42 is more toxic and fibrillogenic than Aβ40 and is the dominant contributor of senile amyloid plaques [486]. AD research for DMTs has focused on the reduction of Aβ42 production, fibrillization and seeding, and Aβ clearance through the use of biologics or small molecules (BACE1 or γ-secretase inhibitors) [487,488]. Despite of the massive efforts and research (Table 11), no drugs have been approved since 2003. Recent failures of anti-amyloid drugs in phase III clinical trials in patients with early, mild, and mild-to-moderate AD include anti-Aβ specific mAbs [488,489,490,491,492,493,494] and γ-secretase small molecule inhibitor [495]. The few remaining anti-amyloid drugs in phase III are aducanumab, BAN2401 [496], and gantenerumab [497]. Although aducanumab did not meet its endpoint in phase III ENGAGE study, the sponsor argued that the subset of patients who received a high dose of the drug showed significant benefits. The biologics license application of aducanumab has been accepted for priority review by FDA [498] and a phase IIIb (NCT04241068) has been opened to evaluate the long-term safety and tolerability in patients who previously had received aducanumab.
Failure of AD clinical trials can be explained by inaccurate choice of the targets, difficulty in disease diagnosis and prediction, lack of connectivity between animal models and human, poor uptake of mAb into the CNS across the BBB, or inadequate understanding of the pathophysiology of AD [499,500,501,502,503]. The lack of success with the chosen targets to treat the disease questions the validity of the amyloid hypothesis. It is unclear if targeting various form of Aβ such as monomeric, oligomeric, aggregates, or plaques will stop or reverse the progress of the disease. It might be already too late to treat patients with mild cognitive impairment as the pathological progression of AD is thought to occur years before diagnosis [500,504]. This brings up the critical question as to what the best treatment window for AD is. To prevent AD, UB-311, a peptide vaccine targeting Aβ1-14 was developed. UB-311 prevented Aβ aggregation in transgenic mice model [505] and progressed to phase IIa in 2018 [506]. However, given the unpredictable onset and the long time needed for the AD to develop, the future of AD vaccine remains unknown. The efficacy data in small animal AD models has failed to be translated in human studies, suggesting the need for developing relevant and multiple preclinical models [503,507,508]. Poor permeability of therapeutic antibodies through the BBB has also been attributed to a lack of efficacy [501]. Engineering antibodies capable of crossing BBB might increase their potency against Aβ in the brain [501,509,510]. RO7126209 is one of the earliest examples of an Aβ mAb with a brain-shuttle technology currently in phase I clinical trials (NCT04023994). RO7126209 is based on transferrin receptor single-chain Fab fragment attached to the C-terminus of gantenerumab Fc. The fusion protein has been shown to be effective in AD mouse models [511].
In the healthy state, tau protein binds to microtubules in cells to stabilize and facilitate neuronal transport system. In AD brain, the hyperphosphorylated tau is released from the microtubules, forms aggregates, and folds into NFTs, inhibiting neuronal transport and microtubule function. Initially the tau hypothesis was thought to be a downstream event of Aβ pathology [512]. However, it is likely that tau and Aβ act through parallel pathways to cause AD with magnified toxic effects [513]. Biologics and small molecules targeting tau-dependent mechanisms include tau assembly inhibitors, tau kinase inhibitors, or microtubule stabilizers (Table 10). Tau protein aggregation inhibitor, leuco-methylthioninium bis (hydromethanesulphonate) (LMTM) is the most advanced entity in clinical development [514]. Similar to aducanumab, LMTM failed to meet its end point in phase III clinical trials but is still moving forward with an expanded access program (NTC03539380) to treat patients with early to mild-moderate AD. Data from the phase III study was re-analyzed and focused on a subset that received a low dose of LMTM to show efficacy [515]. Other small molecule- and mAb-based tau therapies are still in the early clinical trials and need time to assess success. Tau protein contains a large number of potential phosphorylation sites with more than 30 sites associated with the formation of NFT [516,517]. It was reported that O-GlcNAcylation of tau inhibits its phosphorylation. It is also shown that levels of O-GlcNAcylated tau are decreased in the brain of AD patients [518] O-GlcNAcylation was shown to interrupt tau aggregation and slowed down neurodegeneration in mouse model [519,520]. Therefore, increasing tau O-GlcNAcylation may serve as a new direction to delay disease progress.
Tau has also been a target of interest in early on-set AD diagnosis. The high cost, limited availability, and invasive nature of current AD diagnosis using PET or CSF analysis has encouraged development of affordable and accurate alternatives [521,522]. Recently approved blood-based tests for p-tau 181 and p-tau 217 can differentiate AD from other neurodegenerative diseases with accuracy close to PET or CSF methods [523,524].
Neuroinflammation contributes to early AD progression [525]. Microglial cells are first line of defense and use phagocytosis to eliminate foreign substances. They also play an important part in neurogenesis, neuronal plasticity, and regeneration. Activation of microglia is a typical pathophysiological characteristic of AD, resulting in generation of either neuroprotective (anti-inflammatory cytokines IL-4, IL-6, IL-10, IL-11, and IL-13) or cytotoxic (pro-inflammatory cytokines IL-1, TNF-α, IL-6, IL-8, and IFN-γ) effects [526,527]. There are a substantial number of clinical trials investigating the effect of targeting neuroinflammation in AD (Table 8). The most advanced small molecules are ALZT-OP1, Azeliragon, and Masitinib in phase III clinical trials. ALZT-OP1 is a combination of cromolyn and ibuprofen, both approved anti-inflammatory agents. Cromolyn has been shown to reduce Aβ aggregation by promoting microglial phagocytosis in animal studies [528]. Ibuprofen is a NSAID with extensive preclinic and clinical evaluation in AD [529]. Treatment with ALZT-OP1 shifts microglial immune cells from their pro-inflammatory state to their neuroprotective state [530]. Azeliragon is an inhibitor of receptor for advanced glycation end products (RAGE), which plays a role in proinflammation response, induction of oxidative stress, and Aβ clearance [531]. RAGE is upregulated in microglia in AD patients and is thought to mediate the transport of plasma Aβ into the brain [532]. Preclinical studies in small animals have shown that azeliragon reduces Aβ plaque deposition, decreases total Aβ concentration in brain while increasing plasma Aβ levels, reduces levels of inflammatory cytokines, and more importantly slows cognitive decline [533]. Masitinib is a tyrosine kinase inhibitor that targets and blocks the activation of mast cells [534]. In small animal studies, stimulation of mast cells results in the release of proinflammatory signals to microglia, promotes neuroinflammation, and generates Aβ containing fragments due to upregulation of chymotrypsin-like protease [535]. Preclinical studies in small animal AD models with masitinib had no effect on Aβ concentration or neuroinflammation (measured by IL-1β level) but restored cognitive function. Masitinib has shown synapto-protective properties, most possibly due to inhibition of synaptic toxins released by mast cells [536]. Phase II clinical studies with masitinib reduced the rate of cognitive decline in AD patients with acceptable tolerance [537]. Phase II/III study with mastinib in AD is currently on-going. Interestingly, the most recent research has connected the gut bacteria composition to AD-related neuroinflammation [538]. It was reported that the change in gut microbiota population enhances Th1 proliferation and brain-infiltration in AD mice model [539,540]. Sodium oligomannate, GV-971, is approved to down-regulate neuroinflammation by reconstituting the gut bacteria composition. GV-971 has met its phase III endpoint in China and is currently recruiting patients for phase III in the US [539,540].
Additional neuroinflammation targets include triggering receptor expressed on myeloid cells 2 (TREM2), sialic acid binding Ig-like lectin 3 (SIGLEC 3), and TNF-α. AL002 is an agonist mAb against microglial receptor TREM2. TREM2 activation is thought to promote cell migration, survival, proliferation, and enhance phagocytosis of Aβ plaques and tau aggregates [541]. Preclinical studies in small animal AD models show that treatment with AL002 lowers Aβ plaques, reduces abnormal behavior, and enhances microglial response to Aβ [542]. AL002 was safe and well tolerated in phase I trials and expected to enter phase II. AL003, an antagonist of SIGLEC 3, blocks TREM2 signaling and inhibits phagocytosis [543]. SIGLEC 3 Inhibition enhances activity of microglial and its neuroprotective effects. AL003 is still in early phase I clinical trials. Etanercept and XPro1595 target inflammatory cytokine TNF-α. Etanercept contains two copies of TNF-α receptor fused to the Fc of IgG and binds soluble and membrane bound forms of TNF-α. XPro1595 is a non-receptor binding variant of TNF-α that forms heterotrimers with endogenous soluble TNF-α to inhibit neuroinflammation mediated by TNFR1. XPro1595 does not suppress TNFR2 [544]. In preclinical AD animal models, XPro1595 showed reduction in Aβ and improved synaptic function [545,546,547,548]. XPro1595 is currently recruiting patients with mild to moderate AD for phase I clinical trials. Etanercept completed phase II in 2015 with promising results, however further evaluation was halted without any explanation.
Metabolic disorders such as diabetes, obesity, hypertension, and dyslipidemia may worsen neurological symptoms through poorly defined mechanisms [549,550]. An example is the involvement of BACE1 in AD and metabolic disorders. BACE1 levels are typically associated with raised Aβ peptide and increased risk of AD [551,552]. It was shown that the reduction in BACE1 levels protects against diet-induced obesity and increases glucose metabolism and insulin sensitivity in preclinical studies [553]. Increased neuroinflammation, elevated BACE1 levels, and cognitive dysfunction was observed in small animal models with hypercholesterolemia [554]. Additionally, the deficiency of adiponectin (APN), a protective protein hormone, has been associated with insulin resistance, neuroinflammation, and cognitive impairment in rodents and human. APN receptor agonist, adipoRon, results in improved insulin sensitivity, reduced microglial activation, and decreased plaque and Aβ levels in AD small animal models [555,556]. AD has recently been termed “type-3 diabetes” due to the similarity between insulin resistance and insulin-like growth factor dysfunction to AD-like neurodegeneration [557,558]. Multiple AD clinical trials are ongoing with drugs originally approved for diabetes (Table 8). Nasal insulin and metformin are currently in phase II/III clinical trials. The role of insulin in the brain is poorly understood, however, it has been suggested to play a role in glucose uptake and synaptic remodeling [559,560]. Insulin involvement in proteostasis can influence Aβ peptide clearance and tau phosphorylation. Insulin dysregulation is associated with vascular dysfunction, inflammation, and dyslipidaemia [561]. Nasal administration of insulin has the advantage of delivery into the brain through olfactory routes. Since bypassing the peripheral blood system, the undesired side effects of hypoglycemia or insulin resistance can be eliminated [562]. Intranasal insulin improved and preserved short- and long-term memory in small animal AD models [563] and in pilot clinical trial studies. Intranasal administration of insulin has also shown to improve memory and cognition in patients with AD [564,565,566]. Safety and efficacy of intranasal insulin over a twelve month period by two different devices is under evaluation in phase II/III for AD patients with mild cognitive impairment [567]. Inconsistent reliability of one of the devices led to the use of the second device for the remaining participants and designated main cohort. Results showed no cognitive or functional benefits with intranasal insulin treatment compared to the placebo group. It is important to note that issues with insulin delivery device complicated the study protocol and interpretation of the results. Therefore, further research is needed to evaluate the therapeutic benefit of intranasal insulin in AD patients. Metformin is a widely used drug for T2D treatment. It restores the response to insulin, decreases blood sugar production in the liver, and increases intestinal and stomach glucose absorption [568]. Preclinical AD mouse models treated with metformin showed improved cognitive behavior and decreased levels of Aβ and tau phosphorylation. Additionally, pilot clinical trials with metformin showed promising memory improvement in patients with mild cognitive impairment compared to the placebo group [569,570]. Phase II/III trial for patients with mild cognitive impairment and AD is currently underway. Other diabetes drugs studied for AD therapy include GLP-1 analog, Liraglutide [571] (phase II), and two sodium-glucose cotransporter-2 Dapagliflozin and Empagliflozin [572] (phase I). Dyslipidemia metabolic disorder are targets of gemfibrozil (phase I), T3D-959 (phase II), and GSK2647544 (phase I). Gemfibrozil and T3D-959 are agonists of peroxisome proliferator-activated receptor (PPAR) α and PPAR δ/γ, respectively. PPARs are lipid sensors that stimulate breakdown of fatty acids and cholesterol, cause gluconeogenesis, and decrease triglyceride levels to regulate energy homeostasis [573]. PPAR α has been shown to inhibit the Aβ pathway, tau phosphorylation, and neuroinflammation [574], while PPAR δ/γ inhibits inflammatory response and decreases Aβ levels by blocking BACE1 activity [575].
Stem cell therapy provides a new potential for treatment of AD [576] (Table 12). Traditionally, stem cell therapy was used to regenerate injured cells or tissues through stem cell transplantation [577] or induce the activation of endogenous stem cells to improve the disease [578]. Recent progress and developments in preclinical [579] and clinical trials (Table 11) have sparked the interest in using stem cell therapy for AD. Currently there is one phase I and six phase II trials of stem cell therapy for AD. AstroStem are stem cells derived from autologous adipose tissue that are administered intravenously ten times into patients with AD. Phase I/II was completed in mid-2019 and result have yet to be reported (NTC03117738). Human umbilical cord blood-derived mesenchymal stem cells (hUCB-MSC) are being used by two different sponsors. Medipost has two phase I/II clinical trials to test the intra-ventricular administration of hUCB-MSC into patients with AD (NTC02054208) with intent for a long-term follow-up (NTC03172177). The hUCB-MSCs migrate towards injury signals in the brain to promote the degradation of Aβ by stimulating microglia cells that secrete β-amyloid degrading enzymes [580]. The South China Research Center is conducting two phase I/II clinical trials with intravenous administration of 20 million hUCB-MSCs over eight infusions (NTC02513706, NTC02672306). In Stemedica’s phase II study, hMSCs are administered intravenously in patients with mild to moderate AD (NCT02833792) [581]. Trial with low and high dose of Longeveron mesenchymal stem cells (LMSCs) in patients with AD (NCT02600130) is also ongoing [582]. The hope is that stem cell therapy replaces damaged neuronal cells and reverse the progression of the disease as opposed to merely reducing or blocking SPs and NFTs.
Gene therapies are being evaluated in AD with two registered clinical trials ongoing at phase II and one at phase I (Table 13). The goal is to insert the correct copy of the defective gene to restore the function of the protein of interest. Definition of gene therapy has evolved to include recombinant DNA- or RNA-based drugs for repairing, replacing, adding, altering, or blocking a gene sequence [583]. Therefore, understanding what drives manifestation of is critical in determining potential gene therapy targets [584]. In two of the ongoing trials, genes are delivered by the safe and effective AAV vectors [585] or by ASO drug design [586]. CERE-110 is an AAV-based gene therapy used for delivery of NGF to protects cholinergic neurons and improve basal forebrain neurons [587,588]. In phase II clinical trials, CRE-110 was delivered directly by bilateral stereotactic surgery in patients with mild to moderate AD (NCT00876863). CERE-110 was safe and well tolerated, but ineffective. IONIS-MAPTRX (BIIB080) is an ASO drug designed to reduce production of tau protein in the brain [589]. BIIB080 is currently in phase II with multiple ascending dose administered intrathecally in patients with mild AD (NCT03186989). Another AAV-based gene therapy targets apolipoprotein E (APOE) 2 gene for APOE4 associated AD and is currently open for recruitment for phase I clinical trials (NCT03634007). APOE4 gene inheritance is a major risk factor for AD. APOE4 homozygotes have a markedly increase risk of developing AD when compared to APOE3 homozygotes. In contrast APOE2 is a protective gene, reducing AD by 50% and delaying the disease onset even in the presence of APOE4 [590]. The safety and toxicity profile of AAVrh.10hAPOE2 via intracisternal administration will be assessed in AD patients who are APOE4 homozygotes.
Devices are a non-drug alternative modality for treatment of AD (Table S1). Many of the deep brain stimulation (DBS) devices are considered to stimulate cognitive modulation, enhance blood flow to promote amyloid and tau clearance, reduce inflammation and oxidative stress, and disrupt BBB at certain level to allow passage of drugs [488,591]. Use of devices can be invasive (surgically implanted electrodes) or non-invasive (light application, electrical current, ultrasound, electromagnetic, IR-LED and laser therapy). Completed studies have shown no consistent cognitive benefit with DBS [592].
4.2.2. Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide [593], affected by both genetic and environmental risk factors. Eighteen chromosomal regions identified as PARK loci are among PD-related genetic factors. An example is the mutation of α-synuclein coding gene PAK1, which is associated with the increased risks of PD. The primary risk factor for PD is aging. Various environmental elements, such as nitrative stress as well as insecticides that cause mitochondrial dysfunction and oxidative stress [594,595] are associated with the disease. The physical symptoms are combination of motor disabilities such as tremors, slowed movement (bradykinesia), stiffness, difficulty with balance and coordination, and nonmotor symptoms such as apathy, depression, sleep disorders, constipation, loss of sense of smell, and cognitive impairment [596]. At the cellular level, the disease is primarily characterized by the formation of intraneuronal Lewy bodies (LB), consistent of the aggregated α-synuclein (α-syn) and neurofilament proteins, and the irreversible loss of dopaminergic neurons in the substantia nigra in the midbrain [597,598,599]. Other neurotransmitter systems (glutamatergic, cholinergic, adrenergic) are also impacted. [594] but α-syn aggregation and LB formation is the major drivers of neurodegeneration in PD [600,601]. In a healthy state, α-syn at synaptic terminals is involved in vesicle trafficking and recycling [601]. Oligomerization, fibrillization, and aggregation of α-syn results in LB formation and disruption of cellular functions, mitochondria damage, synaptic dysfunctions, and neuronal death [600]. Dopamine is a type of neurotransmitter that interacts with various receptors (D1 and D2 class) in the pre- and post- synaptic space to modulate neuronal excitation or inhibition [602]. The death of dopaminergic neurons in the substantia nigra leads to loss of movement coordination and muscle contraction.
There is no cure for PD. The primary goal is to reduce the symptoms through regulating the dopamine level in the brain or to restore synaptic plasticity. Current drugs alleviate motor symptoms by dopamine substitution (levodopa, dopamine agonist, monoamine oxidase-B inhibitors, catechol-o-methyl transferase inhibitors) and/or by targeting alternative neurotransmitter system (anticholinergics, amantadine and istradefylline) (Table 14). Levodopa is converted to dopamine by DOPA decarboxylase in the brain and is effective for relief of short-term symptoms of PD. Levodopa efficacy is enhanced when used in combination with caribidopa, a decarboxylation inhibitor that prevents the breakdown and metabolism of levodopa in the peripheral blood [603]. Long-term use of levodopa is associated with motor fluctuations and involuntary movements (dyskinesia) [604]. Dopamine receptor activation is another therapeutic strategy. Dopamine agonists bind to D2 receptor family in the brain that are involved in the control of voluntary movement. Side effects associated with dopamine agonists include constipation, nausea, headaches, sedation, hallucination, and impulse control disorders [605]. Monotherapy with dopamine agonists are recommended in young and early PD patients to avoid use of levodopa that leads to motor fluctuations and dyskinesia [606]. MAO-B inhibitors block MAO-B enzyme that metabolizes dopamine. MAO-B inhibitors are taken alone or in combination with levodopa to enhance the endogenous levels of dopamine as well as its production [607]. COMT antagonists inhibit degradation of dopamine, they have no direct effect on PD symptoms, and should be used in combination with other PD drugs [608].
Anticholinergic drugs block function of the neurotransmitter acetylcholine to reduce muscle tremors. They can be taken alone or in combination with levodopa or dopamine agonist in patients with persistent tremors [609]. The pharmacology and mechanism of action of amantadine in PD is poorly understood. Studies suggest that amantadine is an antagonist of NMDA receptor that diminishes sustained stimulation of NMDA receptors and their excitotoxicity effects [610,611]. Amantadine may be taken alone or with levodopa-carbidopa to control dyskinesia. Istradefylline is a newly approved add-on therapy to levodopa-carbidopa for PD. It is an adenosine receptor antagonist with unknown mechanism of action and it is presumed to reduce overactivity of the striatal pathway to restore balance in the basal ganglia [612].
An alternative FDA approved PD therapy is use of surgical deep brain stimulation (DBS) to suppress pathological neuronal oscillations. For that purpose, electrodes are inserted into the motor circuit components of the brain, including ventral intermediate nucleus of the thalamus (VIM), globus pallidus (GPi), and the subthalamic nucleus (STN) [613,614]. The electrodes are connected to an impulse generator battery (IPG) that sends electrical impulses to the brain. Due to its invasive nature, risk versus benefit should be carefully assessed. The procedure is suggested for patients with advanced PD with disabling motor symptoms, poor response to medication, and dyskinesias. DBS might affect synapse function or interfere with pathological events [615,616,617,618]. Even the most recently approved drugs, istradefylline and orphenadrine, are for symptomatic relief and do not offer a cure.
There has been a surge of clinical trials of drugs against PD (Table 15). Current understanding of the molecular mechanisms causing PD have provided insights into new targets as well as novel approaches that are discussed below [619,620,621,622]. The two main strategies involve lysosome clearance by small molecule and gene therapy and neutralization of α-synuclein by mAbs and small molecules.
Lysosome enzyme glucocerebrosidase (GCase) is involved in cellular clearance of waste proteins. In vitro studies show that GCase activation was effective in reducing α-synuclein inclusions and cytotoxicity in neurons [623]. In synucleinopathy mouse models, AAV-mediated overexpression of GCase in brain reduces pathology and memory loss [624]. Ambroxol is a small molecule activating GCase, currently in phase II for PD patients with dementia [625]. PR001 is a gene-based therapy that uses adeno-associated virus 9 (AAV9) to deliver GBA1 gene encoding for GCase to the brain. PR001 is administered directly to the cisterna magna at the base of the brain through a single injection [626]. Phase I and II clinical trials are currently recruiting patients with at least one GBA1 mutation to test PR001′s safety, tolerability, and efficacy.
Immunotherapy drugs have focused on targeting α-synuclein. Because of its prevalence in PD pathology, α-synuclein has become the main target of mAbs. Cinpanemab, binds to α-synuclein residues 1–10 and has a high affinity for aggregated α-synuclein. Cinpanemab attenuates α-synuclein spreading and decreases pathology and motor symptoms in mouse models [627]. Cinpanemab is being evaluated in phase II. Prasinezumab is a humanized IgG1 mAb against aggregated α-synuclein in phase II. LU AF82422 (mAb targeting the C-terminal of α-synuclein) and MEDI1341 (mAb targeting monomeric and aggregated α-synuclein) are in phase I clinical trials. Small molecules NPT200-11 and anle138b also target α-synuclein by preventing its misfolding and aggregation. Treatment with NPT200-11 is shown to reduce α-synuclein pathology and neuroinflammation and improve motor function in small animal PD models that overexpress α-synuclein [628]. Phase Ib was initiated for testing of NPT-200-11 in patients with PD [629]. Anle138b inhibits α-synuclein oligomer formation and improves neuronal function and movement in small animal PD models [630,631]. The phase I results showed that Anle138b was safe and tolerable in healthy adults with no drug specific side effects [632]. Anti-cancer drugs such as Abl kinase inhibitors are potential DMT for PD. Abl kinase phosphorylates α-synuclein and prevents its degradation. Therefore, α-synuclein clearance by autophagy is possible through Abl kinase inhibition. Three Abl kinase inhibitors (Nilotinib and K0706 in phase II and FB-101 in phase I) are currently in clinical trials in PD patients.
Better understanding of what impacts PD pathology has resulted in emergence of novel drugs. Anavex 2–73 is a small molecule sigma-1 receptor (SIGMAR1) agonist. SIGMAR1 activity restores neuronal cell homeostasis and promotes neuroplasticity [633]. Anavex 2–73 is currently in phase II clinical trials for PD patients with dementia. Leucine-rich repeat kinase 2 (LRRK2) activity contributes to PD pathology by affecting vesicle trafficking and lysosome function [634,635]. There are three phase I trials for LRRK2 inhibition, including small molecules DNL151 and DNL201 and gene therapy ASO drug BIIB094. Last on PD DMT list, cerebral dopamine neurotrophic factor (CDNF) is a neurotrophic peptide that promotes neuronal survival by regulating the unfolded protein response (UPR). UPR is involved in a signaling pathway that contributes to ER stress and cell death in multiple neurodegenerative proteinopathies [636]. CDNF protects dopamine neurons from degeneration and restores their function in PD animal studies [637,638]. CDNF was dosed directly into the brain using an implanted investigational drug delivery system and recently completed phase I. It is currently under evaluation for long-term safety and tolerability.
5. Concluding Remarks
The concept of druggability first described in 2002 [639] refers to feasibility of the target to be engaged by a ligand. Advances in science and novel molecular modalities, outlined here, have made many targets “druggable”. Novel targets in diabetes, autoimmune diseases, and neurological diseases are also discussed. Peptide-based therapeutics are dominant in diabetes, whereas small molecules and mAbs are the key modalities for treatment of autoimmune diseases, chronic pain, and neurodegenerative diseases. Gene and cell therapies have evolved for tissue and cell regeneration. The advent of early detection methods, precision intervention, and customized treatments have revolutionized the treatment options in the recent decade. The combination of small molecule, mAbs, peptides, and ONs is an emerging trend to promote efficacy against multiple targets and/or pathways involved in the disease.
Supplementary Materials
Supplementary materials can be found at https://0-www-mdpi-com.brum.beds.ac.uk/1422-0067/22/6/2805/s1.
Author Contributions
Original draft preparation, review and editing: J.L., J.P.T., S.A.-A., Y.D., S.A. These authors have contributed equally to this work: J.P.T., S.A.-A., Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Eli Lilly and Company. The funder provided support in the form of salaries for authors and had no additional role in the manuscript preparation. This research received no external funding.
Conflicts of Interest
At the time of this manuscript preparation, all authors were employees of Eli Lilly and Company. The authors declare no conflict of interest.
Abbreviations
| FDA | U.S. Food and Drug Administration |
| CDER | The Center for Drug Evaluation and Research |
| CBER | The Center for Biologics Evaluation and Research |
| WHO | World health organization |
| mAb | Monoclonal antibody |
| bsAb | Bispecific antibody |
| ADC | Antibody drug conjugate |
| CAR T | Chimeric antigen receptor T cell |
| TNF | Tumor necrosis factor |
| GPCR | G-protein coupled receptor |
| GLP-1 | Glucagon-like peptide-1 |
| GIP | Glucose-dependent insulinotropic polypeptide |
| AAV | Adeno-associated virus |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| ON | Oligonucleotide |
| ASO | Antisense oligonucleotide |
| siRNA | Short interfering RNA |
| GalNac | N-acetylgalactosamine |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| T1D | Type 1 diabetes mellitus |
| T2D | Type 2 diabetes mellitus |
| HbA1c | Hemoglobin A1c |
| HSC | Hematopoietic stem cell |
| MSC | Mesenchymal stem cell |
| NF-κB | Nuclear factor -κB |
| JAK | Janus kinase |
| STAT | Transducer and activator of transcription |
| SYK | Spleen tyrosine kinase |
| TYK2 | Tyrosine kinase 2 |
| BTK | Bruton’s tyrosine kinase |
| AtD | Atopic dermatitis |
| Th | T helper |
| PDE4 | Phosphodiesterase 4 |
| EASI | Eczema area and severity index |
| H4R | Histamine H4 receptor |
| MAdCAM | Mucosal addressin cell adhesion molecule |
| S1P | Sphingosine 1-phosphate |
| CNS | Central nervous system |
| OA | Osteoarthritis |
| NSAID | Nonsteroidal anti-inflammatory drug |
| COX | Cyclooxygenase |
| NGF | Nerve growth factor |
| MMP | Matrix metalloproteinase |
| CGRP | Calcitonin gene-related peptide |
| ND | Neurodegenerative disease |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| AChEIs | Acetylcholinesterase inhibitors |
| NMDA | N-methyl-D-aspartic acid |
| DMTs | Disease-modifying treatments |
References
- FDA. FDA-Regulated Products and Facilities. 2019. Available online: https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance (accessed on 1 August 2020).
- Meier, C.; Cairns-Smith, S.; Schulze, U. Can emerging drug classes improve R&D productivity? Drug Discov. Today 2013, 18, 607–609. [Google Scholar] [CrossRef] [PubMed]
- David, E.; Tramontin, T.; Zemmel, R. Pharmaceutical R&D: The road to positive returns. Nat. Rev. Drug Discov. 2009, 8, 609–610. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Yu, J.X.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef] [Green Version]
- Czechowicz, A.; Palchaudhuri, R.; Scheck, A.; Hu, Y.; Hoggatt, J.; Saez, B.; Pang, W.W.; Mansour, M.K.; Tate, T.A.; Chan, Y.Y.; et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 2019, 10, 617. [Google Scholar] [CrossRef] [Green Version]
- Gillard, G.R.; Proctor, J.; Brooks, M.; Lamothe, T.; Hyzy, S.; McDonough, S.; Palchaudhuri, R.; Bhat, A.; Sarma, G.; Bhattarai, P.; et al. Administration of a CD45 Antibody Drug Conjugate as a Novel, Targeted Approach to Achieve Immune System Reset: A Single Dose of CD45-targeted ADC Safely Conditions for Autologous Transplant and Ameliorates Disease in Multiple Models of Autoimmune Disease [abstract]. Arthritis Rheumatol. 2019, 71 (Suppl. 10), 120. [Google Scholar]
- Jovcevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ting, J.P.; Liu, J.; Al-Azzam, S.; Pandya, P.; Afshar, S. Impact of non-proteinogenic amino acids in the discovery and development of peptide therapeutics. Amino Acids 2020, 52, 1207–1226. [Google Scholar] [CrossRef]
- Valeur, E.; Gueret, S.M.; Adihou, H.; Gopalakrishnan, R.; Lemurell, M.; Waldmann, H.; Grossmann, T.N.; Plowright, A.T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem. Int. Ed. Engl. 2017, 56, 10294–10323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Afshar, S. In Vitro Assays: Friends or Foes of Cell-Penetrating Peptides. Int. J. Mol. Sci. 2020, 21, 4719. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Yang, W.; Gadgil, P.; Krishnamurthy, V.R.; Landis, M.; Mallick, P.; Patel, D.; Patel, P.J.; Reid, D.L.; Sanchez-Felix, M. The Evolving Druggability and Developability Space: Chemically Modified New Modalities and Emerging Small Molecules. AAPS J. 2020, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Njardarson, J.T. Top 200 Brand Name Drugs by Retail Sales in 2019. 2019. Available online: https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed on 2 August 2020).
- McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, H.; Ghosh, D.; Williams, R.O., 3rd. Just how prevalent are peptide therapeutic products? A critical review. Int. J. Pharm. 2020, 587, 119491. [Google Scholar] [CrossRef] [PubMed]
- Ashmore-Harris, C.; Fruhwirth, G.O. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin. Transl. Med. 2020, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Katrekar, D.; Chen, G.; Meluzzi, D.; Ganesh, A.; Worlikar, A.; Shih, Y.R.; Varghese, S.; Mali, P. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat. Methods 2019, 16, 239–242. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, W.; Mei, C.; Chen, X.; Yao, Y.; Liu, Y.; Qin, X.; Yuan, Y. Description of CRISPR/Cas9 development and its prospect in hepatocellular carcinoma treatment. J. Exp. Clin. Cancer Res. 2020, 39, 1–11. [Google Scholar] [CrossRef]
- Stein, R. First U.S. Patients Treated With CRISPR As Human Gene-Editing Trials Get Underway. 2019. Available online: https://www.npr.org/sections/health-shots/2019/04/16/712402435/first-u-s-patients-treated-with-crispr-as-gene-editing-human-trials-get-underway (accessed on 2 August 2020).
- Henderson, H. CRISPR Clinical Trials: A 2019 Update; Innovative Genomics Institute: Berkeley, CA, USA, 2019. [Google Scholar]
- Rosenbaum, L. New Data From First Human Crispr Trials Shows Promising Results. 2019. Available online: https://www.forbes.com/sites/leahrosenbaum/2019/11/19/human-crispr-trials-promising/?sh=7e470f132daa (accessed on 10 August 2020).
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Viscomi, C.; Simard, M.L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, C.; Ding, Y.; Fei, Y.; Lu, B. ATTEC: A potential new approach to target proteinopathies. Autophagy 2020, 16, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fei, Y.; Lu, B. Emerging New Concepts of Degrader Technologies. Trends Pharmacol. Sci. 2020, 41, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, M.; Langer, R.; Traverso, G. Microbial therapeutics: New opportunities for drug delivery. J. Exp. Med. 2019, 216, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Das, P.; Buschmann, M.; Gilbert, J.A. The Future of Microbiome-Based Therapeutics in Clinical Applications. Clin. Pharmacol. Ther. 2020, 107, 123–128. [Google Scholar] [CrossRef]
- Garrett, W.S. Immune recognition of microbial metabolites. Nat. Rev. Immunol. 2020, 20, 91–92. [Google Scholar] [CrossRef]
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Li, B.; Sun, D.; Chen, S. Gut Microbiota: Implications in Alzheimer’s Disease. J. Clin. Med. 2020, 9, 2042. [Google Scholar] [CrossRef]
- Charbonneau, M.R.; Isabella, V.M.; Li, N.; Kurtz, C.B. Developing a new class of engineered live bacterial therapeutics to treat human diseases. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- International Diabetes Federation. IDF Diabetes Atlas 2019. 2019. Available online: https://www.diabetesatlas.org/en/ (accessed on 20 August 2020).
- American Diabetes Association. Economic Costs of Diabetes in the U.S. in 2017. Diabetes Care 2018, 41, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Daneman, D. Type 1 diabetes. Lancet 2006, 367, 847–858. [Google Scholar] [CrossRef]
- Redondo, M.J.; Fain, P.R.; Eisenbarth, G.S. Genetics of type 1A diabetes. Recent Prog. Horm. Res. 2001, 56, 69–89. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- American Diabetes, A. Gestational diabetes mellitus. Diabetes Care 2003, 26 (Suppl. S1), S103–S105. [Google Scholar] [CrossRef] [Green Version]
- Melmer, A.; Laimer, M. Treatment Goals in Diabetes. Endocr. Dev. 2016, 31, 1–27. [Google Scholar] [CrossRef]
- Kahanovitz, L.; Sluss, P.M.; Russell, S.J. Type 1 Diabetes—A Clinical Perspective. Point Care 2017, 16, 37–40. [Google Scholar] [CrossRef]
- Pickup, J.C. Insulin-pump therapy for type 1 diabetes mellitus. N. Engl. J. Med. 2012, 366, 1616–1624. [Google Scholar] [CrossRef]
- Ryan, G.J.; Jobe, L.J.; Martin, R. Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin. Ther 2005, 27, 1500–1512. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1219–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, R.E.; Dickey, R.; Fineman, M.; Maggs, D.G.; Shen, L.; Strobel, S.A.; Weyer, C.; Kolterman, O.G. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long-term glycaemic and weight control in Type 1 diabetes mellitus: A 1-year, randomized controlled trial. Diabet Med. 2004, 21, 1204–1212. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in patients with type 2 diabetes and kidney disease: A systematic review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, A.L.; Travaglini, M.T. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy 2001, 21, 1082–1099. [Google Scholar] [CrossRef] [PubMed]
- Beysen, C.; Murphy, E.J.; Nagaraja, H.; Decaris, M.; Riiff, T.; Fong, A.; Hellerstein, M.K.; Boyle, P.J. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J. Lipid Res. 2008, 49, 2657–2663. [Google Scholar] [CrossRef] [Green Version]
- Tessier, D.; Dawson, K.; Tetrault, J.P.; Bravo, G.; Meneilly, G.S. Glibenclamide vs gliclazide in type 2 diabetes of the elderly. Diabet Med. 1994, 11, 974–980. [Google Scholar] [CrossRef]
- Foster, R.H.; Plosker, G.L. Glipizide. A review of the pharmacoeconomic implications of the extended-release formulation in type 2 diabetes mellitus. Pharmacoeconomics 2000, 18, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Langtry, H.D.; Balfour, J.A. Glimepiride. A review of its use in the management of type 2 diabetes mellitus. Drugs 1998, 55, 563–584. [Google Scholar] [CrossRef]
- Crowley, M.F.; Wolff, F.W.; Bloom, A. Tolbutamide in diabetes; some clinical and biochemical studies. Br. Med. J. 1957, 2, 327–331. [Google Scholar] [CrossRef]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; Group, S.-N.T.R. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Scott, L.J.; Spencer, C.M. Miglitol: A review of its therapeutic potential in type 2 diabetes mellitus. Drugs 2000, 59, 521–549. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hisauchi, I.; Taguchi, I.; Sezai, A.; Toyoda, S.; Tomiyama, H.; Sata, M.; Ueda, S.; Oyama, J.I.; Kitakaze, M.; et al. Effects of canagliflozin in patients with type 2 diabetes and chronic heart failure: A randomized trial (CANDLE). ESC Heart Fail. 2020, 7, 1585–1594. [Google Scholar] [CrossRef]
- Deeks, E.D.; Scheen, A.J. Canagliflozin: A Review in Type 2 Diabetes. Drugs 2017, 77, 1577–1592. [Google Scholar] [CrossRef]
- Dhillon, S. Dapagliflozin: A Review in Type 2 Diabetes. Drugs 2019, 79, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.E. Empagliflozin: A Review in Type 2 Diabetes. Drugs 2018, 78, 1037–1048. [Google Scholar] [CrossRef]
- Lorenz, M.; Evers, A.; Wagner, M. Recent progress and future options in the development of GLP-1 receptor agonists for the treatment of diabesity. Bioorg. Med. Chem. Lett. 2013, 23, 4011–4018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M. Incretin therapies: Highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes. Metab. 2016, 18, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroda, V.R. A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 22–33. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Alogliptin: A review of its use in patients with type 2 diabetes mellitus. Drugs 2015, 75, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Sitagliptin: A Review in Type 2 Diabetes. Drugs 2017, 77, 209–224. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Saxagliptin/Dapagliflozin: A Review in Type 2 Diabetes Mellitus. Drugs 2017, 77, 319–330. [Google Scholar] [CrossRef]
- McGill, J.B. Linagliptin for type 2 diabetes mellitus: A review of the pivotal clinical trials. Ther. Adv. Endocrinol. Metab. 2012, 3, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Linagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2012, 72, 1793–1824. [Google Scholar] [CrossRef] [PubMed]
- Wallia, A.; Molitch, M.E. Insulin therapy for type 2 diabetes mellitus. JAMA 2014, 311, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, E.; Brennan, P. The effectiveness of continuous subcutaneous insulin pumps with continuous glucose monitoring in outpatient adolescents with type 1 diabetes: A systematic review. JBI Libr. Syst. Rev. 2012, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeland, B.; Farber, M.S. A Review of Insulin for the Treatment of Diabetes Mellitus. Home Healthc. Now 2016, 34, 416–423. [Google Scholar] [CrossRef]
- Njardarson Group. Top Pharmaceuticals Poster. Available online: https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed on 21 August 2020).
- Global Diabetes Drugs Market. Available online: https://www.fortunebusinessinsights.com/industry-reports/infographics/diabetes-drugs-market-100570 (accessed on 1 September 2020).
- Saboo, B. Key elements of successful intensive therapy in patients with type 1 diabetes. Indian J. Endocrinol. Metab. 2015, 19, S44–S46. [Google Scholar] [CrossRef]
- Fineberg, S.E.; Kawabata, T.T.; Finco-Kent, D.; Fountaine, R.J.; Finch, G.L.; Krasner, A.S. Immunological responses to exogenous insulin. Endocr. Rev. 2007, 28, 625–652. [Google Scholar] [CrossRef] [PubMed]
- PhRMA. Follow the Dollar Report. Available online: https://www.phrma.org/report/follow-the-dollar-report (accessed on 10 August 2020).
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Cowart, K. Oral Semaglutide: First-in-Class Oral GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus. Ann. Pharmacother. 2020, 54, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Hedrington, M.S.; Davis, S.N. Oral semaglutide for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2019, 20, 133–141. [Google Scholar] [CrossRef]
- Coskun, T.; Sloop, K.W.; Loghin, C.; Alsina-Fernandez, J.; Urva, S.; Bokvist, K.B.; Cui, X.; Briere, D.A.; Cabrera, O.; Roell, W.C.; et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol. Metab. 2018, 18, 3–14. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef]
- Lilly Investors. Lilly’s Tirzepatide Significantly Reduced A1C and Body Weight in People with Type 2 Diabetesdiabetes. 2020. Available online: https://investor.lilly.com/news-releases/news-release-details/lillys-tirzepatide-significantly-reduced-a1c-and-body-weight (accessed on 9 December 2020).
- Willard, F.S.; Douros, J.D.; Gabe, M.B.; Showalter, A.D.; Wainscott, D.B.; Suter, T.M.; Capozzi, M.E.; van der Velden, W.J.; Stutsman, C.; Cardona, G.R.; et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Andersen, A.; Lund, A.; Knop, F.K.; Vilsboll, T. Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 2018, 14, 390–403. [Google Scholar] [CrossRef]
- Samms, R.J.; Coghlan, M.P.; Sloop, K.W. How May GIP Enhance the Therapeutic Efficacy of GLP-1? Trends Endocrinol. Metab. 2020, 31, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sturchler, E.; Zhu, J.; Nieto, A.; Cistrone, P.A.; Xie, J.; He, L.; Yea, K.; Jones, T.; Turn, R.; et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996, 51, 319–351, discussion 352–313. [Google Scholar]
- Kim, S.J.; Nian, C.; McIntosh, C.H. Adipocyte expression of the glucose-dependent insulinotropic polypeptide receptor involves gene regulation by PPARgamma and histone acetylation. J. Lipid Res. 2011, 52, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceperuelo-Mallafre, V.; Duran, X.; Pachon, G.; Roche, K.; Garrido-Sanchez, L.; Vilarrasa, N.; Tinahones, F.J.; Vicente, V.; Pujol, J.; Vendrell, J.; et al. Disruption of GIP/GIPR axis in human adipose tissue is linked to obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2014, 99, E908–E919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulina, G.R.; Rayfield, E.J. The Role of Glucagon in the Pathophysiology and Management of Diabetes. Endocr. Pract. 2016, 22, 612–621. [Google Scholar] [CrossRef]
- Haedersdal, S.; Lund, A.; Knop, F.K.; Vilsboll, T. The Role of Glucagon in the Pathophysiology and Treatment of Type 2 Diabetes. Mayo Clin. Proc. 2018, 93, 217–239. [Google Scholar] [CrossRef] [Green Version]
- Glucagon nasal powder (Baqsimi) for severe hypoglycemia. Med. Lett. Drugs Ther. 2019, 61, 148–149.
- Jall, S.; Sachs, S.; Clemmensen, C.; Finan, B.; Neff, F.; DiMarchi, R.D.; Tschop, M.H.; Muller, T.D.; Hofmann, S.M. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol. Metab. 2017, 6, 440–446. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Holscher, C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Stiller, C.R.; Dupre, J.; Gent, M.; Heinrichs, D.; Jenner, M.R.; Keown, P.A.; Laupacis, A.; Martell, R.; Rodger, N.W.; Wolfe, B.M.; et al. Effects of cyclosporine in recent-onset juvenile type 1 diabetes: Impact of age and duration of disease. J. Pediatr. 1987, 111, 1069–1072. [Google Scholar] [CrossRef]
- Feutren, G.; Papoz, L.; Assan, R.; Vialettes, B.; Karsenty, G.; Vexiau, P.; Du Rostu, H.; Rodier, M.; Sirmai, J.; Lallemand, A.; et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet 1986, 2, 119–124. [Google Scholar] [CrossRef]
- Baekkeskov, S.; Nielsen, J.H.; Marner, B.; Bilde, T.; Ludvigsson, J.; Lernmark, A. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature 1982, 298, 167–169. [Google Scholar] [CrossRef]
- Zimmet, P. Antibodies to glutamic acid decarboxylase in the prediction of insulin dependency. Diabetes Res. Clin. Pract. 1996, 34 (Suppl. S1), S125–S131. [Google Scholar] [CrossRef]
- Tian, J.; Clare-Salzler, M.; Herschenfeld, A.; Middleton, B.; Newman, D.; Mueller, R.; Arita, S.; Evans, C.; Atkinson, M.A.; Mullen, Y.; et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat. Med. 1996, 2, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Tisch, R.; Liblau, R.S.; Yang, X.D.; Liblau, P.; McDevitt, H.O. Induction of GAD65-specific regulatory T-cells inhibits ongoing autoimmune diabetes in nonobese diabetic mice. Diabetes 1998, 47, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, M.J.; Jaramillo, A.; Zipris, D.; Lazarus, A.H.; Serreze, D.V.; Leiter, E.H.; Cyopick, P.; Danska, J.S.; Delovitch, T.L. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J. Exp. Med. 1993, 178, 87–99. [Google Scholar] [CrossRef]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Faresjo, M.; Hjorth, M.; Axelsson, S.; Cheramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes Prevention Trial--Type 1 Diabetes Study, G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1685–1691. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Cheramy, M.; Axelsson, S.; Pihl, M.; Akerman, L.; Casas, R.; Clinical GAD-Study Group in Sweden. GAD-treatment of children and adolescents with recent-onset type 1 diabetes preserves residual insulin secretion after 30 months. Diabetes Metab. Res. Rev. 2014, 30, 405–414. [Google Scholar] [CrossRef]
- Elias, D.; Avron, A.; Tamir, M.; Raz, I. DiaPep277 preserves endogenous insulin production by immunomodulation in type 1 diabetes. Ann. N. Y. Acad. Sci. 2006, 1079, 340–344. [Google Scholar] [CrossRef]
- Lazar, L.; Ofan, R.; Weintrob, N.; Avron, A.; Tamir, M.; Elias, D.; Phillip, M.; Josefsberg, Z. Heat-shock protein peptide DiaPep277 treatment in children with newly diagnosed type 1 diabetes: A randomised, double-blind phase II study. Diabetes Metab. Res. Rev. 2007, 23, 286–291. [Google Scholar] [CrossRef]
- Chatenoud, L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat. Rev. Endocrinol. 2010, 6, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Gitelman, S.; Greenbaum, C.; Puck, J.; Hagopian, W.; Gottlieb, P.; Sayre, P.; Bianchine, P.; Wong, E.; Seyfert-Margolis, V.; et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin. Immunol. 2009, 132, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagopian, W.; Ferry, R.J., Jr.; Sherry, N.; Carlin, D.; Bonvini, E.; Johnson, S.; Stein, K.E.; Koenig, S.; Daifotis, A.G.; Herold, K.C.; et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: Two-year results from the randomized, placebo-controlled Protege trial. Diabetes 2013, 62, 3901–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, K.C.; Gitelman, S.E.; Willi, S.M.; Gottlieb, P.A.; Waldron-Lynch, F.; Devine, L.; Sherr, J.; Rosenthal, S.M.; Adi, S.; Jalaludin, M.Y.; et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: A randomised controlled trial. Diabetologia 2013, 56, 391–400. [Google Scholar] [CrossRef] [Green Version]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J., Jr.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Washington Business Journal. MacroGenics, Lilly Abandon Diabetes Drug 2010: Washington Business Journal. Available online: https://www.bizjournals.com/washington/quick_news/2010/10/macrogenics-lilly-abandon-diabetes-drug.html (accessed on 5 September 2020).
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Mulvey, A. FDA Breakthrough Therapy Designation for Teplizumab—Based on the First Study to Delay the Onset of T1D for 2+ Years. 2019. Available online: https://www.jdrf.org/blog/2019/08/05/fda-breakthrough-therapy-designation-teplizumab-based-first-study-delay-onset-t1d-2-years/ (accessed on 10 August 2020).
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambery, P.; Donner, T.W.; Biswas, N.; Donaldson, J.; Parkin, J.; Dayan, C.M. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med. 2014, 31, 399–402. [Google Scholar] [CrossRef]
- Aronson, R.; Gottlieb, P.A.; Christiansen, J.S.; Donner, T.W.; Bosi, E.; Bode, B.W.; Pozzilli, P.; Group, D.I. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: Results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 2014, 37, 2746–2754. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, R. Diabetes: Immunotherapy for T1DM--still not there yet. Nat. Rev. Endocrinol. 2013, 9, 697–698. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, C.J.; Schatz, D.A.; Haller, M.J.; Sanda, S. Through the fog: Recent clinical trials to preserve beta-cell function in type 1 diabetes. Diabetes 2012, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Greenbaum, C.J.; Rosasco, M.; Presnell, S.; Herold, K.C.; Dufort, M.J. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new-onset type 1 diabetes. Genes Immun. 2019, 20, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.I.; Le Page, M.A.; Maxwell, M.J.; Stolp, J.; Guo, H.; Jayasimhan, A.; Hibbs, M.L.; Santamaria, P.; Miller, J.F.; Plebanski, M.; et al. The CD19 signalling molecule is elevated in NOD mice and controls type 1 diabetes development. Diabetologia 2013, 56, 2659–2668. [Google Scholar] [CrossRef]
- Lee, M.; Park, H.; Youn, J.; Oh, E.T.; Ko, K.; Kim, S.; Park, Y. Interleukin-10 plasmid construction and delivery for the prevention of type 1 diabetes. Ann. N. Y. Acad. Sci. 2006, 1079, 313–319. [Google Scholar] [CrossRef]
- ActoBio Therapeutics. ActoBio Therapeutics Greenlighted by FDA to Commence a Phase Ib/IIa Trial with AG019 for the Treatment of Early Onset Type 1 Diabetes. 2018. Available online: https://www.prnewswire.com/news-releases/actobio-therapeutics-greenlighted-by-fda-to-commence-a-phase-ibiia-trial-with-ag019-for-the-treatment-of-early-onset-type-1-diabetes-300621670.html (accessed on 21 August 2020).
- ActoBio Therapeutics. ActoBio Therapeutics™ Progresses AG019 to Next Stage of a Phase Ib/IIa Clinical Study for the Treatment of Type 1 Diabetes. 2019. Available online: https://www.prnewswire.com/news-releases/actobio-therapeutics-progresses-ag019-to-next-stage-of-a-phase-ibiia-clinical-study-for-the-treatment-of-type-1-diabetes-300878301.html (accessed on 21 August 2020).
- Thrower, S.L.; James, L.; Hall, W.; Green, K.M.; Arif, S.; Allen, J.S.; Van-Krinks, C.; Lozanoska-Ochser, B.; Marquesini, L.; Brown, S.; et al. Proinsulin peptide immunotherapy in type 1 diabetes: Report of a first-in-man Phase I safety study. Clin. Exp. Immunol. 2009, 155, 156–165. [Google Scholar] [CrossRef]
- Gremizzi, C.; Vergani, A.; Paloschi, V.; Secchi, A. Impact of pancreas transplantation on type 1 diabetes-related complications. Curr. Opin. Organ Transplant. 2010, 15, 119–123. [Google Scholar] [CrossRef]
- Gondolesi, G.E.; Aguirre, N.F.; Ramisch, D.A.; Mos, F.A.; Pedraza, N.F.; Fortunato, M.R.; Gutierrez, L.M.; Fraguas, H.; Marrugat, R.; Rabin, G.E.; et al. Pancreas Transplantation at a Single Latin-American Center; Overall Results with Type 1 and Type 2 Diabetes Mellitus. Transplant. Proc. 2018, 50, 1475–1481. [Google Scholar] [CrossRef]
- Al-Qaoud, T.M.; Odorico, J.S.; Redfield, R.R., 3rd. Pancreas transplantation in type 2 diabetes: Expanding the criteria. Curr. Opin. Organ Transplant. 2018, 23, 454–460. [Google Scholar] [CrossRef]
- Stratta, R.J.; Fridell, J.A.; Gruessner, A.C.; Odorico, J.S.; Gruessner, R.W. Pancreas transplantation: A decade of decline. Curr. Opin. Organ Transplant. 2016, 21, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Najarian, J.S.; Sutherland, D.E.; Matas, A.J.; Steffes, M.W.; Simmons, R.L.; Goetz, F.C. Human islet transplantation: A preliminary report. Transplant. Proc. 1977, 9, 233–236. [Google Scholar] [PubMed]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Gala-Lopez, B.; Pepper, A.R.; Abualhassan, N.S.; Shapiro, A.J. Islet cell transplantation for the treatment of type 1 diabetes: Recent advances and future challenges. Diabetes Metab. Syndr. Obes. 2014, 7, 211–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, A.M.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hering, B.J.; Kandaswamy, R.; Ansite, J.D.; Eckman, P.M.; Nakano, M.; Sawada, T.; Matsumoto, I.; Ihm, S.H.; Zhang, H.J.; Parkey, J.; et al. Single-donor, marginal-dose islet transplantation in patients with type 1 diabetes. JAMA 2005, 293, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; Senior, P.; Salam, A.; Kin, T.; Imes, S.; Dinyari, P.; Malcolm, A.; Toso, C.; Nilsson, B.; Korsgren, O.; et al. Insulin-heparin infusions peritransplant substantially improve single-donor clinical islet transplant success. Transplantation 2010, 89, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Takita, M.; Chaussabel, D.; Noguchi, H.; Shimoda, M.; Sugimoto, K.; Itoh, T.; Chujo, D.; SoRelle, J.; Onaca, N.; et al. Improving efficacy of clinical islet transplantation with iodixanol-based islet purification, thymoglobulin induction, and blockage of IL-1beta and TNF-alpha. Cell Transplant. 2011, 20, 1641–1647. [Google Scholar] [CrossRef]
- Faradji, R.N.; Tharavanij, T.; Messinger, S.; Froud, T.; Pileggi, A.; Monroy, K.; Mineo, D.; Baidal, D.A.; Cure, P.; Ponte, G.; et al. Long-term insulin independence and improvement in insulin secretion after supplemental islet infusion under exenatide and etanercept. Transplantation 2008, 86, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Posselt, A.M.; Bellin, M.D.; Tavakol, M.; Szot, G.L.; Frassetto, L.A.; Masharani, U.; Kerlan, R.K.; Fong, L.; Vincenti, F.G.; Hering, B.J.; et al. Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab. Am. J. Transplant. 2010, 10, 1870–1880. [Google Scholar] [CrossRef]
- Turgeon, N.A.; Avila, J.G.; Cano, J.A.; Hutchinson, J.J.; Badell, I.R.; Page, A.J.; Adams, A.B.; Sears, M.H.; Bowen, P.H.; Kirk, A.D.; et al. Experience with a novel efalizumab-based immunosuppressive regimen to facilitate single donor islet cell transplantation. Am. J. Transplant. 2010, 10, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, N.; Sathyamurthy, A.; Elghazi, L.; Gould, A.; Weiss, A.; Shiojima, I.; Walsh, K.; Bernal-Mizrachi, E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J. Biol. Chem. 2009, 284, 7832–7842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, S.; Fung, J.J.; Lu, L.; Qian, S. Tolerance-inducing strategies in islet transplantation. Int. J. Endocrinol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Noorchashm, H.; Sutter, J.A.; Naji, M.; Prak, E.L.; Boyer, J.; Green, T.; Rickels, M.R.; Tomaszewski, J.E.; Koeberlein, B.; et al. B lymphocyte-directed immunotherapy promotes long-term islet allograft survival in nonhuman primates. Nat. Med. 2007, 13, 1295–1298. [Google Scholar] [CrossRef]
- Cabello-Olmo, M.; Arana, M.; Radichev, I.; Smith, P.; Huarte, E.; Barajas, M. New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. Int. J. Mol. Sci. 2019, 20, 4789. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jiang, Z.; Delgado, E.; Li, H.; Zhou, H.; Hu, W.; Perez-Basterrechea, M.; Janostakova, A.; Tan, Q.; Wang, J.; et al. Platelet-Derived Mitochondria Display Embryonic Stem Cell Markers and Improve Pancreatic Islet beta-cell Function in Humans. Stem Cells Transl. Med. 2017, 6, 1684–1697. [Google Scholar] [CrossRef]
- Kang, E.M.; Zickler, P.P.; Burns, S.; Langemeijer, S.M.; Brenner, S.; Phang, O.A.; Patterson, N.; Harlan, D.; Tisdale, J.F. Hematopoietic stem cell transplantation prevents diabetes in NOD mice but does not contribute to significant islet cell regeneration once disease is established. Exp. Hematol. 2005, 33, 699–705. [Google Scholar] [CrossRef]
- Voltarelli, J.C.; Couri, C.E.; Stracieri, A.B.; Oliveira, M.C.; Moraes, D.A.; Pieroni, F.; Coutinho, M.; Malmegrim, K.C.; Foss-Freitas, M.C.; Simoes, B.P.; et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2007, 297, 1568–1576. [Google Scholar] [CrossRef] [Green Version]
- D’Addio, F.; Valderrama Vasquez, A.; Ben Nasr, M.; Franek, E.; Zhu, D.; Li, L.; Ning, G.; Snarski, E.; Fiorina, P. Autologous nonmyeloablative hematopoietic stem cell transplantation in new-onset type 1 diabetes: A multicenter analysis. Diabetes 2014, 63, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Miao, H.; Zhang, J.; Hu, J.; Zhou, W.; Gu, W.; Wang, W.; Ning, G. Clinical benefits of autologous haematopoietic stem cell transplantation in type 1 diabetes patients. Diabetes Metab. 2018, 44, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Snarski, E.; Milczarczyk, A.; Halaburda, K.; Torosian, T.; Paluszewska, M.; Urbanowska, E.; Krol, M.; Boguradzki, P.; Jedynasty, K.; Franek, E.; et al. Immunoablation and autologous hematopoietic stem cell transplantation in the treatment of new-onset type 1 diabetes mellitus: Long-term observations. Bone Marrow Transplant. 2016, 51, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.; Kahlenberg, S.; Hornsby, P. Therapeutic potential of mesenchymal stem cells for diabetes. J. Mol. Endocrinol. 2017, 59, R109–R120. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Yu, X.; Wang, Z.; Wang, F.; Wang, L.; Gao, H.; Chen, Y.; Zhao, W.; Jia, Z.; Yan, S.; et al. Long term effects of the implantation of Wharton’s jelly-derived mesenchymal stem cells from the umbilical cord for newly-onset type 1 diabetes mellitus. Endocr. J. 2013, 60, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, U.G.; Trivedi, H.L.; Vanikar, A.V.; Dave, S.D. Insulin-secreting adipose-derived mesenchymal stromal cells with bone marrow-derived hematopoietic stem cells from autologous and allogenic sources for type 1 diabetes mellitus. Cytotherapy 2015, 17, 940–947. [Google Scholar] [CrossRef]
- Vanikar, A.V.; Dave, S.D.; Thakkar, U.G.; Trivedi, H.L. Cotransplantation of adipose tissue-derived insulin-secreting mesenchymal stem cells and hematopoietic stem cells: A novel therapy for insulin-dependent diabetes mellitus. Stem Cells Int. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Dave, S.D.; Vanikar, A.V.; Trivedi, H.L.; Thakkar, U.G.; Gopal, S.C.; Chandra, T. Novel therapy for insulin-dependent diabetes mellitus: Infusion of in vitro-generated insulin-secreting cells. Clin. Exp. Med. 2015, 15, 41–45. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, P.; Wang, X.; Dai, G.; Cheng, H.; Zhang, Z.; Hua, R.; Niu, X.; Shi, J.; An, Y. A preliminary evaluation of efficacy and safety of Wharton’s jelly mesenchymal stem cell transplantation in patients with type 2 diabetes mellitus. Stem Cell Res. Ther. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhansali, S.; Dutta, P.; Kumar, V.; Yadav, M.K.; Jain, A.; Mudaliar, S.; Bhansali, S.; Sharma, R.R.; Jha, V.; Marwaha, N.; et al. Efficacy of Autologous Bone Marrow-Derived Mesenchymal Stem Cell and Mononuclear Cell Transplantation in Type 2 Diabetes Mellitus: A Randomized, Placebo-Controlled Comparative Study. Stem Cells Dev. 2017, 26, 471–481. [Google Scholar] [CrossRef]
- Jiang, R.; Han, Z.; Zhuo, G.; Qu, X.; Li, X.; Wang, X.; Shao, Y.; Yang, S.; Han, Z.C. Transplantation of placenta-derived mesenchymal stem cells in type 2 diabetes: A pilot study. Front. Med. 2011, 5, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Skyler, J.S.; Fonseca, V.A.; Segal, K.R.; Rosenstock, J.; Investigators, M.-D. Allogeneic Mesenchymal Precursor Cells in Type 2 Diabetes: A Randomized, Placebo-Controlled, Dose-Escalation Safety and Tolerability Pilot Study. Diabetes Care 2015, 38, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Huang, Q.; Xu, X.J.; Shao, Z.L.; Huang, L.H.; Yang, X.Z.; Guo, W.; Li, C.M.; Chen, C. The effect of liraglutide in combination with human umbilical cord mesenchymal stem cells treatment on glucose metabolism and beta cell function in type 2 diabetes mellitus. Zhonghua Nei Ke Za Zhi 2016, 55, 349–354. [Google Scholar] [CrossRef]
- Hildreth, C. Top Companies Developing Cell Therapy Treatments for Diabetes. 2019. Available online: https://bioinformant.com/stem-cells-for-diabetes/ (accessed on 20 August 2020).
- Zhao, Y.; Lin, B.; Darflinger, R.; Zhang, Y.; Holterman, M.J.; Skidgel, R.A. Human cord blood stem cell-modulated regulatory T lymphocytes reverse the autoimmune-caused type 1 diabetes in nonobese diabetic (NOD) mice. PLoS ONE 2009, 4, e4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y. Stem cell educator therapy and induction of immune balance. Curr. Diabetes Rep. 2012, 12, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Zhou, H.; Yin, Z.; Chen, Y.; Zhang, Y.; Wang, S.; et al. Targeting insulin resistance in type 2 diabetes via immune modulation of cord blood-derived multipotent stem cells (CB-SCs) in stem cell educator therapy: Phase I/II clinical trial. BMC Med. 2013, 11, 160. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Yin, Z.; Li, H.; Zhang, Y.; Diao, Y.; Li, Y.; et al. Reversal of type 1 diabetes via islet beta cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. 2012, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Delgado, E.; Perez-Basterrechea, M.; Suarez-Alvarez, B.; Zhou, H.; Revuelta, E.M.; Garcia-Gala, J.M.; Perez, S.; Alvarez-Viejo, M.; Menendez, E.; Lopez-Larrea, C.; et al. Modulation of Autoimmune T-Cell Memory by Stem Cell Educator Therapy: Phase 1/2 Clinical Trial. EBioMedicine 2015, 2, 2024–2036. [Google Scholar] [CrossRef] [Green Version]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Zhang, L.; Sosinowski, T.; Cox, A.R.; Cepeda, J.R.; Sekhar, N.S.; Hartig, S.M.; Miao, D.; Yu, L.; Pietropaolo, M.; Davidson, H.W. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 2019, 96, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Crawford, F.; Yu, L.; Michels, A.; Nakayama, M.; Davidson, H.W.; Kappler, J.W.; Eisenbarth, G.S. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 2656–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, K.; Chera, S.; van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Ko, U.H.; Oh, Y.; Lim, A.; Sohn, J.W.; Shin, J.H.; Kim, H.; Han, Y.M. Islet-like organoids derived from human pluripotent stem cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramli, R.; Reddy, M.; Oliver, N. Artificial Pancreas: Current Progress and Future Outlook in the Treatment of Type 1 Diabetes. Drugs 2019, 79, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- FDA. What Is the Pancreas? What Is an Artificial Pancreas Device System? 2018. Available online: https://www.fda.gov/medical-devices/artificial-pancreas-device-system/what-pancreas-what-artificial-pancreas-device-system (accessed on 10 August 2020).
- Saunders, A.; Messer, L.H.; Forlenza, G.P. MiniMed 670G hybrid closed loop artificial pancreas system for the treatment of type 1 diabetes mellitus: Overview of its safety and efficacy. Expert Rev. Med. Devices 2019, 16, 845–853. [Google Scholar] [CrossRef]
- Brown, S.A.; Kovatchev, B.P.; Raghinaru, D.; Lum, J.W.; Buckingham, B.A.; Kudva, Y.C.; Laffel, L.M.; Levy, C.J.; Pinsker, J.E.; Wadwa, R.P.; et al. Six-Month Randomized, Multicenter Trial of Closed-Loop Control in Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- NIH. Artificial Pancreas System Better Controls Blood Glucose Levels Than Current Technology. 2019. Available online: https://www.nih.gov/news-events/news-releases/artificial-pancreas-system-better-controls-blood-glucose-levels-current-technology (accessed on 9 September 2020).
- Abai, A.M.; Hobart, P.M.; Barnhart, K.M. Insulin delivery with plasmid DNA. Hum. Gene Ther. 1999, 10, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; McLane, B.; Kim, E.H.; Yoon, J.W.; Jun, H.S. Remission of diabetes by insulin gene therapy using a hepatocyte-specific and glucose-responsive synthetic promoter. Mol. Ther. 2011, 19, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.F.; Chatenoud, L. Correction: Antibiotics in Early Life Alter the Gut Microbiome and Increase Disease Incidence in a Spontaneous Mouse Model of Autoimmune Insulin-Dependent Diabetes. PLoS ONE 2016, 11, e0147888. [Google Scholar] [CrossRef] [Green Version]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.F.; Chatenoud, L. Antibiotics in early life alter the gut microbiome and increase disease incidence in a spontaneous mouse model of autoimmune insulin-dependent diabetes. PLoS ONE 2015, 10, e0125448. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Ardissone, A.N.; Dias, R.; Simell, V.; Leonard, M.T.; Kemppainen, K.M.; Drew, J.C.; Schatz, D.; Atkinson, M.A.; Kolaczkowski, B.; et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front. Microbiol. 2014, 5, 678. [Google Scholar] [CrossRef]
- De Goffau, M.C.; Fuentes, S.; van den Bogert, B.; Honkanen, H.; de Vos, W.M.; Welling, G.W.; Hyoty, H.; Harmsen, H.J. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 2014, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Li, Z.; Zhou, Z. Gut microbiome in type 1 diabetes: A comprehensive review. Diabetes Metab. Res. Rev. 2018, 34, e3043. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef]
- Beer, K. The Gut Microbiome in Type 2 Diabetes. Clin. Rev. 2018, 28, 13–14. [Google Scholar]
- Tai, N.; Wong, F.S.; Wen, L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev. Endocr. Metab. Disord. 2015, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS ONE 2014, 9, e100778. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Wang, Z.; Saha, S.; Van Horn, S.; Thomas, E.; Traini, C.; Sathe, G.; Rajpal, D.K.; Brown, J.R. Gut microbiome differences between metformin- and liraglutide-treated T2DM subjects. Endocrinol. Diabetes Metab. 2018, 1, e00009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, O.; Nieuwdorp, M.; Gerdes, V. The Gut Microbiome as a Target for the Treatment of Type 2 Diabetes. Curr. Diabates Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Tripathi, P. Gut microbiome and type 2 diabetes: Where we are and where to go? J. Nutr. Biochem. 2019, 63, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Restrepo, N.; Posso-Osorio, I.; Naranjo-Escobar, J.; Tobón, G.J. Autoimmune diseases and their relation with immunological, neurological and endocrinological axes. Autoimmun. Rev. 2017, 16, 684–692. [Google Scholar] [CrossRef]
- Cooper, G.S.; Stroehla, B.C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2003, 2, 119–125. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- WHO. Chronic Diseases and Health Promotion. Available online: https://www.who.int/chp/topics/rheumatic/en/ (accessed on 4 October 2020).
- Ezerioha, M. Let’s Dig into Everything about RA; Rheumatoid Arthritis Support Network. 2018. Available online: https://www.rheumatoidarthritis.org (accessed on 15 September 2020).
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Giannini, D.; Antonucci, M.; Petrelli, F.; Bilia, S.; Alunno, A.; Puxeddu, I. One year in review 2020: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2020, 38, 387–397. [Google Scholar]
- Ramos, P.S.; Shedlock, A.M.; Langefeld, C.D. Genetics of autoimmune diseases: Insights from population genetics. J. Hum. Genet. 2015, 60, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar] [PubMed]
- Croia, C.; Bursi, R.; Sutera, D.; Petrelli, F.; Alunno, A.; Puxeddu, I. One year in review 2019: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2019, 37, 347–357. [Google Scholar]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Watad, A.; Amital, H. ACPAs Are Much More Than Diagnostic Autoantibodies. Rambam Maimonides Med. J. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Kurowska, W.; Kuca-Warnawin, E.H.; Radzikowska, A.; Maśliński, W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent. Eur. J. Immunol. 2017, 42, 390. [Google Scholar] [CrossRef]
- Harre, U.; Georgess, D.; Bang, H.; Bozec, A.; Axmann, R.; Ossipova, E.; Jakobsson, P.J.; Baum, W.; Nimmerjahn, F.; Szarka, E.; et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J. Clin. Investig. 2012, 122, 1791–1802. [Google Scholar] [CrossRef]
- Nielen, M.M.; van Schaardenburg, D.; Reesink, H.W.; van de Stadt, R.J.; van der Horst-Bruinsma, I.E.; de Koning, M.H.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, S.; Yang, Y.; Zhang, K.; Dong, S.; Wang, X.; Liu, X.; Ren, Y.; Zhang, M.; Yan, X. T helper 17 and T helper 1 cells are increased but regulatory T cells are decreased in subchondral bone marrow microenvironment of patients with rheumatoid arthritis. Am. J. Transl. Res. 2016, 8, 2956. [Google Scholar]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Nakayamada, S.; Kubo, S.; Iwata, S.; Tanaka, Y. Recent Progress in JAK Inhibitors for the Treatment of Rheumatoid Arthritis. BioDrugs 2016, 30, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.; Lisse, J.; Rizzo, W.; Albani, S. Discussion: DMARDs and biologic therapies in the management of inflammatory joint diseases. Expert Rev. Clin. Immunol. 2009, 5, 291–299. [Google Scholar] [CrossRef]
- Gerriets, V.; Bansal, P.; Goyal, A.; Khaddour, K. Tumor Necrosis Factor (TNF) Inhibitors. In StatPearls; StatPearls Publishing Copyright © 2020; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kalden, J.R.; Schulze-Koops, H. Immunogenicity and loss of response to TNF inhibitors: Implications for rheumatoid arthritis treatment. Nat. Rev. Rheumatol. 2017, 13, 707–718. [Google Scholar] [CrossRef] [PubMed]
- AbbVie. Novel Antibody Drug Conjugate ABBV-3373 Shows Improvement in Disease Activity in Phase 2a Study of Patients with Rheumatoid Arthritis. 2020. Available online: https://news.abbvie.com/news/press-releases/novel-antibody-drug-conjugate-abbv-3373-shows-improvement-in-disease-activity-in-phase-2a-study-patients-with-rheumatoid-arthritis.htm (accessed on 10 September 2020).
- Xu, Z.; Bouman-Thio, E.; Comisar, C.; Frederick, B.; Van Hartingsveldt, B.; Marini, J.C.; Davis, H.M.; Zhou, H. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br. J. Clin. Pharmacol. 2011, 72, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.; Fleischmann, R.; Fiore, S.; Radin, A.; Fan, C.; Huizinga, T. SAT0117 sarilumab, a subcutaneously-administered, fully-human monoclonal antibody inhibitor of the IL-6 receptor: Relationship between eular responses and change from baseline of selected clinical parameters. Ann. Rheum. Dis. 2013, 72, A620. [Google Scholar] [CrossRef]
- Rafique, A.; Martin, J.; Blome, M.; Huang, T.; Ouyang, A.; Papadopoulos, N. AB0037 Evaluation of the binding kinetics and functional bioassay activity of sarilumab and tocilizumab to the human il-6 receptor (il-6r) alpha. Ann. Rheum. Dis. 2013, 72, A797. [Google Scholar] [CrossRef]
- Bae, S.-C.; Lee, Y.H. Comparison of the efficacy and tolerability of tocilizumab, sarilumab, and sirukumab in patients with active rheumatoid arthritis: A Bayesian network meta-analysis of randomized controlled trials. Clin. Rheumatol. 2018, 37, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C. Rituximab for the treatment of rheumatoid arthritis: An update. Drug Des. Dev. Ther. 2014, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Autoimmun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A.; Deeks, E.D. Abatacept: A review in rheumatoid arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef]
- Avci, A.B.; Feist, E.; Burmester, G.R. Targeting GM-CSF in rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 39–44. [Google Scholar]
- Crotti, C.; Raimondo, M.G.; Becciolini, A.; Biggioggero, M.; Favalli, E.G. Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: Evidence to date. Drug Des. Dev. Ther. 2017, 11, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.C.; Saurigny, D.; Vencovsky, J.; Takeuchi, T.; Nakamura, T.; Matsievskaia, G.; Hunt, B.; Wagner, T.; Souberbielle, B. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: A randomized, controlled trial. Arthritis Res. Ther. 2019, 21, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.C.; van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 46–55. [Google Scholar] [CrossRef]
- Whang, J.A.; Chang, B.Y. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov. Today 2014, 19, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) Affects Markers of B Cell Activation, Chemotaxis, and Osteoclasts in Patients with Rheumatoid Arthritis: Results from a Mechanistic Study. Rheumatol. Ther. 2020, 7, 101–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillooly, K.M.; Pulicicchio, C.; Pattoli, M.A.; Cheng, L.; Skala, S.; Heimrich, E.M.; McIntyre, K.W.; Taylor, T.L.; Kukral, D.W.; Dudhgaonkar, S.; et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS ONE 2017, 12, e0181782. [Google Scholar] [CrossRef] [Green Version]
- Watterson, S.H.; Liu, Q.; Beaudoin Bertrand, M.; Batt, D.G.; Li, L.; Pattoli, M.A.; Skala, S.; Cheng, L.; Obermeier, M.T.; Moore, R.; et al. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A. Fenebrutinib Versus Placebo or Adalimumab in Rheumatoid Arthritis: A Randomized, Double-Blind, Phase II Trial. Arthritis Rheumatol. 2020, 72, 1435–1446. [Google Scholar] [CrossRef]
- Norman, P. Investigational Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin. Investig. Drugs 2016, 25, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, E.; Gevorkyan, H.; Faessel, H.; Zhao, Z.; Hanna, K.; Smithson, G.; Wagner, J.; Fedyk, E.R.; Mclean, L. SAT0226 A phase 1, randomized, double-blind, placebo-controlled, single- and multiple-rising dose study of the btk inhibitor tak-020 in healthy volunteers. Ann. Rheum. Dis. 2018, 77, 974. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Wang, L.; Huang, S.; Li, S.; Li, M.; Shi, J.; Bai, W.; Wang, Q.; Zheng, L.; Liu, Y. Efficacy and Safety of Umbilical Cord Mesenchymal Stem Cell Therapy for Rheumatoid Arthritis Patients: A Prospective Phase I/II Study. Drug Des. Dev. Ther. 2019, 13, 4331–4340. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Ann. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Bae, Y.-S. Dendritic cell-based immunotherapy for rheumatoid arthritis: From bench to bedside. Immune Netw. 2016, 16, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 227–234. [Google Scholar] [CrossRef]
- Álvaro-Gracia, J.M.; Jover, J.A.; García-Vicuña, R.; Carreño, L.; Alonso, A.; Marsal, S.; Blanco, F.; Martínez-Taboada, V.M.; Taylor, P.; Martín-Martín, C.; et al. Intravenous administration of expanded allogeneic adipose-derived mesenchymal stem cells in refractory rheumatoid arthritis (Cx611): Results of a multicentre, dose escalation, randomised, single-blind, placebo-controlled phase Ib/IIa clinical trial. Ann. Rheum. Dis. 2017, 76, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, H.; Wang, Y.; Chen, S.; Liu, J.; Li, W.; Dou, H.; Hou, W.; Meng, M. Study of the adoptive immunotherapy on rheumatoid arthritis with Thymus-derived invariant natural killer T cells. Int. Immunopharmacol. 2019, 67, 427–440. [Google Scholar] [CrossRef]
- Wu, X.; He, B.; Liu, J.; Feng, H.; Ma, Y.; Li, D.; Guo, B.; Liang, C.; Dang, L.; Wang, L. Molecular insight into gut microbiota and rheumatoid arthritis. Int. J. Mol. Sci. 2016, 17, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Pan, H.; Guo, R.; Ju, Y.; Wang, Q.; Zhu, J.; Xie, Y.; Zheng, Y.; Li, T.; Liu, Z.; Lu, L.; et al. A single bacterium restores the microbiome dysbiosis to protect bones from destruction in a rat model of rheumatoid arthritis. Microbiome 2019, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Marietta, E.V.; Murray, J.A.; Luckey, D.H.; Jeraldo, P.R.; Lamba, A.; Patel, R.; Luthra, H.S.; Mangalam, A.; Taneja, V. Suppression of inflammatory arthritis by human gut-derived Prevotella histicola in humanized mice. Arthritis Rheumatol. 2016, 68, 2878–2888. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.F.; Pan, H.; Yan, H.; Sandell, L.J.; Pham, C.T.N.; Wickline, S.A. Applications of RNA interference in the treatment of arthritis. Transl. Res. 2019, 214, 1–16. [Google Scholar] [CrossRef]
- Cohen, J.L.; Shen, Y.; Aouadi, M.; Vangala, P.; Tencerova, M.; Amano, S.U.; Nicoloro, S.M.; Yawe, J.C.; Czech, M.P. Peptide- and Amine-Modified Glucan Particles for the Delivery of Therapeutic siRNA. Mol. Pharm. 2016, 13, 964–978. [Google Scholar] [CrossRef] [Green Version]
- Plavcová, Z.; Šalamúnová, P.; Saloň, I.; Štěpánek, F.; Hanuš, J.; Hošek, J. Curcumin encapsulation in yeast glucan particles promotes its anti-inflammatory potential in vitro. Int. J. Pharm. 2019, 568, 118532. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, Y.P.; Wu, J.; Jie, L.G.; Deng, J.X.; Zhao, D.B.; Yu, Q.H. Downregulated microRNA-135a ameliorates rheumatoid arthritis by inactivation of the phosphatidylinositol 3-kinase/AKT signaling pathway via phosphatidylinositol 3-kinase regulatory subunit 2. J. Cell. Physiol. 2019, 234, 17663–17676. [Google Scholar] [CrossRef] [PubMed]
- Nutten, S. Atopic dermatitis: Global epidemiology and risk factors. Ann. Nutr. Metab. 2015, 66 (Suppl. S1), 8–16. [Google Scholar] [CrossRef]
- Spergel, J.M. Epidemiology of atopic dermatitis and atopic march in children. Immunol. Allergy Clin. 2010, 30, 269–280. [Google Scholar] [CrossRef]
- Badloe, F.M.S.; De Vriese, S.; Coolens, K.; Schmidt-Weber, C.B.; Ring, J.; Gutermuth, J.; Krohn, I.K. IgE autoantibodies and autoreactive T cells and their role in children and adults with atopic dermatitis. Clin. Transl. Allergy 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Sullivan, M.; Silverberg, N.B. Current and emerging concepts in atopic dermatitis pathogenesis. Clin. Dermatol. 2017, 35, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Reszka, E.; Gutowska-Owsiak, D.; Trzeciak, M.; Lange, M.; Jarczak, J.; Niedoszytko, M.; Jablonska, E.; Romantowski, J.; Strapagiel, D. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 6484. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2020, 48, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.S.; McPhee, R.; Arruda, L.K.; Howell, M.D. Targeting the T helper 2 inflammatory axis in atopic dermatitis. Int. Arch. Allergy Immunol. 2016, 171, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.-C.; Meller, S.; Rieker, J. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Wollina, U. Microbiome in atopic dermatitis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renert-Yuval, Y.; Guttman-Yassky, E. New treatments for atopic dermatitis targeting beyond IL-4/IL-13 cytokines. Ann. Allergy Asthma Immunol. 2020, 124, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyle, M.; Cevikbas, F.; Harden, J.L.; Guttman-Yassky, E. Understanding the immune landscape in atopic dermatitis: The era of biologics and emerging therapeutic approaches. Exp. Dermatol. 2019, 28, 756–768. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Hanifin, J.M.; Boguniewicz, M.; Wollenberg, A.; Bissonnette, R.; Purohit, V.; Kilty, I.; Tallman, A.M.; Zielinski, M.A. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp. Dermatol. 2019, 28, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, J.; Zhang, X.; Zhang, Y.; Qin, Z.-l.; Wang, H.; Luo, X.-y. Application of topical phosphodiesterase 4 inhibitors in mild to moderate atopic dermatitis: A systematic review and meta-analysis. JAMA Dermatol. 2019, 155, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.L.; Imafuku, S.; Poulin, Y.; Ungar, B.; Zhou, L.; Malik, K.; Wen, H.C.; Xu, H.; Estrada, Y.D.; Peng, X.; et al. A Phase 2 Randomized Trial of Apremilast in Patients with Atopic Dermatitis. J. Invest. Dermatol. 2019, 139, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Ulzii, D.; Nakahara, T.; Tsuji, G.; Furue, K.; Hashimoto-Hachiya, A.; Kido-Nakahara, M. Implications of IL-13Rα2 in atopic skin inflammation. Allergol. Int. 2020, 69, 412–416. [Google Scholar] [CrossRef]
- D’Ippolito, D.; Pisano, M. Dupilumab (Dupixent): An Interleukin-4 Receptor Antagonist for Atopic Dermatitis. Pharm. Ther. 2018, 43, 532–535. [Google Scholar]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Guttman-Yassky, E.; Blauvelt, A.; Eichenfield, L.F.; Paller, A.S.; Armstrong, A.W.; Drew, J.; Gopalan, R.; Simpson, E.L. Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.G.; Narayana, P.K.; Pouliquen, I.J.; Lopez, M.C.; Ferreira-Cornwell, M.C.; Getsy, J.A. Efficacy and safety of mepolizumab administered subcutaneously for moderate to severe atopic dermatitis. Allergy 2020, 75, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I.; Pinter, A.; Pulka, G.; Poulin, Y.; Bouaziz, J.-D.; Wollenberg, A.; Murrell, D.F.; Alexis, A.; Lindsey, L.; Ahmad, F. Phase 2B randomized study of nemolizumab in adults with moderate-to-severe atopic dermatitis and severe pruritus. J. Allergy Clin. Immunol. 2020, 145, 173–182. [Google Scholar] [CrossRef] [Green Version]
- McKeage, K.; Duggan, S. Risankizumab: First Global Approval. Drugs 2019, 79, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Brunner, P.M.; Neumann, A.U.; Khattri, S.; Pavel, A.B.; Malik, K.; Singer, G.K.; Baum, D.; Gilleaudeau, P.; Sullivan-Whalen, M.; et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol. 2018, 78, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.L.; Lacour, J.P.; Spelman, L.; Galimberti, R.; Eichenfield, L.F.; Bissonnette, R.; King, B.A.; Thyssen, J.P.; Silverberg, J.I.; Bieber, T.; et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: Results from two randomized monotherapy phase III trials. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Kim, B.S.; Howell, M.D.; Sun, K.; Papp, K.; Nasir, A.; Kuligowski, M.E. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J. Allergy Clin. Immunol. 2020, 145, 572–582. [Google Scholar] [CrossRef] [Green Version]
- AbbVie. RINVOQ™(Upadacitinib) Monotherapy Shows Improvement in Skin Clearance and Itch in First Phase 3 Study for Atopic Dermatitis. 2020. Available online: https://news.abbvie.com/news/press-releases/rinvoq-upadacitinib-monotherapy-shows-improvement-in-skin-clearance-and-itch-in-first-phase-3-study-for-atopic-dermatitis.htm (accessed on 4 September 2020).
- Silverberg, J.I.; Simpson, E.L.; Thyssen, J.P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; DiBonaventura, M.; Nduaka, C.; et al. Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol. 2020, 156, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Maari, C.; Forman, S.; Bhatia, N.; Lee, M.; Fowler, J.; Tyring, S.; Pariser, D.; Sofen, H.; Dhawan, S.; et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: Results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 2019, 181, 733–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Guttman-Yassky, E. JAK Inhibitors for Atopic Dermatitis: An Update. Am. J. Clin. Dermatol. 2019, 20, 181–192. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hirasawa, N. The role of histamine H1 and H4 receptors in atopic dermatitis: From basic research to clinical study. Allergol. Int. 2014, 63, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werfel, T.; Layton, G.; Yeadon, M.; Whitlock, L.; Osterloh, I.; Jimenez, P.; Liu, W.; Lynch, V.; Asher, A.; Tsianakas, A.; et al. Efficacy and safety of the histamine H(4) receptor antagonist ZPL-3893787 in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 1830–1837. [Google Scholar] [CrossRef]
- Simpson, E.L.; Parnes, J.R.; She, D.; Crouch, S.; Rees, W.; Mo, M.; van der Merwe, R. Tezepelumab, an anti–thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J. Am. Acad. Dermatol. 2019, 80, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Pavel, A.B.; Zhou, L.; Estrada, Y.D.; Zhang, N.; Xu, H.; Peng, X.; Wen, H.C.; Govas, P.; Gudi, G.; et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 144, 482–493. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Iizuka, H.; Nemoto, O.; Shimabe, M.; Furukawa, Y.; Kikuta, N.; Ootaki, K. Safety, tolerability and efficacy of repeated intravenous infusions of KHK4083, a fully human anti-OX40 monoclonal antibody, in Japanese patients with moderate to severe atopic dermatitis. J. Dermatol. Sci. 2020, 99, 82–89. [Google Scholar] [CrossRef]
- MorphoSys. MorphoSys AG: MOR106 Clinical Development in Atopic Dermatitis Stopped. 2019. Available online: https://www.morphosys.com/media-investors/media-center/morphosys-ag-mor106-clinical-development-in-atopic-dermatitis-stopped (accessed on 28 September 2020).
- Ungar, B.; Pavel, A.B.; Li, R.; Kimmel, G.; Nia, J.; Hashim, P.; Kim, H.J.; Chima, M.; Vekaria, A.S.; Estrada, Y. Phase 2 randomized, double-blind study of IL-17 targeting with secukinumab in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 147, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.L.; Gutowska-Owsiak, D.; Hardman, C.S.; Westmoreland, M.; MacKenzie, T.; Cifuentes, L.; Waithe, D.; Lloyd-Lavery, A.; Marquette, A.; Londei, M.; et al. Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.R.; Costa, S.K.; Zaramela, L.S.; Khalil, S.; Todd, D.A.; Winter, H.L.; Sanford, J.A.; O’Neill, A.M.; Liggins, M.C.; Nakatsuji, T.; et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Myles, I.A.; Williams, K.W.; Reckhow, J.D.; Jammeh, M.L.; Pincus, N.B.; Sastalla, I.; Saleem, D.; Stone, K.D.; Datta, S.K. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight 2016, 1, e86955. [Google Scholar] [CrossRef] [Green Version]
- Myles, I.; Earland, N.; Anderson, E.; Moore, I.; Kieh, M.; Williams, K. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight 2018, 3, e120608. [Google Scholar] [CrossRef] [Green Version]
- Itano, A.; Cormack, T.; Ramani, K.; Barth, K.; Wang, I.; Mukherjee, A.; Ponichtera, H.; McKenna, C.; Jahic, M.; Bodmer, M. Orally-Administered EDP1815, a Single Strain of Prevotella histicola, Has Potent Systemic Anti-Inflammatory Effects in Type 1, Type 2, and Type 3 Inflammatory Models; Evelo Biosciences: Cambridge, MA, USA, 2020. [Google Scholar]
- Lee, W.-R.; Shen, S.-C.; Chen, W.-Y.; Aljuffali, I.A.; Suen, S.-Y.; Fang, J.-Y. Noninvasive delivery of siRNA and plasmid DNA into skin by fractional ablation: Erbium: YAG laser versus CO2 laser. Eur. J. Pharm. Biopharm. 2014, 86, 315–323. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Kurano, T.; Oogi, C.; Takashima, Y.; Seta, Y. Anti-RelA siRNA-Encapsulated Flexible Liposome with Tight Junction-Opening Peptide as a Non-invasive Topical Therapeutic for Atopic Dermatitis. Biol. Pharm. Bull. 2019, 42, 1216–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Transdermal anti-nuclear kappaB siRNA therapy for atopic dermatitis using a combination of two kinds of functional oligopeptide. Int. J. Pharm. 2018, 542, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Development of an Innovative Intradermal siRNA Delivery System Using a Combination of a Functional Stearylated Cytoplasm-Responsive Peptide and a Tight Junction-Opening Peptide. Molecules 2016, 21, 1279. [Google Scholar] [CrossRef]
- Liew, W.C.; Sundaram, G.M.; Quah, S.; Lum, G.G.; Tan, J.S.; Ramalingam, R.; Common, J.E.; Tang, M.B.; Lane, E.B.; Thng, S.T.G. Belinostat resolves skin barrier defects in atopic dermatitis by targeting the dysregulated miR-335: SOX6 axis. J. Allergy Clin. Immunol. 2020, 146, 606–620. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Feuerstein, J.D.; Cheifetz, A.S. Crohn Disease: Epidemiology, Diagnosis, and Management. Mayo Clin. Proc. 2017, 92, 1088–1103. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.; Panaccione, R. Anti-adhesion molecule strategies for Crohn disease. BioDrugs 2008, 22, 259–264. [Google Scholar] [CrossRef]
- Zundler, S.; Becker, E.; Weidinger, C.; Siegmund, B. Anti-Adhesion Therapies in Inflammatory Bowel Disease-Molecular and Clinical Aspects. Front. Immunol. 2017, 8, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagnini, C.; Arseneau, K.O.; Cominelli, F. Natalizumab in the treatment of Crohn’s disease patients. Expert Opin. Biol. Ther. 2017, 17, 1433–1438. [Google Scholar] [CrossRef]
- Dotan, I.; Allez, M.; Danese, S.; Keir, M.; Tole, S.; McBride, J. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 2020, 40, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Protagonist Therapeutics Inc. Oral Alpha-4-Beta-7 Integrin Antagonist PN-943 Demonstrates Sustained Dose-Related Target Engagement Activity in a Multiple Ascending Dose Phase 1 Study. 2019. Available online: www.prnewswire.com (accessed on 20 August 2020).
- Sandborn, W.J.; Lee, S.D.; Tarabar, D.; Louis, E.; Klopocka, M.; Klaus, J.; Reinisch, W.; Hébuterne, X.; Park, D.I.; Schreiber, S.; et al. Phase II evaluation of anti-MAdCAM antibody PF-00547659 in the treatment of Crohn’s disease: Report of the OPERA study. Gut 2018, 67, 1824–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in Inflammatory bowel disease: New avenues for modulating intestinal leukocyte migration. J. Crohns Colitis 2018, 12, S678–S686. [Google Scholar] [CrossRef] [Green Version]
- Peyrin-Biroulet, L.; Christopher, R.; Behan, D.; Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun. Rev. 2017, 16, 495–503. [Google Scholar] [CrossRef]
- Ishii, I.; Ye, X.; Friedman, B.; Kawamura, S.; Contos, J.J.; Kingsbury, M.A.; Yang, A.H.; Zhang, G.; Brown, J.H.; Chun, J. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P2/LPB2/EDG-5 and S1P3/LPB3/EDG-3. J. Biol. Chem. 2002, 277, 25152–25159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feagan, B.G.; Sandborn, W.J.; Danese, S.; Wolf, D.C.; Liu, W.J.; Hua, S.Y.; Minton, N.; Olson, A.; D’Haens, G. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: A single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol. Hepatol. 2020, 5, 819–828. [Google Scholar] [CrossRef]
- Gerstenberger, B.S.; Banker, M.E.; Clark, J.D.; Dowty, M.E.; Fensome, A.; Gifford, R.; Griffor, M.C.; Hegen, M.; Hollingshead, B.D.; Knafels, J.D.; et al. Demonstration of In Vitro to In Vivo Translation of a TYK2 Inhibitor That Shows Cross Species Potency Differences. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011, 3, 535–545. [Google Scholar] [CrossRef]
- Feagan, B.G.; Panés, J.; Ferrante, M.; Kaser, A.; D’Haens, G.R.; Sandborn, W.J.; Louis, E.; Neurath, M.F.; Franchimont, D.; Dewit, O.; et al. Risankizumab in patients with moderate to severe Crohn’s disease: An open-label extension study. Lancet Gastroenterol. Hepatol. 2018, 3, 671–680. [Google Scholar] [CrossRef]
- Xiao, Q.; Boushey, R.P.; Cino, M.; Drucker, D.J.; Brubaker, P.L. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1057–R1063. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, M.; McKay, D.; Yang, P.; Cameron, H.; Perdue, M. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut 2000, 47, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Buchman, A.L.; Katz, S.; Fang, J.C.; Bernstein, C.N.; Abou-Assi, S.G.; Group, T.S. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 962–973. [Google Scholar] [CrossRef]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.J.; Sartor, R.B. Therapeutic Manipulation of the Microbiome in IBD: Current Results and Future Approaches. Curr. Treat. Options Gastroenterol. 2015, 13, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Massier, S.; Darfeuille-Michaud, A.; Billard, E.; Barnich, N. Understanding host-adherent-invasive Escherichia coli interaction in Crohn’s disease: Opening up new therapeutic strategies. BioMed. Res. Int. 2014, 2014, 567929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivignon, A.; Bouckaert, J.; Bernard, J.; Gouin, S.G.; Barnich, N. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin. Ther. Targets 2017, 21, 837–847. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Giorgio, M.E.; Saviano, A.; Scaldaferri, F.; Gasbarrini, A.; Cammarota, G. Bacteriocins and bacteriophages: Therapeutic weapons for gastrointestinal diseases? Int. J. Mol. Sci. 2019, 20, 183. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Galtier, M.; De Sordi, L.; Sivignon, A.; de Vallée, A.; Maura, D.; Neut, C.; Rahmouni, O.; Wannerberger, K.; Darfeuille-Michaud, A.; Desreumaux, P.; et al. Bacteriophages Targeting Adherent Invasive Escherichia coli Strains as a Promising New Treatment for Crohn’s Disease. J. Crohns Colitis 2017, 11, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laveissière, A.; Morra, R.; Paillarse, J.M.; Bonny, C.; Patel, J. EB8018, a First-In-Class Fimh Blocker, Reduces Proinflammatory Cytokines TNFÎʻ, Il6 and Il8 in Surgical Explants and May Represent a Potential New Therapeutic Approach for the Treatment of Crohn’s Disease. Gastroenterology 2017, 152, 989. [Google Scholar] [CrossRef]
- Brown, C.L.; Smith, K.; Wall, D.M.; Walker, D. Activity of Species-specific Antibiotics Against Crohn’s Disease–Associated Adherent-invasive Escherichia coli. Inflamm. Bowel Dis. 2015, 21, 2372–2382. [Google Scholar]
- McKay, R.; Ghodasra, M.; Schardt, J.; Quan, D.; Pottash, A.E.; Shang, W.; Jay, S.M.; Payne, G.F.; Chang, M.W.; March, J.C.; et al. A platform of genetically engineered bacteria as vehicles for localized delivery of therapeutics: Toward applications for Crohn’s disease. Bioeng. Transl. Med. 2018, 3, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Suzuki, K.; Watanabe, M.; Okamoto, R. Stem cell-based therapy for inflammatory bowel disease. Intest. Res. 2019, 17, 311–316. [Google Scholar] [CrossRef]
- Barnhoorn, M.C.; Wasser, M.; Roelofs, H.; Maljaars, P.W.J.; Molendijk, I.; Bonsing, B.A.; Oosten, L.E.M.; Dijkstra, G.; van der Woude, C.J.; Roelen, D.L.; et al. Long-term Evaluation of Allogeneic Bone Marrow-derived Mesenchymal Stromal Cell Therapy for Crohn’s Disease Perianal Fistulas. J. Crohns Colitis 2020, 14, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.C.; Chang, C.; Boschetti, G.; Ungaro, R.; Giri, M.; Grout, J.A.; Gettler, K.; Chuang, L.S.; Nayar, S.; Greenstein, A.J.; et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 2019, 178, 1493–1508. [Google Scholar] [CrossRef]
- Grand View Research. Inflammatory Bowel Disease Treatment Market. Size, Share & Trends Analysis Report By Type (Ulcerative Colitis, Crohn’s Disease), By Route of Administration, By Distribution Channel, And Segment Forecasts 2019–2026. 2019. Available online: https://www.grandviewresearch.com/industry-analysis/inflammatory-bowel-disease-ibd-treatment-market (accessed on 10 September 2020).
- GlobalData. Crohn’s Disease Market Growth to 2026 Fuelled by Interleukin Inhibitor and Anti-Integrin Therapy Launches. 2017. Available online: https://www.globaldata.com/crohns-disease-market-growth-to-2026-fuelled-by-interleukin-inhibitor-and-anti-integrin-therapy-launches/ (accessed on 10 September 2020).
- Dahlhamer, J.; Lucas, J.; Zelaya, C.; Nahin, R.; Mackey, S.; DeBar, L.; Kerns, R.; Von Korff, M.; Porter, L.; Helmick, C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults—United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. [Google Scholar] [CrossRef]
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.J.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef]
- Watson, J.C. Treatment of Pain. 2020. Available online: https://www.merckmanuals.com/home/brain,-spinal-cord,-and-nerve-disorders/pain/treatment-of-pain (accessed on 2 October 2020).
- Binder, A.; Baron, R. The Pharmacological Therapy of Chronic Neuropathic Pain. Dtsch. Arztebl. Int. 2016, 113, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, K.E. Management of neuropathic pain. J. Am. Osteopath. Assoc. 2005, 105, S12–S19. [Google Scholar] [PubMed]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Bijlsma, J.W.; Berenbaum, F.; Lafeber, F.P. Osteoarthritis: An update with relevance for clinical practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- Fu, K.; Robbins, S.R.; McDougall, J.J. Osteoarthritis: The genesis of pain. Rheumatology 2018, 57, iv43–iv50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuca, B.; Silberstein, S.D.; Wietecha, L.; Berg, P.H.; Dozier, G.; Lipton, R.B.; Group, C.M.-S. Lasmiditan is an effective acute treatment for migraine: A phase 3 randomized study. Neurology 2018, 91, e2222–e2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felson, D.T.; Zhang, Y.; Hannan, M.T.; Naimark, A.; Weissman, B.N.; Aliabadi, P.; Levy, D. The incidence and natural history of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum. 1995, 38, 1500–1505. [Google Scholar] [CrossRef]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.V.; Raphael, J.H.; Dimitroulas, T.; Sparkes, E.; Southall, J.L.; Ashford, R.L.; Kitas, G.D. Osteoarthritis pain has a significant neuropathic component: An exploratory in vivo patient model. Rheumatol. Int. 2014, 34, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Dickenson, A.H.; Baron, R. Osteoarthritis pain: Nociceptive or neuropathic? Nat. Rev. Rheumatol. 2014, 10, 374–380. [Google Scholar] [CrossRef]
- Chen, Q.; Heinricher, M.M. Descending Control Mechanisms and Chronic Pain. Curr. Rheumatol. Rep. 2019, 21, 1–7. [Google Scholar] [CrossRef]
- Chimenti, R.L.; Frey-Law, L.A.; Sluka, K.A. A Mechanism-Based Approach to Physical Therapist Management of Pain. Phys. Ther. 2018, 98, 302–314. [Google Scholar] [CrossRef]
- Chang, D.S.; Hsu, E.; Hottinger, D.G.; Cohen, S.P. Anti-nerve growth factor in pain management: Current evidence. J. Pain Res. 2016, 9, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, Y.Y.; Huebner, J.L.; Haaland, B.; Wong, S.B.S.; Kraus, V.B. Synovial fluid pro-inflammatory profile differs according to the characteristics of knee pain. Osteoarthr. Cartil. 2017, 25, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Mora, J.C.; Przkora, R.; Cruz-Almeida, Y. Knee osteoarthritis: Pathophysiology and current treatment modalities. J. Pain Res. 2018, 11, 2189–2196. [Google Scholar] [CrossRef] [Green Version]
- Dray, A.; Read, S.J. Arthritis and pain. Future targets to control osteoarthritis pain. Arthritis Res. Ther. 2007, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- OARSI. Osteoarthritis: A Serious Disease 2016. Available online: https://oarsi.org/sites/default/files/docs/2016/oarsi_white_paper_oa-serious-disease.pdf (accessed on 10 August 2020).
- Kloppenburg, M.; Berenbaum, F. Osteoarthritis year in review 2019: Epidemiology and therapy. Osteoarthr. Cartil. 2020, 28, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeed, M.H.; Sherazi, S.A.A.; Bacon, D.; Bajwa, Z.H. Pharmacological Treatment of Pain in Osteoarthritis: A Descriptive Review. Curr. Rheumatol. Rep. 2018, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of Pharmacological Treatments With Long-term Pain Control in Patients With Knee Osteoarthritis: A Systematic Review and Meta-analysis. JAMA 2018, 320, 2564–2579. [Google Scholar] [CrossRef] [Green Version]
- Sarzi-Puttini, P.; Cimmino, M.A.; Scarpa, R.; Caporali, R.; Parazzini, F.; Zaninelli, A.; Atzeni, F.; Canesi, B. Osteoarthritis: An overview of the disease and its treatment strategies. Semin. Arthritis Rheum. 2005, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pertusi, R.M. Selective cyclooxygenase inhibition in pain management. J. Am. Osteopath Assoc. 2004, 104, S19–S24. [Google Scholar]
- Citrome, L.; Weiss-Citrome, A. A systematic review of duloxetine for osteoarthritic pain: What is the number needed to treat, number needed to harm, and likelihood to be helped or harmed? Postgrad. Med. 2012, 124, 83–93. [Google Scholar] [CrossRef]
- Lee, B.; Sodhi, N.; Anis, H.K.; Ehiorobo, J.O.; Mont, M.A. Injection Alternatives for the Management of Knee Osteoarthritis Pain. Surg. Technol. Int. 2019, 34, 513–519. [Google Scholar] [PubMed]
- Miller, R.E.; Block, J.A.; Malfait, A.M. What is new in pain modification in osteoarthritis? Rheumatology 2018, 57, iv99–iv107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, S.L.; Li, Y.; Ning, G.Z.; Yuan, Z.F.; Chen, L.X.; Bi, M.C.; Sun, J.C.; Feng, S.Q. Tanezumab for Patients with Osteoarthritis of the Knee: A Meta-Analysis. PLoS ONE 2016, 11, e0157105. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, J.; Li, R.; Wang, H.; Yang, J.; Xu, J.; Zha, Z. Efficacy and Safety of Tanezumab on Osteoarthritis Knee and Hip Pains: A Meta-Analysis of Randomized Controlled Trials. Pain Med. 2017, 18, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnitzer, T.J.; Ekman, E.F.; Spierings, E.L.; Greenberg, H.S.; Smith, M.D.; Brown, M.T.; West, C.R.; Verburg, K.M. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann. Rheum. Dis 2015, 74, 1202–1211. [Google Scholar] [CrossRef]
- Miller, R.E.; Block, J.A.; Malfait, A.M. Nerve growth factor blockade for the management of osteoarthritis pain: What can we learn from clinical trials and preclinical models? Curr. Opin. Rheumatol. 2017, 29, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Lane, N.E.; Corr, M. Osteoarthritis in 2016: Anti-NGF treatments for pain—Two steps forward, one step back? Nat. Rev. Rheumatol. 2017, 13, 76–78. [Google Scholar] [CrossRef]
- Kloppenburg, M.; Peterfy, C.; Haugen, I.K.; Kroon, F.; Chen, S.; Wang, L.; Liu, W.; Levy, G.; Fleischmann, R.M.; Berenbaum, F.; et al. Phase IIa, placebo-controlled, randomised study of lutikizumab, an anti-interleukin-1alpha and anti-interleukin-1beta dual variable domain immunoglobulin, in patients with erosive hand osteoarthritis. Ann. Rheum. Dis. 2019, 78, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischmann, R.M.; Bliddal, H.; Blanco, F.J.; Schnitzer, T.J.; Peterfy, C.; Chen, S.; Wang, L.; Feng, S.; Conaghan, P.G.; Berenbaum, F.; et al. A Phase II Trial of Lutikizumab, an Anti-Interleukin-1alpha/beta Dual Variable Domain Immunoglobulin, in Knee Osteoarthritis Patients With Synovitis. Arthritis Rheumatol. 2019, 71, 1056–1069. [Google Scholar] [CrossRef]
- Chevalier, X.; Ravaud, P.; Maheu, E.; Baron, G.; Rialland, A.; Vergnaud, P.; Roux, C.; Maugars, Y.; Mulleman, D.; Lukas, C.; et al. Adalimumab in patients with hand osteoarthritis refractory to analgesics and NSAIDs: A randomised, multicentre, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2015, 74, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Aitken, D.; Laslett, L.L.; Pan, F.; Haugen, I.K.; Otahal, P.; Bellamy, N.; Bird, P.; Jones, G. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis—The HUMOR trial. Osteoarthr. Cartil. 2018, 26, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Bainbridge, C.; Berkowitz, M. A Phase IIa Study of Anti-GM-CSF Antibody GSK3196165 in Subjects with Inflammatory Hand Osteoarthritis [abstract]. Arthritis Rheumatol. 2018, 70 (Suppl. 10), 1365. [Google Scholar]
- Jin, Y.; Smith, C.; Monteith, D.; Brown, R.; Camporeale, A.; McNearney, T.A.; Deeg, M.A.; Raddad, E.; Xiao, N.; de la Pena, A.; et al. CGRP blockade by galcanezumab was not associated with reductions in signs and symptoms of knee osteoarthritis in a randomized clinical trial. Osteoarthr. Cartil. 2018, 26, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017, 16, 545–564. [Google Scholar] [CrossRef] [Green Version]
- Bagal, S.M.C.; Brady, P.; Stauffer, J. A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Titration-to-Effect Study of Orally Administered CR845 in Patients with Osteoarthritis of the Hip or Knee [abstract]. Arthritis Rheumatol. 2017, 69 (Suppl. 10), 15L. [Google Scholar]
- Vertex. Vertex Reports Full-Year and Fourth-Quarter 2016 Financial Results. 2017. Available online: https://investors.vrtx.com/news-releases/news-release-details/vertex-reports-full-year-and-fourth-quarter-2016-financial (accessed on 22 September 2020).
- Stevens, R.M.; Ervin, J.; Nezzer, J.; Nieves, Y.; Guedes, K.; Burges, R.; Hanson, P.D.; Campbell, J.N. Randomized, Double-Blind, Placebo-Controlled Trial of Intraarticular Trans-Capsaicin for Pain Associated With Osteoarthritis of the Knee. Arthritis Rheumatol. 2019, 71, 1524–1533. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, P.; Chiche, D.; Brown, W.; Miller, J.; Treister, R.; Leff, R.; Walker, P.; Katz, N. NEO6860, modality-selective TRPV1 antagonist: A randomized, controlled, proof-of-concept trial in patients with osteoarthritis knee pain. Pain Rep. 2018, 3, e696. [Google Scholar] [CrossRef]
- Lories, R.J.; Monteagudo, S. Review Article: Is Wnt Signaling an Attractive Target for the Treatment of Osteoarthritis? Rheumatol. Ther. 2020, 7, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Li, Y.; Tang, Y.; Ding, C.; Hunter, D.J. Pharmacotherapy for knee osteoarthritis: Current and emerging therapies. Expert Opin. Pharmacother. 2020, 21, 797–809. [Google Scholar] [CrossRef]
- Wu, Y.; Goh, E.L.; Wang, D.; Ma, S. Novel treatments for osteoarthritis: An update. Open Access Rheumatol. 2018, 10, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Ha, C.W.; In, Y.; Cho, S.D.; Choi, E.S.; Ha, J.K.; Lee, J.H.; Yoo, J.D.; Bin, S.I.; Choi, C.H.; et al. A Multicenter, Double-Blind, Phase III Clinical Trial to Evaluate the Efficacy and Safety of a Cell and Gene Therapy in Knee Osteoarthritis Patients. Hum. Gene Ther. Clin. Dev. 2018, 29, 48–59. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-beta Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a022269. [Google Scholar] [CrossRef] [Green Version]
- Maheshwer, B.; Polce, E.M.; Paul, K.; Williams, B.T.; Wolfson, T.S.; Yanke, A.; Verma, N.N.; Cole, B.J.; Chahla, J. Regenerative Potential of Mesenchymal Stem Cells for the Treatment of Knee Osteoarthritis and Chondral Defects: A Systematic Review and Meta-Analysis. Arthroscopy 2020, 37, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Suo, H.; Wang, Z.; Feng, W. Progress in the treatment of osteoarthritis with umbilical cord stem cells. Hum. Cell 2020, 33, 470–475. [Google Scholar] [CrossRef]
- Pourakbari, R.; Khodadadi, M.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Yousefi, M. The potential of exosomes in the therapy of the cartilage and bone complications; emphasis on osteoarthritis. Life Sci. 2019, 236, 116861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Cai, Y.; Jiang, Y.; Lin, X. Exosomes in osteoarthritis and cartilage injury: Advanced development and potential therapeutic strategies. Int. J. Biol. Sci. 2020, 16, 1811–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.H.; Gouze, J.N.; Gouze, E.; Robbins, P.D.; Ghivizzani, S.C. Osteoarthritis gene therapy. Gene Ther. 2004, 11, 379–389. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, J.; Fan, Y.; Li, J.; You, T.; He, S.; Xiao, G.; Chen, D. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann. Rheum. Dis. 2019, 78, 676–682. [Google Scholar] [CrossRef]
- Zhang, W. The powerful placebo effect in osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S120), 118–123. [Google Scholar]
- Grandi, F.C.; Bhutani, N. Epigenetic Therapies for Osteoarthritis. Trends Pharmacol. Sci. 2020, 41, 557–569. [Google Scholar] [CrossRef]
- Migraine Research Foundation. About Migraine. Available online: https://migraineresearchfoundation.org/about-migraine/migraine-facts/ (accessed on 10 October 2020).
- American Headache Society. Migraine. Available online: https://americanheadachesociety.org/wp-content/uploads/2019/03/GENERALMIGRIANE_11x14.pdf (accessed on 10 October 2020).
- Zameel Cader, M. The molecular pathogenesis of migraine: New developments and opportunities. Hum. Mol. Genet. 2013, 22, R39-44. [Google Scholar] [CrossRef] [Green Version]
- Andreou, A.P.; Edvinsson, L. Mechanisms of migraine as a chronic evolutive condition. J. Headache Pain 2019, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.J. Acute Migraine Treatment in Adults. Headache 2015, 55, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Burstein, R.; Ashina, M.; Tfelt-Hansen, P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009, 8, 679–690. [Google Scholar] [CrossRef]
- Charbit, A.R.; Akerman, S.; Goadsby, P.J. Dopamine: What’s new in migraine? Curr. Opin. Neurol. 2010, 23, 275–281. [Google Scholar] [CrossRef]
- Barbanti, P.; Aurilia, C.; Egeo, G.; Fofi, L.; Palmirotta, R. Serotonin receptor targeted therapy for migraine treatment: An overview of drugs in phase I and II clinical development. Expert Opin. Investig. Drugs 2017, 26, 269–277. [Google Scholar] [CrossRef]
- Taylor, F.R.; Kaniecki, R.G. Symptomatic treatment of migraine: When to use NSAIDs, triptans, or opiates. Curr. Treat. Options Neurol. 2011, 13, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, D.; Adornetto, A.; Rombola, L.; Naturale, M.D.; Morrone, L.A.; Bagetta, G.; Tonin, P.; Corasaniti, M.T. New Trends in Migraine Pharmacology: Targeting Calcitonin Gene-Related Peptide (CGRP) With Monoclonal Antibodies. Front. Pharmacol. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, I.; Brown, M.D.; Innes, G.D.; Grafstein, E.; Roberts, T.E.; Rowe, B.H. Parenteral dihydroergotamine for acute migraine headache: A systematic review of the literature. Ann. Emerg. Med. 2005, 45, 393–401. [Google Scholar] [CrossRef]
- Dahlof, C.; Maassen Van Den Brink, A. Dihydroergotamine, ergotamine, methysergide and sumatriptan—Basic science in relation to migraine treatment. Headache 2012, 52, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.E.; Vinge, E. Beta-adrenoceptor blockers and calcium antagonists in the prophylaxis and treatment of migraine. Drugs 1990, 39, 355–373. [Google Scholar] [CrossRef]
- Diener, H.C.; Kaube, H.; Limmroth, V. Migraine: Clinical aspects, diagnosis and pharmacotherapy. Internist 2000, 41, 390–398. [Google Scholar] [CrossRef]
- Akerman, S.; Goadsby, P.J. Dopamine and migraine: Biology and clinical implications. Cephalalgia 2007, 27, 1308–1314. [Google Scholar] [CrossRef]
- Peroutka, S.J.; Wilhoit, T.; Jones, K. Clinical susceptibility to migraine with aura is modified by dopamine D2 receptor (DRD2) NcoI alleles. Neurology 1997, 49, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Markley, H.G.; Cheronis, J.C.; Piepho, R.W. Verapamil in prophylactic therapy of migraine. Neurology 1984, 34, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Arceri, S.; Tronconi, L.; Tassorelli, C. Chronic migraine and Botulinum Toxin Type A: Where do paths cross? Toxicon 2020, 178, 69–76. [Google Scholar] [CrossRef]
- Sprenger, T.; Viana, M.; Tassorelli, C. Current Prophylactic Medications for Migraine and Their Potential Mechanisms of Action. Neurotherapeutics 2018, 15, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Oswald, J.C.; Schuster, N.M. Lasmiditan for the treatment of acute migraine: A review and potential role in clinical practice. J. Pain Res. 2018, 11, 2221–2227. [Google Scholar] [CrossRef] [Green Version]
- Clemow, D.B.; Johnson, K.W.; Hochstetler, H.M.; Ossipov, M.H.; Hake, A.M.; Blumenfeld, A.M. Lasmiditan mechanism of action—Review of a selective 5-HT1F agonist. J. Headache Pain 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Deen, M.; Correnti, E.; Kamm, K.; Kelderman, T.; Papetti, L.; Rubio-Beltran, E.; Vigneri, S.; Edvinsson, L.; Maassen Van Den Brink, A.M. Blocking CGRP in migraine patients—A review of pros and cons. J. Headache Pain 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, B.J.; Wattiez, A.S.; Waite, J.S.; Castonguay, W.C.; Schmidt, C.M.; Fairbanks, A.M.; Robertson, B.R.; Brown, C.J.; Mason, B.N.; Moldovan-Loomis, M.C.; et al. Peripherally administered calcitonin gene-related peptide induces spontaneous pain in mice: Implications for migraine. Pain 2018, 159, 2306–2317. [Google Scholar] [CrossRef] [PubMed]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in migraine. Cephalalgia 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Bucklan, J.; Ahmed, Z. CGRP antagonists for decreasing migraine frequency: New options, long overdue. Clevel. Clin. J. Med. 2020, 87, 211–218. [Google Scholar] [CrossRef]
- Moreno-Ajona, D.P.-R.A.; Goadsby, P.J. Small-molecule CGRP receptor antagonists: A new approach to the acute and preventive treatment of migraine. Med. Drug Discov. 2020, 7, 100053. [Google Scholar] [CrossRef]
- Karuna Therapeutics. Pipline. 2020. Available online: https://karunatx.com/programs/ (accessed on 12 October 2020).
- Alzheimer’s Association. Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 12 September 2020).
- Parkinson’s Foundation. Statistics. Available online: https://www.parkinson.org/Understanding-Parkinsons/Statistics (accessed on 12 September 2020).
- Alzheimer’s Disease International; Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. 2018. Available online: https://www.alzint.org/resource/world-alzheimer-report-2018/ (accessed on 13 September 2020).
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10 (Suppl. S1), S10–S17. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Smith, A.D. Imaging the progression of Alzheimer pathology through the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 4135–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo Clinic. Alzheimer’s Stages: How the Disease Progresses. Available online: https://www.mayoclinic.org/diseases-conditions/alzheimers-disease/in-depth/alzheimers-stages/art-20048448#:~:text=There%20are%20five%20stages%20associated,dementia%20due%20to%20Alzheimer’s%20disease (accessed on 11 October 2020).
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. 2020. Available online: https://www.alz.org/media/Documents/facts2020_report_1.pdf (accessed on 1 October 2020).
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Perez, S.E.; Chen, K.; Mufson, E.J. Braak Stage, Cerebral Amyloid Angiopathy, and Cognitive Decline in Early Alzheimer’s Disease. J. Alzheimers Dis. 2020, 74, 189–197. [Google Scholar] [CrossRef]
- Markesbery, W.R. Neuropathologic alterations in mild cognitive impairment: A review. J. Alzheimers Dis. 2010, 19, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, G.; Kovari, E.; Corte, G.; Herrmann, F.R.; Canuto, A.; Bussiere, T.; Hof, P.R.; Bouras, C.; Giannakopoulos, P. Clinical validity of A beta-protein deposition staging in brain aging and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Sarter, M.; Bruno, J.P. Cognitive functions of cortical acetylcholine: Toward a unifying hypothesis. Brain Res. Brain Res. Rev. 1997, 23, 28–46. [Google Scholar] [CrossRef]
- Hakansson, L. Mechanism of action of cholinesterase inhibitors in Alzheimer’s disease. Acta Neurol. Scand. Suppl. 1993, 149, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Rissel, M.; Le Bot, M.A.; Guillouzo, A. Toxic effects of tacrine on primary hepatocytes and liver epithelial cells in culture. Cell Biol. Toxicol. 1998, 14, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Touchon, J.; Bergman, H.; Gambina, G.; He, Y.; Rapatz, G.; Nagel, J.; Lane, R. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer’s disease over a 2-year period. Curr. Med. Res. Opin. 2005, 21, 1317–1327. [Google Scholar] [CrossRef]
- Wilcock, G.; Howe, I.; Coles, H.; Lilienfeld, S.; Truyen, L.; Zhu, Y.; Bullock, R.; Kershaw, P.; Group, G.-G.-S. A long-term comparison of galantamine and donepezil in the treatment of Alzheimer’s disease. Drugs Aging 2003, 20, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Gauthier, S.; Ng, K.P.; Pascoal, T.A.; Zhang, H.; Rosa-Neto, P. Targeting Alzheimer’s Disease at the Right Time and the Right Place: Validation of a Personalized Approach to Diagnosis and Treatment. J. Alzheimers Dis. 2018, 64, S23–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, A. The therapeutic potential of tacrine. J. Neural. Transm. Suppl. 1998, 54, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, B. Donepezil: A review. Expert Opin. Drug Metab. Toxicol. 2005, 1, 527–536. [Google Scholar] [CrossRef]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Razay, G.; Wilcock, G.K. Galantamine in Alzheimer’s disease. Expert Rev. Neurother. 2008, 8, 9–17. [Google Scholar] [CrossRef]
- Van Marum, R.J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2009, 5, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, T.; Bieger, S.C.; Bruhl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef]
- Atri, A. Current and Future Treatments in Alzheimer’s Disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Biogen and Eisai to Discontinue Phase 3 ENGAGE and EMERGE Trials of Aducanumab in Alzheimer’s Disease. 2019. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-eisai-discontinue-phase-3-engage-and-emerge-trials (accessed on 22 September 2020).
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Roche. Roche to Discontinue Phase III CREAD 1 and 2 Clinical Studies of Crenezumab in Early Alzheimer’s Disease (AD)—Other Company Programmes in AD Continue. 2019. Available online: https://www.roche.com/media/releases/med-cor-2019-01-30.htm (accessed on 10 October 2020).
- Lancet Neurology. Solanezumab: Too late in mild Alzheimer’s disease? Lancet Neurol. 2017, 16, 97. [Google Scholar] [CrossRef] [Green Version]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Mullard, A. Alzheimer prevention failure rattles field, anew. Nat. Rev. Drug Discov. 2019, 18, 656–657. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Initiation of Phase III Clinical Trial of BAN2401 in Early Alzheimer’s Disease. 2019. Available online: https://www.eisai.com/news;https://www.eisai.com/news/2019/news201919.html (accessed on 22 September 2020).
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Biogen. FDA Accepts Biogen’s Aducanumab Biologics License Application for Alzheimer’s Disease with Priority Review. Available online: http://media.biogen.com/news-releases/news-release-details/fda-accepts-biogens-aducanumab-biologics-license-application (accessed on 21 November 2020).
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Pardridge, W.M. Alzheimer’s disease: Future drug development and the blood-brain barrier. Expert Opin. Investig. Drugs 2019, 28, 569–572. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Inacio, P. UB-311 Vaccine Safe in Mild Alzheimer’s Patients, Phase 2a Trial Shows. 2019. Available online: https://alzheimersnewstoday.com/2019/04/03/ub-311-vaccine-safe-mild-alzheimers-patients-phase-2-study/ (accessed on 2 September 2020).
- Salloway, S.P.; Sevingy, J.; Budur, K.; Pederson, J.T.; DeMattos, R.B.; Von Rosenstiel, P.; Paez, A.; Evans, R.; Weber, C.J.; Hendrix, J.A.; et al. Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement. 2020, 6, e12073. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Pardridge, W.M. Re-engineering therapeutic antibodies for Alzheimer’s disease as blood-brain barrier penetrating bi-specific antibodies. Expert Opin. Biol. Ther. 2016, 16, 1455–1468. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, K.; Singer, T.; et al. Brain Shuttle Antibody for Alzheimer’s Disease with Attenuated Peripheral Effector Function due to an Inverted Binding Mode. Cell Rep. 2018, 22, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E. Open questions for Alzheimer’s disease immunotherapy. Alzheimers Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kingston, A.; Comas-Herrera, A.; Jagger, C.; MODEM Project. Forecasting the care needs of the older population in England over the next 20 years: Estimates from the Population Ageing and Care Simulation (PACSim) modelling study. Lancet Public Health 2018, 3, e447–e455. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997, 323 Pt 3, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Cheung, A.H.; Okon, M.; McIntosh, L.P.; Vocadlo, D.J. O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J. Mol. Biol. 2014, 426, 1736–1752. [Google Scholar] [CrossRef]
- Duits, F.H.; Martinez-Lage, P.; Paquet, C.; Engelborghs, S.; Lleo, A.; Hausner, L.; Molinuevo, J.L.; Stomrud, E.; Farotti, L.; Ramakers, I.; et al. Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study. Alzheimers Dement. 2016, 12, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, R.; Knapp, M.; Karagiannidou, M.; Dickson, J.; Schott, J. Economic impacts of introducing diagnostics for mild cognitive impairment Alzheimer’s disease patients. Alzheimers Dement. 2019, 5, 382–387. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs. Other Neurodegenerative Disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.; Elmaleh, D.R.; et al. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid beta-Protein by Promoting Microglial Phagocytosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008, 70, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AZTherapies. Advancing A Broad Pipeline Targeting Neurodegenerative Disease. Available online: https://aztherapies.com/pipeline/ (accessed on 9 November 2020).
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Yan, S.D.; Stern, D.M.; Walker, D.G. Preventing activation of receptor for advanced glycation endproducts in Alzheimer’s disease. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 249–266. [Google Scholar] [CrossRef]
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedros, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.B.; Siman, R.; Iqbal, M.A.; Potter, H. Identification of a chymotrypsin-like mast cell protease in rat brain capable of generating the N-terminus of the Alzheimer amyloid beta-protein. J. Neurochem. 1993, 61, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Tengfei Lia, E.M.; Abada, Y.; Boucher, C.; Ces, A.; Youssef, I.; Fenaux, G.; Forand, Y.; Legrand, A.; Nachiket, N.; Dhenain, M.; et al. Effects of Chronic Masitinib Treatment in APPswe/PSEN1dE9 Transgenic Mice Modeling Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1339–1345. [Google Scholar]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 1–11. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- NIH U.S. National Library of Medicine. A Study of Sodium Oligomannate (GV-971) in Participants With Mild to Moderate Alzheimer’s Disease (GREEN MEMORY). 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04520412 (accessed on 12 November 2020).
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; Desjarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, C.; Tse, Y.C.; Nguyen, H.B.; Krantic, S.; Breitner, J.C.; Quirion, R.; Wong, T.P. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 2016, 47, 41–49. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, N.A.; van der Marck, M.A.; Pijl, H.; Olde Rikkert, M.G.; Bloem, B.R.; Roos, R.A. Weight loss in neurodegenerative disorders. J. Neurol. 2008, 255, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, S.; Ellul, J.; Argyriou, A.A.; Talelli, P.; Chroni, E.; Papapetropoulos, T. The effect of vascular disease on late onset Parkinson’s disease. Eur. J. Neurol. 2004, 11, 231–235. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Lindholm, K.; Yang, L.B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Harper, A.J.; Hamilton, D.L.; Gallagher, J.; McNeilly, A.D.; Burgess, L.A.; Vaanholt, L.M.; Bannon, K.A.; Latcham, J.; Hussain, I.; et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 2012, 441, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Ng, R.C.; Jian, M.; Ma, O.K.; Bunting, M.; Kwan, J.S.; Zhou, G.J.; Senthilkumar, K.; Iyaswamy, A.; Chan, P.K.; Li, M.; et al. Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol. Psychiatry 2020, 1–21. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Wang, J.G.; Liu, C.L.; Yan, H.J. AdipoRon improves cognitive dysfunction of Alzheimer’s disease and rescues impaired neural stem cell proliferation through AdipoR1/AMPK pathway. Exp. Neurol. 2020, 327, 113249. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Clarke, D.W.; Boyd, F.T., Jr.; Kappy, M.S.; Raizada, M.K. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 1984, 259, 11672–11675. [Google Scholar] [CrossRef]
- Raizada, M.K.; Shemer, J.; Judkins, J.H.; Clarke, D.W.; Masters, B.A.; LeRoith, D. Insulin receptors in the brain: Structural and physiological characterization. Neurochem. Res. 1988, 13, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Kirpichnikov, D.; McFarlane, S.I.; Sowers, J.R. Metformin: An update. Ann. Intern. Med. 2002, 137, 25–33. [Google Scholar] [CrossRef]
- Craig, A.; Parvez, F.; Issberner, J. A systematic literature review of the effect of insulin sensitizers on the cognitive symptoms of Alzheimer’s Disease in transgenic mice. Behav. Brain Res. 2019, 372, 112015. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Perna, S.; Mainardi, M.; Astrone, P.; Gozzer, C.; Biava, A.; Bacchio, R.; Spadaccini, D.; Solerte, S.B.; Rondanelli, M. 12-month effects of incretins versus SGLT2-Inhibitors on cognitive performance and metabolic profile. A randomized clinical trial in the elderly with Type-2 diabetes mellitus. Clin. Pharmacol. 2018, 10, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- D’Orio, B.; Fracassi, A.; Ceru, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Reyes-Irisarri, E.; Hull, M.; Kummer, M.P. Impact and Therapeutic Potential of PPARs in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 643–650. [Google Scholar] [CrossRef]
- Kumar, V.; Jahan, S.; Singh, S.; Khanna, V.K.; Pant, A.B. Progress toward the development of in vitro model system for chemical-induced developmental neurotoxicity: Potential applicability of stem cells. Arch. Toxicol. 2015, 89, 265–267. [Google Scholar] [CrossRef]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.M.; Ayala, M.; Zhang, S.C. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, J.S.; Sakowski, S.A.; Hur, J.; Feldman, E.L. Stem cell technology for neurodegenerative diseases. Ann. Neurol. 2011, 70, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.S.; Lee, S.R.; Kim, S.U.; Lee, H.J. Alzheimer’s disease and stem cell therapy. Exp. Neurobiol. 2014, 23, 45–52. [Google Scholar] [CrossRef]
- Medipost. NEUROSTEM. Available online: http://www.medi-post.com/neurostem/ (accessed on 13 September 2020).
- Stemedica. Stemedica Begins First Clinical Trials In The U.S. Using Adult Allogeneic Stem Cells To Treat Alzheimer’s Disease. 2016. Available online: https://www.stemedica.com/stemedica-begins-first-clinical-trials-in-the-u-s-using-adult-allogeneic-stem-cells-to-treat-alzheimers-disease/ (accessed on 10 August 2020).
- Longeveron. Open Clinical Trials: Allogeneic Human Mesenchymal Stem Cell Infusion Versus Placebo in Patients With Alzheimer’s Disease. Available online: http://longeveron.com/clinical-trials/ (accessed on 11 October 2020).
- Klug, B.; Celis, P.; Carr, M.; Reinhardt, J. Regulatory structures for gene therapy medicinal products in the European Union. Methods Enzymol. 2012, 507, 337–354. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Piras, A.; Ismail, M.A.M.; Manchanda, S.; Eyjolfsdottir, H.; Saido, T.C.; Johansson, J.; Eriksdotter, M.; Winblad, B.; Nilsson, P. Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 2018, 284, 2–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penaud-Budloo, M.; Francois, A.; Clement, N.; Ayuso, E. Pharmacology of Recombinant Adeno-associated Virus Production. Mol. Ther. Methods Clin. Dev. 2018, 8, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [Green Version]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef]
- Fischer, W.; Wictorin, K.; Bjorklund, A.; Williams, L.R.; Varon, S.; Gage, F.H. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 1987, 329, 65–68. [Google Scholar] [CrossRef]
- Ionis. Ionis Innovation Pipeline. Available online: https://www.ionispharma.com/ionis-innovation/pipeline/ (accessed on 14 October 2020).
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain Stimulation in Alzheimer’s Disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Yan, H.; Zhou, J.; Yang, X.; Lu, Y.; Han, Y. A circuit view of deep brain stimulation in Alzheimer’s disease and the possible mechanisms. Mol. Neurodegener. 2019, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tarsy, D. Treatment of Parkinson disease: A 64-year-old man with motor complications of advanced Parkinson disease. JAMA 2012, 307, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.; Diederich, N.J.; Kruger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson’s Foundation. Understanding Parkinson’s. Available online: https://www.parkinson.org/understanding-parkinsons/what-is-parkinsons (accessed on 8 September 2020).
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s Diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. Parkinson’s disease and other alpha-synucleinopathies. Clin. Chem. Lab. Med. 2001, 39, 308–312. [Google Scholar] [CrossRef]
- Sonne, J.; Goyal, A.; Bansal, P.; Lopez-Ojeda, W. Dopamine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Boshes, B. Sinemet and the treatment of Parkinsonism. Ann. Intern. Med. 1981, 94, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Thanvi, B.R.; Lo, T.C. Long term motor complications of levodopa: Clinical features, mechanisms, and management strategies. Postgrad. Med. J. 2004, 80, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Borovac, J.A. Side effects of a dopamine agonist therapy for Parkinson’s disease: A mini-review of clinical pharmacology. Yale J. Biol. Med. 2016, 89, 37–47. [Google Scholar]
- Clarke, C.E.; Guttman, M. Dopamine agonist monotherapy in Parkinson’s disease. Lancet 2002, 360, 1767–1769. [Google Scholar] [CrossRef]
- MAO-B inhibitors for the treatment of Parkinson’s disease. Mov. Disord. 2002, 17 (Suppl. S4), S38–S44. [CrossRef]
- Kaakkola, S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs 2000, 59, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci. 2005, 25, 3312–3322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.K.; Yung, K.K. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, Y.; Kondo, T.; Japanese Istradefylline Study Group. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [Green Version]
- Groiss, S.J.; Wojtecki, L.; Sudmeyer, M.; Schnitzler, A. Deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamani, C.; Florence, G.; Heinsen, H.; Plantinga, B.R.; Temel, Y.; Uludag, K.; Alho, E.; Teixeira, M.J.; Amaro, E.; Fonoff, E.T. Subthalamic Nucleus Deep Brain Stimulation: Basic Concepts and Novel Perspectives. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, C.C.; Thakor, N.V. Uncovering the mechanisms of deep brain stimulation for Parkinson’s disease through functional imaging, neural recording, and neural modeling. Crit. Rev. Biomed. Eng. 2002, 30, 249–281. [Google Scholar] [CrossRef] [PubMed]
- Dostrovsky, J.O.; Levy, R.; Wu, J.P.; Hutchison, W.D.; Tasker, R.R.; Lozano, A.M. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J. Neurophysiol. 2000, 84, 570–574. [Google Scholar] [CrossRef]
- Beurrier, C.; Bioulac, B.; Audin, J.; Hammond, C. High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. J. Neurophysiol. 2001, 85, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Mehdizadeh, A.R. Deep Brain Stimulation and Gene Expression Alterations in Parkinson’s Disease. J. Biomed. Phys. Eng. 2016, 6, 47–50. [Google Scholar] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Shihabuddin, L.S.; Brundin, P.; Greenamyre, J.T.; Stephenson, D.; Sardi, S.P. New Frontiers in Parkinson’s Disease: From Genetics to the Clinic. J. Neurosci. 2018, 38, 9375–9382. [Google Scholar] [CrossRef] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Prevail Therapeutics. PR001. 2020. Available online: https://www.prevailtherapeutics.com/programs/#:~:text=PR001,the%20same%20gene%2C%20called%20GBA1. (accessed on 20 September 2020).
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuropore. Inhibit the Formation of Toxic Aggregates of Misfolded Proteins. 2020. Available online: https://www.neuropore.com/programs/anti-oligomerization.htm (accessed on 21 September 2020).
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Botzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Wegrzynowicz, M.; Bar-On, D.; Calo, L.; Anichtchik, O.; Iovino, M.; Xia, J.; Ryazanov, S.; Leonov, A.; Giese, A.; Dalley, J.W.; et al. Depopulation of dense alpha-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 2019, 138, 575–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MODAG Neuroscience Solutions. MODAG Successfully Completes Phase 1 Study of their Lead Candidate Anle138b and Receives Additional USD 1.4 Million from Michael J. Fox Foundation. Available online: https://www.modag.net/index.php/en/press-releases (accessed on 21 September 2020).
- Smith, S.B. Introduction to Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Adv. Exp. Med. Biol. 2017, 964, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Alessi, D.R. Advances in elucidating the function of leucine-rich repeat protein kinase-2 in normal cells and Parkinson’s disease. Curr. Opin. Cell Biol. 2020, 63, 102–113. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Saarma, M. CDNF Protein Therapy in Parkinson’s Disease. Cell Transplant. 2019, 28, 349–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, K.; Renko, J.M.; Matlik, K.; Airavaara, M.; Voutilainen, M.H. Cerebral Dopamine Neurotrophic Factor Diffuses Around the Brainstem and Does Not Undergo Anterograde Transport After Injection to the Substantia Nigra. Front. Neurosci. 2019, 13, 590. [Google Scholar] [CrossRef]
- Chen, Y.C.; Baronio, D.; Semenova, S.; Abdurakhmanova, S.; Panula, P. Cerebral Dopamine Neurotrophic Factor regulates multiple neuronal subtypes and behavior. J. Neurosci. 2020, 40, 6146–6164. [Google Scholar] [CrossRef] [PubMed]
- Abi Hussein, H.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A.C. Global vision of druggability issues: Applications and perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A highlight of therapeutics for T1D and T2D discussed in this review. T1D: Type 1 diabetes; T2D: Type 2 diabetes.
Figure 1.
A highlight of therapeutics for T1D and T2D discussed in this review. T1D: Type 1 diabetes; T2D: Type 2 diabetes.

Figure 2.
Therapeutic targets and modalities discussed here to inhibit autoimmunity in rheumatoid arthritis, atopic dermatitis, and Crohn’s disease. Targets shown in back represent corresponding drugs in the market. Targets and approaches in shown orange and blue represent corresponding drugs in clinical and clinical development, respectively. RA: Rheumatoid arthritis; AtD: Atopic dermatitis; CD: Crohn’s disease; TNF: Tumor necrosis factor; PDE4: Phosphodiesterase 4; JAK: Janus kinase; BTK: Bruton’s tyrosine kinase; SYK: Spleen tyrosine kinase; H4R: Histamine H4 receptor; S1P: Sphingosine 1-phosphate; TYK2: Tyrosine kinase 2; GM-CSF: Granulocyte-macrophage colony stimulating factor; TSLP: Thymic stromal lymphopoietin; MAdCAM: Mucosal addressin cell adhesion molecule; GLP-2: Glucagon-like peptide 2; MSCs: Mesenchymal stem cells; TolDCs: Tolerogenic dendritic cells; INTCs: Invariant natural killer T cells; siRNA: Short interfering RNA.
Figure 2.
Therapeutic targets and modalities discussed here to inhibit autoimmunity in rheumatoid arthritis, atopic dermatitis, and Crohn’s disease. Targets shown in back represent corresponding drugs in the market. Targets and approaches in shown orange and blue represent corresponding drugs in clinical and clinical development, respectively. RA: Rheumatoid arthritis; AtD: Atopic dermatitis; CD: Crohn’s disease; TNF: Tumor necrosis factor; PDE4: Phosphodiesterase 4; JAK: Janus kinase; BTK: Bruton’s tyrosine kinase; SYK: Spleen tyrosine kinase; H4R: Histamine H4 receptor; S1P: Sphingosine 1-phosphate; TYK2: Tyrosine kinase 2; GM-CSF: Granulocyte-macrophage colony stimulating factor; TSLP: Thymic stromal lymphopoietin; MAdCAM: Mucosal addressin cell adhesion molecule; GLP-2: Glucagon-like peptide 2; MSCs: Mesenchymal stem cells; TolDCs: Tolerogenic dendritic cells; INTCs: Invariant natural killer T cells; siRNA: Short interfering RNA.

Table 1.
Expedited novel drug approvals by CDER in 2011–2020.
| Year | Total Approval | Fast Track (% of Total) | Breakthrough (% of Total) | Priority Review (% of Total) | Accelerated Approval (% of Total) | Used One or More Expedited Pathway (% of Total) |
|---|---|---|---|---|---|---|
| 2011 | 30 | 14 (47%) | N/A | 15 (50%) | 3 (10%) | 17 (57%) |
| 2012 | 39 | 14 (36%) | N/A | 16 (41%) | 4 (10%) | 22 (56%) |
| 2013 | 27 | 10 (37%) | 3 (11%) | 10 (37%) | 2 (7%) | 13 (48%) |
| 2014 | 41 | 17 (41%) | 9 (22%) | 25 (61%) | 8 (20%) | 27 (66%) |
| 2015 | 45 | 14 (31%) | 10 (22%) | 24 (53%) | 6 (13%) | 27 (60%) |
| 2016 | 22 | 8 (36%) | 7 (32%) | 15 (68%) | 6 (27%) | 16 (73%) |
| 2017 | 46 | 18 (39%) | 17 (37%) | 28 (61%) | 6 (13%) | 28 (61%) |
| 2018 | 59 | 24 (41%) | 14 (24%) | 43 (73%) | 4 (7%) | 43 (73%) |
| 2019 | 48 | 17 (35%) | 13 (27%) | 28 (58%) | 9 (19%) | 29 (60%) |
| 9/2020 | 40 | 0 | 0 | 19 (48%) | 0 | 17 (48%) |
Source: Drugs@FDA.
Table 2.
Overview of approved therapeutic modalities.
| Modality | Target | Target Site | Delivery | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DNA | RNA | Protein | Extracellular | Plasma Membrane | Intracellular | Oral | Injection | Inhaled | |
| Small molecules | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Antibody-based agents | Yes | Yes | Yes | Yes | |||||
| Peptides | Yes | Yes | Yes | Yes | Yes | Yes | |||
| Oligonucleotide therapy | Yes | Yes | Yes | Yes | |||||
| Gene and Cell therapy | Yes | Yes | Yes | Yes | Yes | ||||
Table 3.
Novel therapeutic modalities approved in 2015–2020.
| Year | Small Molecule | rProtein | mAb/bsAb/Nanobody | ADC | Peptide | Oligonucleotide | Cellular & Gene Therapy | Other |
|---|---|---|---|---|---|---|---|---|
| 2015 | 31 | 3 | 8 (mAb), 1 (Fab) | 0 | 1 | 0 | 1 (oncolytic virus) | 1 (oligosaccharides) |
| 2016 | 11 | 0 | 7 (mAb) | 0 | 1 | 3 | 0 | 0 |
| 2017 | 29 | 2 | 8 (mAb), 1 (bsAb) | 1 | 5 | 0 | 2 (CAR T), 1 (gene) | 0 |
| 2018 | 38 | 5 | 11 (mAb) | 1 | 1 | 2 | 0 | 1 (diagnostic agent) |
| 2019 | 32 | 2 | 3 (mAb), 1 (scFv), 1 (NB) | 3 | 3 | 2 | 1 (gene) | 1 (fatty acid) |
| 9/2020 | 28 | 1 | 6 (mAb) | 2 | 1 | 1 | 1 (CAR T) | 1 (fatty acid) |
| Total | 169 | 13 | 47 | 7 | 12 | 8 | 6 | 4 |
rProtein: Recombinant protein; mAb: Monoclonal antibody; Fab: Fragment antigen-binding; bsAb: Bispecific anybody; scFv: Single-chain v.
Table 4.
Gene therapies approved by CBER and CDER of FDA.
| Strategy | Approval Year | Trade Name | Drug Name | Sponsor | Properties | Indication for Use |
|---|---|---|---|---|---|---|
| Viral vectors | 2015 | Imlygic | talimogene laherparepvec | Amgen | Genetically modified oncolytic virus | Melanoma |
| 2017 | Luxturna | voretigene neparvovec-rzyl | Spark Therapeutics | AAV-based RPE65 gene therapy | Confirmed biallelic RPE65 mutation-associated retinal dystrophy | |
| 2019 | Zolgensma | onasemnogene abeparvovec-xioi | AveXis/Novartis | AAV- based SMN gene therapy | Spinal muscular atrophy (SMA) with bi-allelic mutations in the survival motor neuron 1 (SMN1) gene | |
| Oligonucleotides | 1998 | Vitravene * | fomivirsen | Novartis | ASO designed to inhibit human cytomegalovirus replication | Cytomegalovirus (CMV) retinitis |
| 2004 | Macugen * | pegaptanib | Valeant Pharmas | Aptamar designed to target VEGF | Neovascular age-related macular degeneration | |
| 2013 | Kynamro * | mipomersen | Kastle Theraps | Oligonucleotide inhibitor of apolipoprotein B-100 synthesis | Homozygous familial hypercholesterolemia | |
| 2016 | Defitelio | defibrotide | Gentium | Oligonucleotide mixture with Profibrinolytic properties | Hepatic veno-occlusive disease with additional kidney or lung abnormalities after receiving a hematopoietic stem cell transplantation | |
| 2016 | Exondys 51 | eteplirsen | Sarepta Therapeutics | ASO designed to target dystrophin pre-mRNA | Duchenne muscular dystrophy | |
| 2016 | Spinraza | nusinersen | Biogen/Ionis Pharmaceuticals | ASO designed to target SMN2 pre-mRNA | Spinal muscular atrophy (SMA) | |
| 2018 | Onpattro | patisiran | Alnylam Pharmaceuticals | TTR-directed siRNA | Polyneuropathy of hereditary transthyretin-mediated amyloidosis | |
| 2018 | Tegsedi | inotersen | Ionis Pharmaceuticals | TTR-directed antisense oligonucleotide | Polyneuropathy of hereditary transthyretin-mediated amyloidosis | |
| 2019 | Givlaari | givosiran sodium | Alnylam | AL AS1-directed siRNA (GalNac conjugation) | Acute hepatic porphyria | |
| 2019 | Vyondys 53 | golodirsen | Sarepta | Exon 53 skipping antisense | Duchenne muscular dystrophy |
CBER: Center for Biologics Evaluation and Research; CDER: Center for Drug Evaluation and Research; CAR T: Chimeric antigen receptor T cell; RPE65: retinal pigment epithelium-specific; AAV: adeno-associated virus; SMN: survival of motor neuron 1; VEGF: vascular endothelial growth factor; TTR: transthyretin; AL AS1: aminolevulinate synthase 1. *, Vitravene, Macugen, and Kynamro are discontinued. Viral-based drugs are regulated by CBER whereas oligonucleotide-based drugs are reviewed by CDER. Injection is the dosage form for all the drugs listed in this table. Source: Drugs@FDA.
Table 5.
Approved and investigational biologics and small molecules for RA.
| Target | Modality | Drug Name | Sponsor | Status | NCT | Route of Administration |
|---|---|---|---|---|---|---|
| TNF | mAb | Enbrel (etanercept) | Amgen | Approved, 2002 | Subcutaneous | |
| mAb | Remicade (infliximab) | Janssen Biotech | Approved, 1999 | Intravenous | ||
| mAb | Humira (adalimumab) | AbbVie Inc | Approved, 2002 | Subcutaneous | ||
| mAb | CIMZIA (certolizumab pegol) | UCB | Aproved, 2009 | Subcutaneous | ||
| mAb | Simponi (golimumab) | Centocor, Inc. | Aproved, 2009 | Subcutaneous | ||
| mAb | ABBV-154 | AbbVie | Phase II | n/a | Intravenous | |
| mAb | ABBV-3373 | AbbVie | Phase II | NCT03823391 | Intravenous | |
| IL-6 | mAb | Actemra (tocilizumab) | Genentech | Approved, 2010 | Intravenous or Subcutaneous | |
| mAb | Plivensia (sirukumab) | Janssen Biotech | Withdrawn | Subcutaneous | ||
| mAb | Kevzara (sarilumab) | Sanofi and Regeneron Pharmaceuticals | Approved, 2017 | Subcutaneous | ||
| CD20 | mAb | Rituxan (rituximab) | Genentech, Inc. | Approved, 2006 | Subcutaneous | |
| mAb | Ocrelizumab | Genentech, Inc. | Phase III (terminated) | NCT02720120 | Intravenous | |
| mAb | Veltuzumab | Takeda | Phase II (terminated) | NCT01390545 | Subcutaneous | |
| mAb | Ofatumumab | GlaxoSmithKline | Phase III (terminated) | NCT00611455 | Intravenous | |
| CD80/CD86 | mAb | Orencia (abatacept), | Bristol-Myers Squibb | Approved, 2011 | Intravenous | |
| JAK | SM | XELJANZ (tofacitinib) | Pfizer | Approved, 2016 | Oral | |
| SM | Olumiant (Baricitinib) | Eli Lilly | Approved, 2018 | Oral | ||
| SM | Rinvoq (upadacitinib) | AbbVie | Approved, 2019 | Oral | ||
| SM | Abrocitinib | Pfizer | Phase III | n/a | Oral | |
| SM | Filgotinib | Galapagos NV/Gilead | Phase III | NCT02886728 (request additional data by FDA, approved in Japan) | Oral | |
| SM | Decernotinib | Aclaris Therapeutics | Phase II | n/a | Oral | |
| GM-CSF | mAb | Mavrilimumab | Kiniksa | Phase II | NCT01715896 | Subcutaneous |
| mAb | Namilumab | Takeda | Phase | NCT02379091, NCT02393378 | Subcutaneous | |
| mAb | Otilimab | GSK | Phase III | NCT04134728 | Subcutaneous | |
| BTK | SM | Spebrutinib (CC-292) | Celgene | Phase IIb | NCT01975610 | Oral |
| SM | BMS-986142 | Bristol-Myers Squibb | Phase II | NCT02638948 | Oral | |
| SM | Branebrutinib | Bristol-Myers Squibb | Phase IIb | NCT04186871 | Oral | |
| SM | Evobrutinib | Merk | Phase IIb | NCT03233230 | Oral | |
| SM | TK-020 | Takeda | Phase I | NCT02413255 | Oral | |
| SM | HM71224 | Eli Lilly/Hamni | Phase II (terminated) | NCT01765478 | Oral | |
| SM | Fenebrutinib | Roche | Phase II | n/a | ||
| BTK/JAK1 | SM | ABBV-599 | AbbVie | Phase II (terminated) | NCT03823378 | Oral |
mAb: Monoclonal antibody; SM: Small molecule.
Table 6.
Approved and investigational biologics and small molecules for AtD.
| Target | Modality | Drug | Sponsor | Status | NCT | Route of Admiration |
|---|---|---|---|---|---|---|
| PDE4 | SM | Eucrisa (crisaborole) | Pfizer | Approved, 2016 | Topical Ointment | |
| SM | Apremilast | Amgen | Phase II | NCT02087943 | Oral | |
| SM | E6005 | Elsai | Phase II | NCT01461941 | Ointment | |
| SM | DRM02 | QLT | Phase II | NCT01993420 | Ointment | |
| IL-13 and IL-4 | mAb | Dupixent (dupilumab) | Sanofi/Regeneron | Approved, 2017 | Subcutaneous | |
| mAb | Tralokinumab | LEO Pharma | phase IIb | NCT03562377 | Subcutaneous | |
| mAb | Lebrikizumab | Eli Lilly | Phase III | NCT04392154 | Subcutaneous | |
| IL-5 | mAb | Mepolizumab | GlaxoSmithKline | Phase II | NCT03055195 | Subcutaneous |
| mAb | benralizumab | AstraZeneca | Phase II | NCT03563066 | Subcutaneous | |
| IL-31 | mAb | Nemolizumab | Chugai Pharmaceutical Company | Phase III | NCT03985943 | n/a |
| IL-23 | mAb | Risankizumab | AbbVie | Phase II | NCT03706040 | Subcutaneous |
| IL-22 | mAb | Fezakinumab | Pfizer | Phase Iia | n/a | |
| IL-17C | mAb | MOR106 | MorphoSys and Galapagos | phase II (terminated) | NCT03864627 | Subcutaneous |
| IL-17A | mAb | Secukinumab | GWT-TUD GmbH | Phase II | NCT03568136 | subcutaneous |
| JAK | SM | Olumiant (Baricitinib) | Eli Lilly | Phase III | NCT03334422 | Oral |
| SM | ruxolitinib | Incyte Corporation | Phase II | NCT03011892 | Ointment | |
| SM | RINVOQ (upadacitinib) | AbbVie | Phase III | NCT03569293 | Oral | |
| SM | abrocitinib | Pfizer | Phase III | NCT04345367 | Oral | |
| SM | Gusacitinib | Asana Biosciences | Phase IIb (terminated) | NCT03654755 | Oral | |
| HRH4 | SM | ZPL-3893787 | Ziarco Pharma | Phase II | NCT02424253 | Oral |
| SM | JNJ39758979 | Janssen Pharmaceutical | Phase II (terminated) | NCT01497119 | Oral | |
| TSLP | mAb | Tezepelumab | AstraZeneca | Phase IIa | NCT03809663 | Subcutaneous |
| mAb | MK8226 | Merck Sharp & Dohme Corp. | Phase I (terminated) | NCT01732510 | Intravenous | |
| OX40 | mAb | GBR 830 KHK4083 | Glenmark Pharmaceuticals Kyowa Kirin Pharmaceutical Development, Inc. | Phase II | NCT02683928 NCT03703102 | Intravenous |
| IL-33 | mAb | Etokinumab | AnaptysBio | Phase II (failed) | NCT03533751 | n/a |
| mAb | PF-06817024 | Pfizer | Phase I | NCT02743871 | Intravenous | |
| mAb | REGN3500 | Regeneron/Sanofi | Phase II | NCT03736967 | Subcutaneous |
mAb: monoclonal antibody; SM: small molecule.
Table 7.
Approved and investigational biologics and small molecules for CD.
| Target | Modality | Drug | Sponsor | Status | NCT | Route of Admiration |
|---|---|---|---|---|---|---|
| Integrin | mAb | Natalizumab | Biogen Idec/Elan Corporation | Approved, 2008 | Intravenous | |
| mAb | Entyvio (vedolizumab) | Takeda | Approved, 2014 | Subcutaneous | ||
| mAb | Etrolizumab | Roche | Phase III | NCT02394028 | Subcutaneous | |
| S1P1 | SM | ZEPOSIA (ozanimod) | Bristol-Myers Squibb | Phase III | NCT03440372 | Oral |
| SM | Amiselim (MT-1303) | Biogen | Phase II (terminated) | NCT02378688 | Oral | |
| MAdCAM | mAb | PF-00547659 | Pfizer | Phase II | NCT03283085 | Subcutaneous |
| TYK2 | SM | BMS-986165 | Bristol-Myers Squibb | Phase II | NCT03599622 | Oral |
| TYK2/JAK1 | SM | PF-06700841 | Pfizer | Phase II | NCT03395184 | Oral |
| Pan-JAK/TYK2 | SM | TD-1473 | J&J/Theravance | Phase II | NCT03635112 | Oral |
| IL-23 and IL-12 | mAb | Ustekinumab | Janssen Biotech | Phase I | NCT02968108 | Intravenous |
| mAb | Risankizumab | AbbVie | Phase III | NCT03105128 | Subcutaneous | |
| IL-23R | peptide | PTG-200 | Protagonist Therapeutics/J&J | Phase II | Oral |
mAb: monoclonal antibody; SM: small molecule.
Table 8.
Conventional therapeutics for OA.
| Category | Drugs | Mechanism of Action | Analgesic Action Level |
|---|---|---|---|
| NSAIDs | Aspirin, naproxen, ibuprofen, diclofenac, celecoxib, piroxicam, indomethacin, meloxicam, ketoprofen, sulindac, diflunisal, nabumetone, oxaprozin, tolmetin, salsalate, etodolac, fenoprofen, flurbiprofen, ketorolac, meclofenamate, mefenamic acid, etoricoxib, and rofecoxib. | inhibit COX enzymes leading to decreased prostaglandin synthesis. increasing serotonin in central sites | Peripheral and central effects |
| Analgesics | Acetaminophen | Peripheral: COX1 and 2 inhibition Central: descending serotonergic neuronal pathways, inhibition of L-arginine/NO pathway, stimulation of endocannabinoid system, and anti-nociception mechanisms | Peripheral and central effects |
| Duloxetine | Serotonin and nonrepinephrine reuptake inhibitor | Central effects | |
| Opioids and opioid receptor ligands | Morphine, codeine, acetaminophen with codeine, fentanyl, hydrocodone, acetaminophen with hydrocodone, hydromorphone, meperidine, oxycodone | Activate opioids receptors to hyperpolarizes sensory neurons and attenuate nerve hyperexcitability | Peripheral and central effects |
| Corticosteroids | Prednisone, betamethasone, cortisone, dexamethasone, hydrocortisone, methylprednisolone, prednisolone, triamcinolone acetonide | Immunosuppressive and anti-inflammatory by interrupting the inflammatory cascade | Peripheral |
| Joint modifying treatments | Chondroitin and glucosamine | Increase proteoglycan synthesis in articular cartilage | Peripheral/local |
| Hyaluronic acid | Enhance chondrocyte synthesis of endogenous hyaluronic acid and proteoglycans | Peripheral/local |
Table 9.
Approved therapeutics for migraine.
| Class | Drugs | Mechanism of Action |
|---|---|---|
| NSAIDs | Aspirin, naproxen, ibuprofen, tolfenamic acid, diclofenac, piroxicam, ketoprofen, and ketorolac | Inhibit prostaglandin synthesis |
| Analgesics | Acetaminophen | Inhibit prostaglandin synthesis |
| Triptans | Sumatriptan, eletriptan, naratriptan, zolmitriptan, rizatriptan, frovatriptan, and almotriptan | Serotonin 5-HT1B/1D receptor agonists |
| Ergotamines | Ergotamines, Dihydroergotamine | Serotonin 5-HT1B/1D/1F receptor agonists |
| β-blockers | Propranolol, timolol | Unclear, inhibit noradrenaline release or serotonergic blockade |
| Anti-depressants | Tricyclic antidepressant (TCA) amitriptyline and the selective serotonin reuptake inhibitor (SSRI) fluoxetine | Increase amounts of serotonin and norepinephrine. |
| Anti-emetics | Metoclopramide, prochlorperazine, Domperidone, promethazine, chlorpromazine | Dopamine antagonists |
| Calcium channel blockers | Verapamil, cinnarizine | Unclear, preventing the constriction of the blood vessels prior to the migraine attack. |
| Botox | Botulinum toxin A | Unclear, peripheral and central system sensitization, inactivation of trigeminovascular system |
| Anti-epileptics | Topiramate and divalproex | Unclear, inhibit glutamate-mediated excitation, GABAergic inhibition and reduce CGRP. |
| Ditans | Lasmiditan | Serotonin 5-HT1F receptor agonists |
| Anti-CGRP peptide | mAbs: erenumab, fremanezumab, galcanezumab, eptinezumabSM: ubrogepant, rimegepant | Blocks CGRP binding to receptors |
mAb: monoclonal antibody; SM: small molecule.
Table 11.
Status of select AD drugs in clinical trials.
| Drug | Sponsor | Modality | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| AAB-003 | Janssen, Pfizer | mAb | Anti-Aβ antibody | Phase I (terminated) | NCT01193608 |
| Aducanumab | Biogen, Neurimmune | mAb | Anti-Aβ antibody | Phase III (terminated) Phase IIIc | NCT02484547 NCT04241068 |
| BAN2401 | Biogen, Eisai Co., Ltd. | mAb | Anti-Aβ antibody | Phase III | NCT03887455 |
| Bapineuzumab | Janssen, r | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT00998764 |
| Crenezumab | AC Immune SA, Genentech, Hoffmann-La Roche | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT02670083 |
| Donanemab | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase II | NCT03367403 |
| GSK933776 | GlaxoSmithKline (GSK) | mAb | Anti-Aβ antibody | Phase I (terminated) | NCT00459550 |
| Gantenerumab | Chugai Pharmaceutical Co., Ltd., Hoffmann-La Roche | mAb | Anti-Aβ antibody | Phase III | NCT03444870 |
| LY2599666 | Eli Lilly and Co. | Fc-less, antigen-binding fragment of a monoclonal anti-Aβ antibody linked to polyethylene glycol | Anti-Aβ antibody | Phase I (terminated) | NCT02614131 |
| LY3372993 | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase I | NCT03720548 |
| MEDI1814 | AstraZeneca, Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase I | NCT02036645 |
| Ponezumab | Pfizer | mAb | Anti-Aβ antibody | Phase II (terminated) | NCT00945672 |
| RO7126209 | Hoffmann-La Roche | mAb with “brain shuttle” technology | Anti-Aβ antibody | Phase I | NCT04023994 |
| SAR228810 | Sanofi | mAb | Anti-Aβ antibody | Phase I | NCT01485302 |
| Solanezumab | Eli Lilly and Co. | mAb | Anti-Aβ antibody | Phase III (terminated) | NCT02760602 |
| Atabecestat | Janssen, Shionogi Pharma | SM | BACE inhibitor | Phase III (terminated) | NCT02569398 |
| BI 1181181 | Boehringer Ingelheim, Vitae Pharmaceuticals | SM | BACE inhibitor | Phase I (terminated) | NCT02106247 |
| Elenbecestat | Biogen, Eisai Co., Ltd. | SM | BACE inhibitor | Phase I (terminated) | NCT01600859 |
| LY2886721 | Eli Lilly and Co. | SM | BACE inhibitor | Phase II (terminated) | NCT01561430 |
| Lanabecestat | AstraZeneca, Eli Lilly & Co. | SM | BACE inhibitor | Phase III (terminated) | NCT02783573 |
| PF-06751979 | Pfizer | SM | BACE inhibitor | Phase I (terminated) | NCT02509117 |
| RG7129 | Roche | SM | BACE inhibitor | Phase I (terminated) | NCT01461967 |
| Umibecestat | Amgen, Inc., Novartis Pharmaceuticals Corporation | SM | BACE inhibitor | Phase II/III (terminated) | NCT02565511 |
| Verubecestat | Merck | SM | BACE inhibitor | Phase II/III (terminated | NCT01739348 |
| Avagacestat | Bristol-Myers Squibb | SM | γ-secretase inhibitor | Phase II (terminated) | NCT00890890 |
| Semagacestat | Eli Lilly and Co. | SM | γ-secretase inhibitor | Phase III (terminated) | NCT01035138 |
| ABBV-8E12 | AbbVie, C2N Diagnostics, LLC | mAb | Anti-tau antibody | Phase II (terminated) | NCT02880956 |
| BIIB076 | Biogen, Neurimmune | mAb | Anti-tau antibody | Phase II | NCT03056729 |
| Gosuranemab | Biogen, Bristol-Myers Squibb | mAb | Anti-tau antibody | Phase II | NCT03352557 |
| JNJ-63733657 | Janssen | mAb | Anti-tau antibody | Phase I | NCT03375697 |
| Lu AF87908 | H. Lundbeck | mAb | Anti-tau antibody | Phase I | NCT04149860 |
| PNT001 | Pinteon Therapeutics | mAb | Anti-tau antibody | Phase I | NCT04096287 |
| RG7345 | Roche | mAb | Anti-tau antibody | Phase I (terminated) | NCT02281786 |
| Semorinemab | AC Immune SA, Genentech, Hoffmann-La Roche | mAb | Anti-tau antibody | Phase II | NCT03828747 |
| Zagotenemab | Eli Lilly and Co. | mAb | Anti-tau antibody | Phase II | NCT03518073 |
| LMTM | TauRx Therapeutics Ltd. | SM | tau protein aggregation inhibitor | Phase III | NCT01689246 |
| Epothilone D | Bristol-Myers Squibb | SM | microtubule stabilizer | Phase I (terminated) | NCT01492374 |
| TPI 287 | Cortice Biosciences | SM | microtubule stabilizer | Phase I (terminated) | NCT01966666 |
| Tideglusib | Zeltia Group | SM | glycogen synthase kinase 3 (GSK3-β) inhibitor | Phase II (terminated) | NCT01350362 |
| AL002 | AbbVie, Alector | mAb | TREM2 agonist | Phase I | NCT03635047 |
| AL003 | AbbVie, Alector | mAb | SIGLEC3 antagonist | Phase I | NCT03822208 |
| ALZT-OP1 | AZTherapies, Inc. | SM | NSAID, anti-inflammatory | Phase III | NCT02547818 |
| Azeliragon | Pfizer, TransTech Pharma, Inc., vTv Therapeutics LLC | SM | RAGE antagonist | Phase II/III | NCT03980730 |
| Etanercept | Amgen, Inc., Pfizer | Receptor-Fc fusion | TNF-α antagonist | Phase II | NCT01068353 |
| Masitinib | AB Science | SM | Protein tyrosine kinase antagonist | Phase III | NCT01872598 |
| XPro1595 | INmune Bio Inc. | Heterotrimer biologic | TNF-α antagonist | Phase I | NCT03943264 |
| Dapagliflozin | AstraZeneca, Bristol-Myers Squibb | SM | SGLT2 inhibitor | Phase I/II | NCT04120623 |
| Empagliflozin | Boehringer Ingelheim, Eli Lilly and Co. | SM | SGLT2 inhibitor | Phase I | NCT03852901 |
| Gemfibrozil | Gregory Jicha, 323–5550 | SM | PPARα agonist | Phase I | NCT02045056 |
| Liraglutide | Novo Nordisk A/S | SM | GLP-1R agonist | Phase II | NCT01843075 |
| Metformin | Columbia University | SM | Glucose lowering | Phase II/III | NCT04098666 |
| Nasal Insulin | University of Southern California | SM | unknown | Phase II/III | NCT01767909 |
| T3D-959 | T3D Therapeutics, Inc. | SM | PPARδ/γ agonist | Phase II | NCT04251182 |
mAb: monoclonal antibody; SM: small molecule.
Table 12.
Current Stem Cell Therapies for AD [488].
Table 12.
Current Stem Cell Therapies for AD [488].
| Drug | Sponsor | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| AstroStem | Nature Cell Co. | Regenerative | Phase II | NCT03117738 |
| hUCB-MSCs | Medipost Co | Regenerative | Phase II | NCT02054208 |
| hUCB-MSCs | Medipost Co. | Regenerative | Phase II | NCT03172117 |
| hUCB-MSCs | South China Research Center | Regenerative | Phase II | NCT02513706 |
| hUCB-MSCs | South China Research Center | Regenerative | Phase II | NCT02672306 |
| hMSCs | Stemedica Cell | Regenerative | Phase II | NCT02833792 |
| LMSCs | Longeveron | Regenerative | Phase I | NCT02600130 |
Table 13.
Current gene therapies for AD [488].
Table 13.
Current gene therapies for AD [488].
| Drug | Sponsor | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| CERE-110 | Sangamo Therapeutics | Adeno-associated virus-based gene delivery of NGF | Phase II (terminated) | NCT00876863 |
| IONIS MAPTRX (BIIB080) | Ioni Pharmaceuticals, Biogen | MAPt RNA inhibitor ASO | Phase II | NCT03186989 |
| AAVrh.10hAPOE2 | Cornell University | Serotype rh. 10 adeno-associated virus gene delivery of ApoE2 | Phase I | NCT03634007 |
Table 14.
Current therapeutics for PD.
| Drug | Brand Name/FDA Approval | Modality | MOA |
|---|---|---|---|
| Levodopa and Carbidopa | Sinemet/1975, Parcopa/2004, Rytary/2015 | SM | Dopamine precursor-dopamine decarboxylase inhibitor |
| Selegiline | Eldepryl/1989, Emsam/2006, Zelapar/2006 | SM | MAO B inhibitors |
| Rasagiline | Azilect/2006 | SM | MAO B inhibitors |
| Safinamide | Xadago/2017 | SM | MAO B inhibitors |
| Bromocriptine | Parlodel/2005 | SM | Dopamine agonist |
| Pramipexole | Mirapex/1997 | SM | Dopamine agonist |
| Ropinirole | Requip/1997 | SM | Dopamine agonist |
| Rotigotine | Neupro/2007 | SM | Dopamine agonist |
| Apomorphine | Apokyn/2004 | SM | Dopamine agonist |
| Tolcapone | Tasmar/1998 | SM | COMT inhibitors |
| Entacapone | Comtan/1999 | SM | COMT inhibitors |
| Opicapone | Ongentys/2020 | SM | COMT inhibitors |
| Amantadine | Symmetrel/2003 | SM | Weak, non-competitive NMDA receptor antagonist |
| Istradefylline | Nourianz/2019 | SM | Adenosine receptor antagonist (A2A) |
| Deep brain stimulation | n/a/1997, 2002, 2003 | Device | Electric stimulation |
mAb: monoclonal antibody; SM: small molecule.
Table 15.
Status of select PD DMT drugs in clinical trials.
| Drug | Sponsor | Modality | Mechanism of Action | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| Ambroxol | University College, London | SM | GCase activation | Phase II | NCT02914366 |
| Anavex 2-73 (blarcamesine) | Anavex Life Science Corp. | SM | Sigma-1 receptor (SIGMAR1) agonist | Phase II | NCT03774459 |
| DNL151 | Denali Therapeutics Inc. | SM | LRRK2 inhibitor | Phase I | NCT04056689 |
| DNL201 | Denali Therapeutics Inc. | SM | LRRK2 inhibitor | Phase I | NCT03710707 |
| Nilotinib | Novartis Pharmaceuticals Corporation | SM | c-Abl kinase inhibitor | Phase II | NCT02954978 |
| K0706 | Sun Pharma Advanced Research Company | SM | c-Abl kinase inhibitor | Phase II | NCT03655236 |
| FB-101 | 1ST Biotherapeutics, Inc. | SM | c-Abl kinase inhibitor | Phase I | NCT04165837 |
| BIIB094 | Biogen, IONIS Pharmaceuticals | Gene Therapy | LRRK2 inhibitor | Phase I | NCT03976349 |
| PR001 | Prevail Therapeutics | Gene Therapy | GBA1 (encodes for Gcase) | Phase II | NCT04127578 |
| ABBV-0805 | AbbVie, BioArctic AB | mAb | α-synuclein | Phase I (withdrawn due to strategic considerations) | NCT04127695 |
| Cinpanemab | Biogen, Neurimmune | mAb | α-synuclein | Phase II | NCT03318523 |
| LU AF82422 | Genmab A/S, H. Lundbeck | mAb | α-synuclein | Phase I | NCT03611569 |
| MEDI1341 | AstraZeneca, Takeda Pharmaceutical Company | mAb | α-synuclein | Phase I | NCT04449484 |
| Prasinezumab | Hoffmann-La Roche, Prothena | mAb | α-synuclein | Phase II | NCT02157714 |
| NPT200-11 | Neuropore Therapies Inc. | SM | α-synuclein | Phase I | NCT02606682 |
| anle138b | MODAG GmbH | SM | α-synuclein | Phase I | NCT04208152 |
| CDNF (cerebral dopamine neurotrophic factor) | Herantis Pharma Plc, Renishaw plc. | Peptide | promotes survival of midbrain dopaminergic neurons | Phase II | NCT03295786 |
mAb: monoclonal antibody; SM: small molecule.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, J.; Ting, J.P.; Al-Azzam, S.; Ding, Y.; Afshar, S. Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 2805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062805
AMA Style
Liu J, Ting JP, Al-Azzam S, Ding Y, Afshar S. Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases. International Journal of Molecular Sciences. 2021; 22(6):2805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062805
Chicago/Turabian StyleLiu, Jinsha, Joey Paolo Ting, Shams Al-Azzam, Yun Ding, and Sepideh Afshar. 2021. "Therapeutic Advances in Diabetes, Autoimmune, and Neurological Diseases" International Journal of Molecular Sciences 22, no. 6: 2805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062805
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.