
Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results

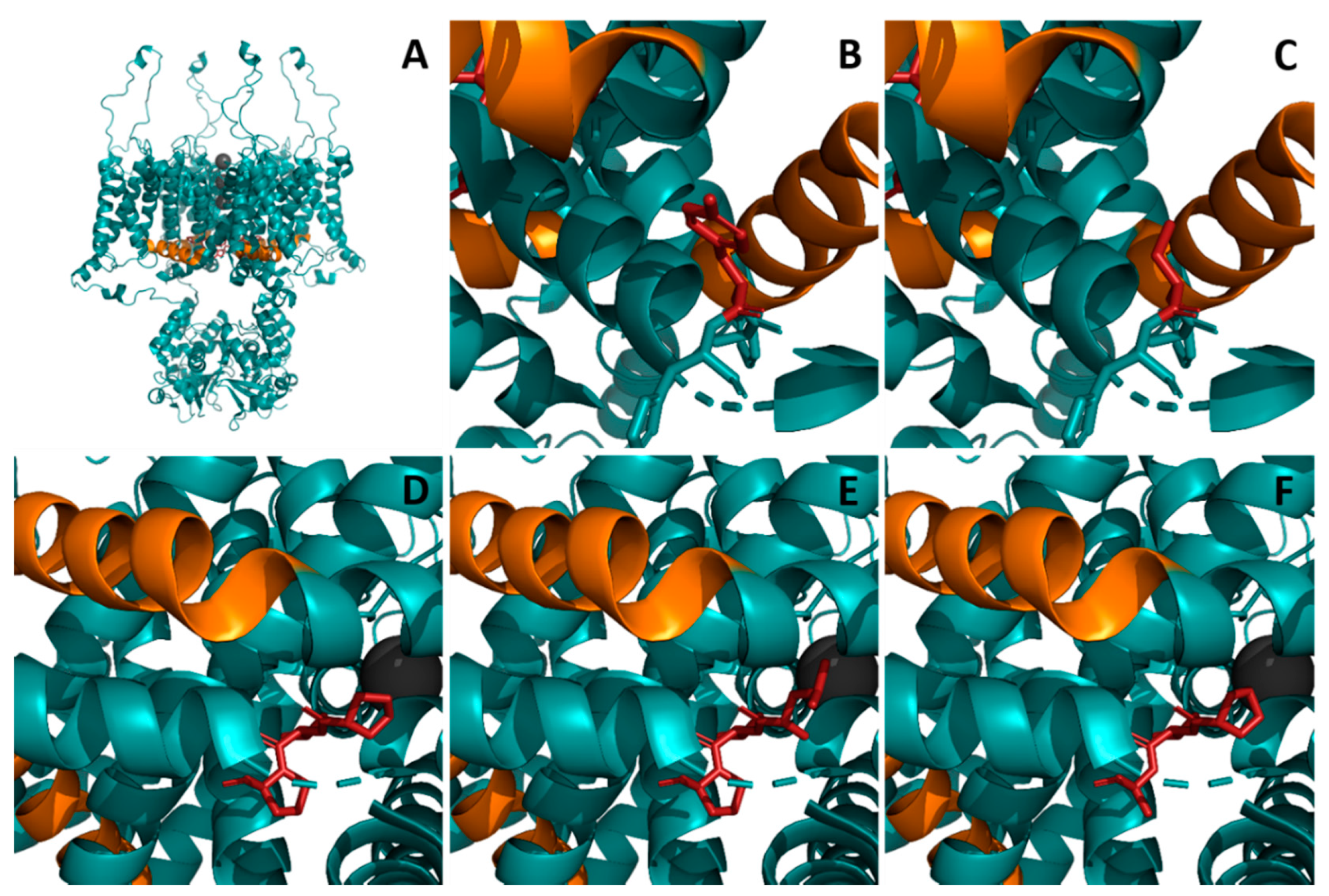{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Change | Protein Change | Functional Consequence | Phenotype | Number of Patients | Reference |
|---|---|---|---|---|---|
| heterozygous variants: | |||||
| missense | |||||
| c.469G > A | p.(Glu157Lys) | GOF | epileptic encephalopathy | 1 | [16] |
| c.676G > A | p.(Glu226Lys) | no data | epilepsy, childhood-onset | 1 | [17] |
| c.788T > C | p.(Ile263Thr) | LOF | epileptic encephalopathy | 1 | [13,16] |
| c.869T > G | p.(Leu290Arg) | GOF/LOF | epileptic encephalopathy | 2 | [16,18,19] |
| c.878T > A | p.(Leu293His) | GOF/LOF | epileptic encephalopathy | 1 | [16] |
| c.881G > A | p.(Arg294His) | LOF | hereditary spastic paraplegia | 7 | [15,20] |
| c.889C > T | p.(Arg297Trp) | no data | developmental and epileptic encephalopathy | 3 | [21,22,23] |
| c.890G > A | p.(Arg297Gln) | GOF | ataxia & myoclonic epilepsy | 15 | [3,13,14,16,24,25,26,27,28,29] |
| c.894G > T | p.(Leu298Phe) | GOF | epileptic encephalopathy | 1 | [13,16] |
| c.959C > T | p.(Thr320Ile) | no data | epilepsy, mild ataxia | 1 | [30] |
| c.971G > C | p.(Ser324Thr) | no data | epilepsy, drug-resistant | 1 | [31] |
| c.982T > G | p.(Leu328Val) | GOF/LOF | epileptic encephalopathy | 2 | [16,32] |
| c.1070G > A | p.(Gln357Arg) | no data | Lennox-Gastaut syndrome | 1 | [25] |
| c.1120A > G | p.(Thr374Ala) | GOF/LOF | epileptic encephalopathy, early onset | 8 | [16,26,33,34,35] |
| c.1192G > T | p.(Gly398Cys) | LOF | epileptic encephalopathy | 1 | [16] |
| c.1195G > A | p.(Val399Met) | no data | epileptic encephalopathy | 1 | [23] |
| c.1202C > T | p.(Thr401Ile) | no data | epileptic encephalopathy | 1 | [36] |
| c.1214C > T | p.(Pro405Leu) | LOF | epileptic encephalopathy | 14 | [13,16,23,34,37,38] |
| c.1223T > C | p.(Val408Ala) | no data | Rett-like syndrome with infantile onset seizures | 1 | [39] |
| truncating | |||||
| c.637C > T | p.(Gln213*) | LOF | epileptic encephalopathy | 1 | [16] |
| c.193C > T | p.(Arg65*) | predicted LOF | epilepsy | 1 | [17] |
| c.1265_1266delAG | p.(Glu422Glyfs*21) | predicted LOF | epileptic encephalopathy | 1 | [26] |
| in-frame deletion | |||||
| c.765_773del | p.(Met255_Ile257del) | LOF | episodic ataxia & pharmacoresponsive epilepsy | 7 | [3] |
| deletion | |||||
| c.110606081_111393713del~788 kb | no data | generalized epilepsy | 1 | [40] | |
| homozygous variants: | |||||
| c.193C > T | p.(Arg65*) | no data | intellectual disability, autosomal-recessive | 4 | [41] |
2.1. Novel Cases with KCNA2 Variants in this Study
2.2. Clinical Characteristics of Individuals with Reported and Novel KCNA2 Variants
2.3. Phenotypic Features of Reported and Novel Patients, Grouped According to Known Functional Consequences on Kv1.2
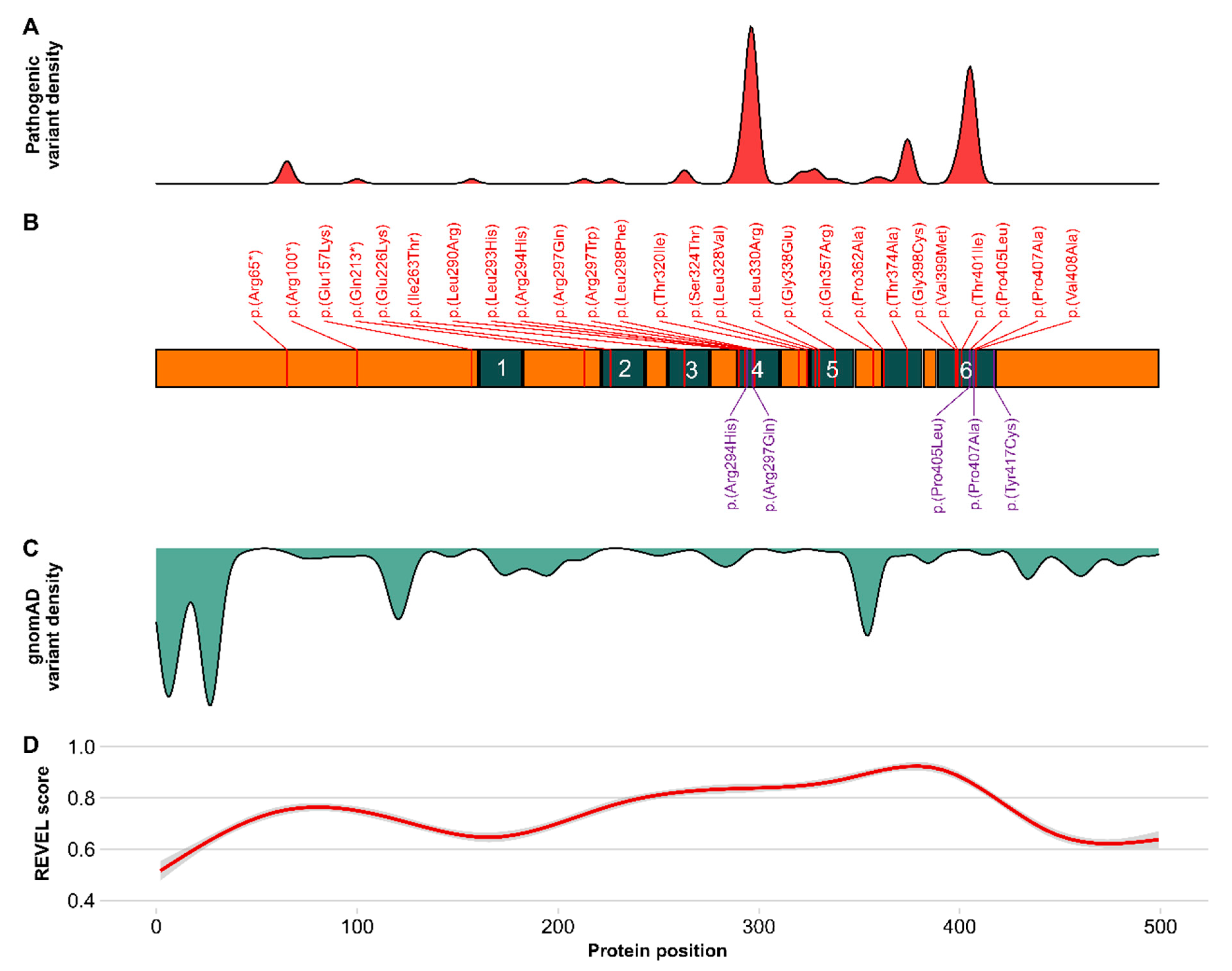
2.4. Prediction of Functional Relevance of KCNA2 Variants
3. Discussion
3.1. Novel Cases of KCNA2 Variants and Clinical Presentation
3.2. Functional Classification and Phenotypic Spectrum of Published KCNA2 Variants
3.3. Prediction of Functional Relevance and Modelling of KCNA2 Variants
4. Materials and Methods
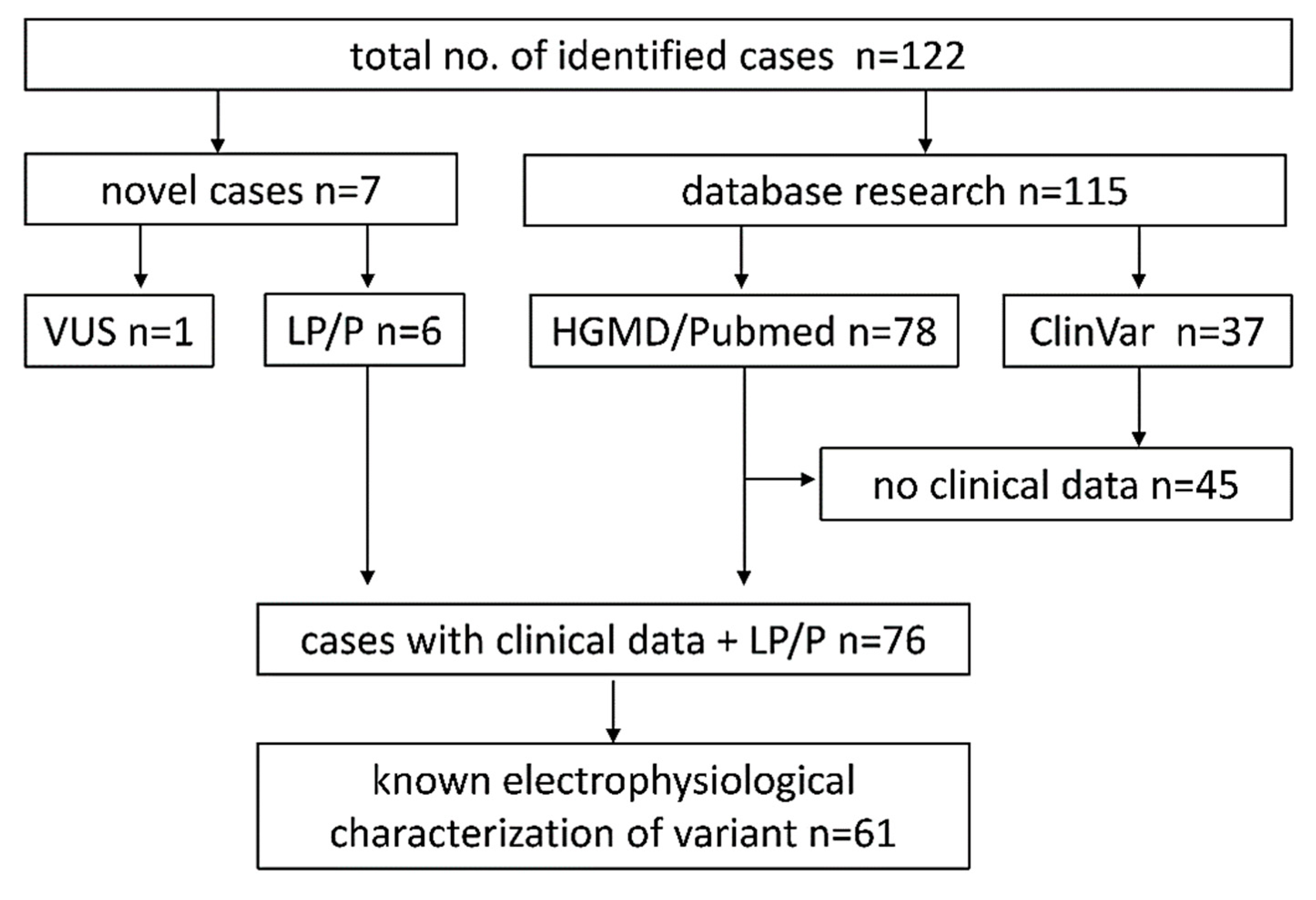
4.1. Database Research
4.2. Computational Pathogenicity Analysis and 3D Protein Structure Modeling
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuo, M.M.; Haynes, W.J.; Loukin, S.H.; Kung, C.; Saimi, Y. Prokaryotic K(+) channels: From crystal structures to diversity. FEMS Microbiol. Rev. 2005, 29, 961–985. [Google Scholar] [CrossRef] [Green Version]
- Strong, M.; Chandy, K.G.; Gutman, G.A. Molecular evolution of voltage-sensitive ion channel genes: On the origins of electrical excitability. Mol. Biol. Evol. 1993, 10, 221–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, M.A.; Bellows, S.T.; Li, M.; Carroll, R.; Micallef, S.; Carvill, G.L.; Myers, C.T.; Howell, K.B.; Maljevic, S.; Lerche, H.; et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology 2016, 87, 1975–1984. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Jan, Y.N.; Jan, L.Y. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science 2003, 301, 646–649. [Google Scholar] [CrossRef] [Green Version]
- Lorincz, A.; Nusser, Z. Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 2008, 28, 14329–14340. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Dendorfer, A.; Finol-Urdaneta, R.K.; Terlau, H.; Olivera, B.M. Biochemical characterization of kappaM-RIIIJ, a Kv1.2 channel blocker: Evaluation of cardioprotective effects of kappaM-conotoxins. J. Biol. Chem. 2010, 285, 14882–14889. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [Green Version]
- Tombola, F.; Pathak, M.M.; Isacoff, E.Y. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron 2005, 45, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Kirschstein, T.; Sadkiewicz, E.; Hund-Goschel, G.; Becker, J.; Guli, X.; Muller, S.; Rohde, M.; Hubner, D.C.; Brehme, H.; Kolbaske, S.; et al. Stereotactically Injected Kv1.2 and CASPR2 Antisera Cause Differential Effects on CA1 Synaptic and Cellular Excitability, but Both Enhance the Vulnerability to Pro-epileptic Conditions. Front. Synaptic. Neurosci. 2020, 12, 13. [Google Scholar] [CrossRef]
- Syrbe, S.; Stettner, G.M.; Bally, J.; Borggraefe, I.; Bien, C.I.; Ferfoglia, R.I.; Huppke, P.; Kern, J.; Polster, T.; Probst-Muller, E.; et al. CASPR2 autoimmunity in children expanding to mild encephalopathy with hypertension. Neurology 2020, 94, e2290–e2301. [Google Scholar] [CrossRef]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djemie, T.; Muller, S.; Moller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Pena, S.D.; Coimbra, R.L. Ataxia and myoclonic epilepsy due to a heterozygous new mutation in KCNA2: Proposal for a new channelopathy. Clin. Genet. 2015, 87, e1–e3. [Google Scholar] [CrossRef]
- Helbig, K.L.; Hedrich, U.B.; Shinde, D.N.; Krey, I.; Teichmann, A.C.; Hentschel, J.; Schubert, J.; Chamberlin, A.C.; Huether, R.; Lu, H.M.; et al. A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann. Neurol. 2016, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symonds, J.D.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, N.M.; Conroy, J.; Shahwan, A.; Lynch, B.; Correa, R.G.; Pena, S.D.; McCreary, D.; Magalhaes, T.R.; Ennis, S.; Lynch, S.A.; et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 2016, 57, e12–e17. [Google Scholar] [CrossRef]
- Bruel, A.L.; Nambot, S.; Quere, V.; Vitobello, A.; Thevenon, J.; Assoum, M.; Moutton, S.; Houcinat, N.; Lehalle, D.; Jean-Marcais, N.; et al. Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. Eur. J. Hum. Genet. 2019, 27, 1519–1531. [Google Scholar] [CrossRef]
- Manole, A.; Mannikko, R.; Hanna, M.G.; Kullmann, D.M.; Houlden, H. De novo KCNA2 mutations cause hereditary spastic paraplegia. Ann. Neurol. 2017, 81, 326–328. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routier, L.; Verny, F.; Barcia, G.; Chemaly, N.; Desguerre, I.; Colleaux, L.; Nabbout, R. Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: Is there a major genetic factor? Clin. Genet. 2019, 96, 254–260. [Google Scholar] [CrossRef]
- Sachdev, M.; Gainza-Lein, M.; Tchapyjnikov, D.; Jiang, Y.H.; Loddenkemper, T.; Mikati, M.A. Novel clinical manifestations in patients with KCNA2 mutations. Seizure 2017, 51, 74–76. [Google Scholar] [CrossRef] [Green Version]
- Canafoglia, L.; Castellotti, B.; Ragona, F.; Freri, E.; Granata, T.; Chiapparini, L.; Gellera, C.; Scaioli, V.; Franceschetti, S.; DiFrancesco, J.C. Progressive myoclonus epilepsy caused by a gain-of-function KCNA2 mutation. Seizure 2019, 65, 106–108. [Google Scholar] [CrossRef] [Green Version]
- Moller, R.S.; Larsen, L.H.; Johannesen, K.M.; Talvik, I.; Talvik, T.; Vaher, U.; Miranda, M.J.; Farooq, M.; Nielsen, J.E.; Svendsen, L.L.; et al. Gene Panel Testing in Epileptic Encephalopathies and Familial Epilepsies. Mol. Syndromol. 2016, 7, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nashabat, M.; Al Qahtani, X.S.; Almakdob, S.; Altwaijri, W.; Ba-Armah, D.M.; Hundallah, K.; Al Hashem, A.; Al Tala, S.; Maddirevula, S.; Alkuraya, F.S.; et al. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure 2019, 69, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Ngo, K.J.; Rexach, J.E.; Lee, H.; Petty, L.E.; Perlman, S.; Valera, J.M.; Deignan, J.L.; Mao, Y.; Aker, M.; Posey, J.E.; et al. A diagnostic ceiling for exome sequencing in cerebellar ataxia and related neurological disorders. Hum. Mutat. 2020, 41, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Uysal, B.; Rosa, F.; Loffler, H.; Mau-Holzmann, U.A.; Liebau, S.; Lerche, H. Establishment of a human induced pluripotent stem cell (iPSC) line (HIHDNEi002-A) from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p.Arg297Gln) mutation. Stem Cell Res. 2019, 37, 101445. [Google Scholar] [CrossRef]
- Fernandez-Marmiesse, A.; Roca, I.; Diaz-Flores, F.; Cantarin, V.; Perez-Poyato, M.S.; Fontalba, A.; Laranjeira, F.; Quintans, S.; Moldovan, O.; Felgueroso, B.; et al. Rare Variants in 48 Genes Account for 42% of Cases of Epilepsy With or Without Neurodevelopmental Delay in 246 Pediatric Patients. Front. Neurosci. 2019, 13, 1135. [Google Scholar] [CrossRef] [Green Version]
- Parrini, E.; Marini, C.; Mei, D.; Galuppi, A.; Cellini, E.; Pucatti, D.; Chiti, L.; Rutigliano, D.; Bianchini, C.; Virdo, S.; et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum. Mutat. 2017, 38, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, N.; Uysal, B.; Rosa, F.; Loffler, H.; Mau-Holzmann, U.A.; Liebau, S.; Lerche, H. Generation of an induced pluripotent stem cell (iPSC) line from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p.Leu328Val) mutation. Stem Cell Res. 2018, 33, 6–9. [Google Scholar] [CrossRef]
- Uysal, B.; Loffler, H.; Rosa, F.; Lerche, H.; Schwarz, N. Generation of an induced pluripotent stem cell (iPSC) line (HIHDNEi003-A) from a patient with developmental and epileptic encephalopathy carrying a KCNA2 (p.Thr374Ala) mutation. Stem Cell Res. 2019, 40, 101543. [Google Scholar] [CrossRef]
- Gong, P.; Xue, J.; Jiao, X.R.; Zhang, Y.H.; Yang, Z.X. [Genotype and phenotype of children with KCNA2 gene related developmental and epileptic encephalopathy]. Zhonghua Er Ke Za Zhi 2020, 58, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hundallah, K.; Alenizi, A.; AlHashem, A.; Tabarki, B. Severe early-onset epileptic encephalopathy due to mutations in the KCNA2 gene: Expansion of the genotypic and phenotypic spectrum. Eur. J. Paediatr. Neurol. 2016, 20, 657–660. [Google Scholar] [CrossRef]
- Zhou, P.; He, N.; Zhang, J.W.; Lin, Z.J.; Wang, J.; Yan, L.M.; Meng, H.; Tang, B.; Li, B.M.; Liu, X.R.; et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018, 17, e12456. [Google Scholar] [CrossRef]
- Miao, P.; Feng, J.; Guo, Y.; Wang, J.; Xu, X.; Wang, Y.; Li, Y.; Gao, L.; Zheng, C.; Cheng, H. Genotype and phenotype analysis using an epilepsy-associated gene panel in Chinese pediatric epilepsy patients. Clin. Genet. 2018, 94, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; AlSayed, M.; Alhassnan, Z.; Alotaibi, M.; Kayyali, H.; Al-Owain, M.; Shah, A.; Rahbeeni, Z.; Al-Muhaizea, M.A.; et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum. Genet. 2017, 136, 921–939. [Google Scholar] [CrossRef] [Green Version]
- Allou, L.; Julia, S.; Amsallem, D.; El Chehadeh, S.; Lambert, L.; Thevenon, J.; Duffourd, Y.; Saunier, A.; Bouquet, P.; Pere, S.; et al. Rett-like phenotypes: Expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin. Genet. 2017, 91, 431–440. [Google Scholar] [CrossRef]
- Lal, D.; Ruppert, A.K.; Trucks, H.; Schulz, H.; de Kovel, C.G.; Kasteleijn-Nolst Trenite, D.; Sonsma, A.C.; Koeleman, B.P.; Lindhout, D.; Weber, Y.G.; et al. Burden analysis of rare microdeletions suggests a strong impact of neurodevelopmental genes in genetic generalised epilepsies. PLoS Genet. 2015, 11, e1005226. [Google Scholar] [CrossRef]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J. Med. Genet 2020, 107003. [Google Scholar] [CrossRef]
- Li, J.; Zhao, T.; Zhang, Y.; Zhang, K.; Shi, L.; Chen, Y.; Wang, X.; Sun, Z. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018, 46, 7793–7804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Yamagata, T.; Kubota, M.; Arai, H.; Yamashita, S.; Nakagawa, T.; Fujii, T.; Sugai, K.; Imai, K.; Uster, T.; et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013, 54, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Paulhus, K.; Ammerman, L.; Glasscock, E. Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int. J. Mol. Sci. 2020, 21, 2802. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ding, S.; Horn, R. Tail end of the s6 segment: Role in permeation in shaker potassium channels. J. Gen. Physiol. 2002, 120, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Horn, R. Effect of S6 tail mutations on charge movement in Shaker potassium channels. Biophys. J. 2003, 84, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Doring, J.H.; Saffari, A.; Bast, T.; Brockmann, K.; Ehrhardt, L.; Fazeli, W.; Janzarik, W.G.; Kluger, G.; Muhle, H.; Moller, R.S.; et al. The Phenotypic Spectrum of PRRT2-Associated Paroxysmal Neurologic Disorders in Childhood. Biomedicines 2020, 8, 456. [Google Scholar] [CrossRef] [PubMed]
- Herlenius, E.; Heron, S.E.; Grinton, B.E.; Keay, D.; Scheffer, I.E.; Mulley, J.C.; Berkovic, S.F. SCN2A mutations and benign familial neonatal-infantile seizures: The phenotypic spectrum. Epilepsia 2007, 48, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Becker, F.; Moller, R.S.; Schubert, J.; Lemke, J.R.; Larsen, L.H.; Eiberg, H.; Nothnagel, M.; Thiele, H.; Altmuller, J.; et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann. Neurol. 2016, 79, 428–436. [Google Scholar] [CrossRef]
- Rogers, A.; Golumbek, P.; Cellini, E.; Doccini, V.; Guerrini, R.; Wallgren-Pettersson, C.; Thuresson, A.C.; Gurnett, C.A. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am. J. Med. Genet. A 2018, 176, 1748–1752. [Google Scholar] [CrossRef]
- Hackos, D.H.; Chang, T.H.; Swartz, K.J. Scanning the intracellular S6 activation gate in the shaker K+ channel. J. Gen. Physiol. 2002, 119, 521–532. [Google Scholar] [CrossRef]
- Imbrici, P.; Jaffe, S.L.; Eunson, L.H.; Davies, N.P.; Herd, C.; Robertson, R.; Kullmann, D.M.; Hanna, M.G. Dysfunction of the brain calcium channel CaV2.1 in absence epilepsy and episodic ataxia. Brain 2004, 127, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Shore, A.N.; Colombo, S.; Tobin, W.F.; Petri, S.; Cullen, E.R.; Dominguez, S.; Bostick, C.D.; Beaumont, M.A.; Williams, D.; Khodagholy, D.; et al. Reduced GABAergic Neuron Excitability, Altered Synaptic Connectivity, and Seizures in a KCNT1 Gain-of-Function Mouse Model of Childhood Epilepsy. Cell Rep. 2020, 33, 108303. [Google Scholar] [CrossRef] [PubMed]
- Starace, D.M.; Bezanilla, F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 2004, 427, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, M.; Huffmeier, U.; Trollmann, R.; Hehr, U.; Uebe, S.; Ekici, A.B.; Kraus, C.; Krumbiegel, M.; Reis, A.; Thiel, C.T.; et al. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet. J. Rare Dis. 2019, 14, 38. [Google Scholar] [CrossRef] [Green Version]




| cDNA Change | Protein Change | Interpretation | Phenotype | Number of Reported Patients |
|---|---|---|---|---|
| missense | ||||
| c.788T > C | p.(Ile263Thr) | LP | epileptic encephalopathy | 2 |
| c.869T > C | p.(Leu290Pro) | LP | epileptic encephalopathy | 1 |
| c.881G > A | p.(Arg294His) | LP | epileptic encephalopathy | 4 |
| c.890G > A | p.(Arg297Gln) | P | epileptic encephalopathy | 6 |
| c.894G > T | p.(Leu298Phe) | P | epileptic encephalopathy | 2 |
| c.959C > T | p.(Thr320Ile) | P | epileptic encephalopathy | 1 |
| c.989T > G | p.(Leu330Arg) | LP | not provided | 1 |
| c.1013G > A | p.(Gly338Glu) | LP | epileptic encephalopathy | 1 |
| c.1084C > G | p.(Pro362Ala) | LP | not provided | 1 |
| c.1120A > G | p.(Thr374Ala) | P | epileptic encephalopathy | 2 |
| c.1195G > A | p.(Val399Met) | LP | epileptic encephalopathy | 2 |
| c.1202C > T | p.(Thr401Ile) | LP | not provided | 1 |
| c.1214C > T | p.(Pro405Leu) | P | epileptic encephalopathy | 8 |
| c.1219C > G | p.(Pro407Ala) | LP | not provided | 2 |
| c.1223T > C | p.(Val408Ala) | P | epileptic encephalopathy | 1 |
| truncating | ||||
| c.298C > T | p.(Arg100*) | LP | epileptic encephalopathy | 1 |
| deletion | ||||
| g.(?_110593873)_(110604802_?)del | P | epileptic encephalopathy | 1 |
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| cDNA Change | c.1250A > G | c.1219C > G | c.1214C > T | c.1214C > T | c.890G > A | c.881G > A | c.881G > A |
| Protein Change | p.(Tyr417Cys) | p.(Pro407Ala) | p.(Pro405Leu) | p.(Pro405Leu) | p.(Arg297Gln) | p.(Arg294His) | p.(Arg294His) |
| Inheritance | parental | de novo | de novo | de novo | de novo | Familial (sister of #7) | familial (brother of #6) |
| Age at inclusion/Gender | 3 y/m | 3 y/f | 8 y/f | 2 y/m | 9 y/m | 8 y/f | 8 y/m |
| Development before seizure onset | normal | na | normal | normal | normal | mildly delayed motor development | delayed speech and motor development |
| Epilepsy onset (months) | 7.5 | 0.25 | 21 | 7 | 8 | 72 | 24 |
| Seizure types | bilateral tonic–clonic | generalized onset tonic and hypomotor seizures with cyanosis | prolonged febrile motor seizure | prolonged focal motor onset seizures and bilateral tonic–clonic seizures, febrile seizures, postictal hemiplegia | generalized onset bilateral tonic–clonic, atypical absences from 2y3m | febrile seizures; generalized onset myoclonic and absence seizures, status epilepticus | febrile seizures; focal and generalized onset, absence seizures, myoclonic and tonic seizures, status epilepticus |
| Seizure outcome | seizure free at 10 months | seizure-free at 2 years | ongoing | seizure free at 2 years | ongoing | ongoing | ongoing |
| ID | normal | severe ID | moderate ID | normal | learning disability | mild ID | moderate ID |
| Movement disorders | no | no | no | episodic ataxia | ataxia | mild tremor | ataxia, tremor, worsening on VPA |
| EEG features | normal at 3y | multifocal spikes and reduced physiologic sleep characteristics | multifocal spikes in both centro-temporal regions, evolving to hemi-CSWS during follow-up | multifocal spikes, activation during sleep | na | multifocal spikes and polyspikes frontal photoparoxysmal reaction, 3–4 Hz spike waves | generalized slowing, activation of epileptiform activity by photostimulation, multifocal spikes, 3–4 Hz spike waves |
| Magnetic resonance imaging | normal | normal | normal | left sided hippocampal sclerosis | normal | cystic lesion subcortical frontal left DD DNET | cerebellar atrophy (vermis) |
| Additional findings | SGA/dystrophy | dysarthria | polydactyly left | dysarthria and stuttering | muscular hypotonia | dystrophy, microcephaly, muscular hypotonia | |
| Comments | reported in ClinVar | reported in ClinVar |
| n = 61 | LOF (n = 35) | GOF (n = 16) | GOF/LOF (n = 10) |
|---|---|---|---|
| Variants | Pro405Leu (16) | Arg297Gln (14) | Thr374Ala (7) |
| Arg294His (9) | Glu157Lys (1) | Leu290Arg (1) | |
| Met255_Ile257del (7) | Leu298Phe (1) | Leu293His (1) | |
| Gln213 * (1) | Leu328Val (1) | ||
| Ile263Thr (1) | |||
| Gly398Cys (1) | |||
| Development before seizure onset | normal (29/34) | normal (9/12) | normal (3/8) |
| impaired (5/34) | impaired (3/12) | impaired (5/8) | |
| Epilepsy onset age mean/median (SD) (months) | early childhood 17.4/8.75 (SD 31.2) (1 outlier with 156 m) | infantile or early childhood 11.6/9.5 (SD 8.6) | neonatal or early infantile 3.1/0.5 (SD 5.6) (1 outlier with 18 m) |
| Seizure types | focal only (5/30) | focal only (0/13) | focal only (5/10) |
| generalized only (14/30) | generalized only (9/13) | generalized only (3/10) | |
| both (11/30) | both (4/13) | both (2/10) | |
| Febrile seizures | yes (13/27) | yes (5/12) | yes (1/9) |
| no (14/27) | no (7/12) | no (8/9) | |
| Seizure outcome | never seizures (5/32) ** | ||
| seizure-free (13/32) | seizure-free (2/13) | seizure-free (0/9) | |
| uncontrolled (14/32) | uncontrolled (11/13) | uncontrolled (9/9) | |
| Intellectual disability | normal (9/35) *** | normal (0/14) | normal (0/10) |
| mild (9/35) | mild (3/14) | mild (1/10) | |
| moderate (12/35) | moderate (7/14) | moderate (0/10) | |
| severe (5/35) | severe (4/14) | severe (9/10) | |
| Movement disorders | ataxia (18/28) | ataxia (16/16) | ataxia (4/4) |
| tremor (7/28) | tremor (7/16) | tremor (1/5) | |
| spasticity (6/28) ** | spasticity (0/16) | spasticity (4/7) | |
| episodic ataxia (6/28) *** | |||
| CSWS | (9/20) | (0/11) | (0/7) |
| cMRI | normal (20/26) | normal (5/14) | normal (5/10) ## |
| cerebellar atrophy (2/26) | cerebellar atrophy (8/14) | cerebellar atrophy (5/10) | |
| hippocampal sclerosis (1/26) | subcortical white matter lesions (1/14) | ||
| DNET (1/26), other (2/26) | |||
| Additional findings |
|
|
|
|
|
| |
|
|
| |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Döring, J.H.; Schröter, J.; Jüngling, J.; Biskup, S.; Klotz, K.A.; Bast, T.; Dietel, T.; Korenke, G.C.; Christoph, S.; Brennenstuhl, H.; et al. Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 2824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062824
Döring JH, Schröter J, Jüngling J, Biskup S, Klotz KA, Bast T, Dietel T, Korenke GC, Christoph S, Brennenstuhl H, et al. Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders. International Journal of Molecular Sciences. 2021; 22(6):2824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062824
Chicago/Turabian StyleDöring, Jan H., Julian Schröter, Jerome Jüngling, Saskia Biskup, Kerstin A. Klotz, Thomas Bast, Tobias Dietel, G. Christoph Korenke, Sophie Christoph, Heiko Brennenstuhl, and et al. 2021. "Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders" International Journal of Molecular Sciences 22, no. 6: 2824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062824