
Activin A and Cell-Surface GRP78 Are Novel Targetable RhoA Activators for Diabetic Kidney Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Protein Extraction and Immunoblotting
2.3. RhoA Activity
2.4. Animal Studies
2.5. Transfection, Luciferase and siRNA
2.6. RNA Extraction and qPCR
2.7. Statistical Analysis
3. Results
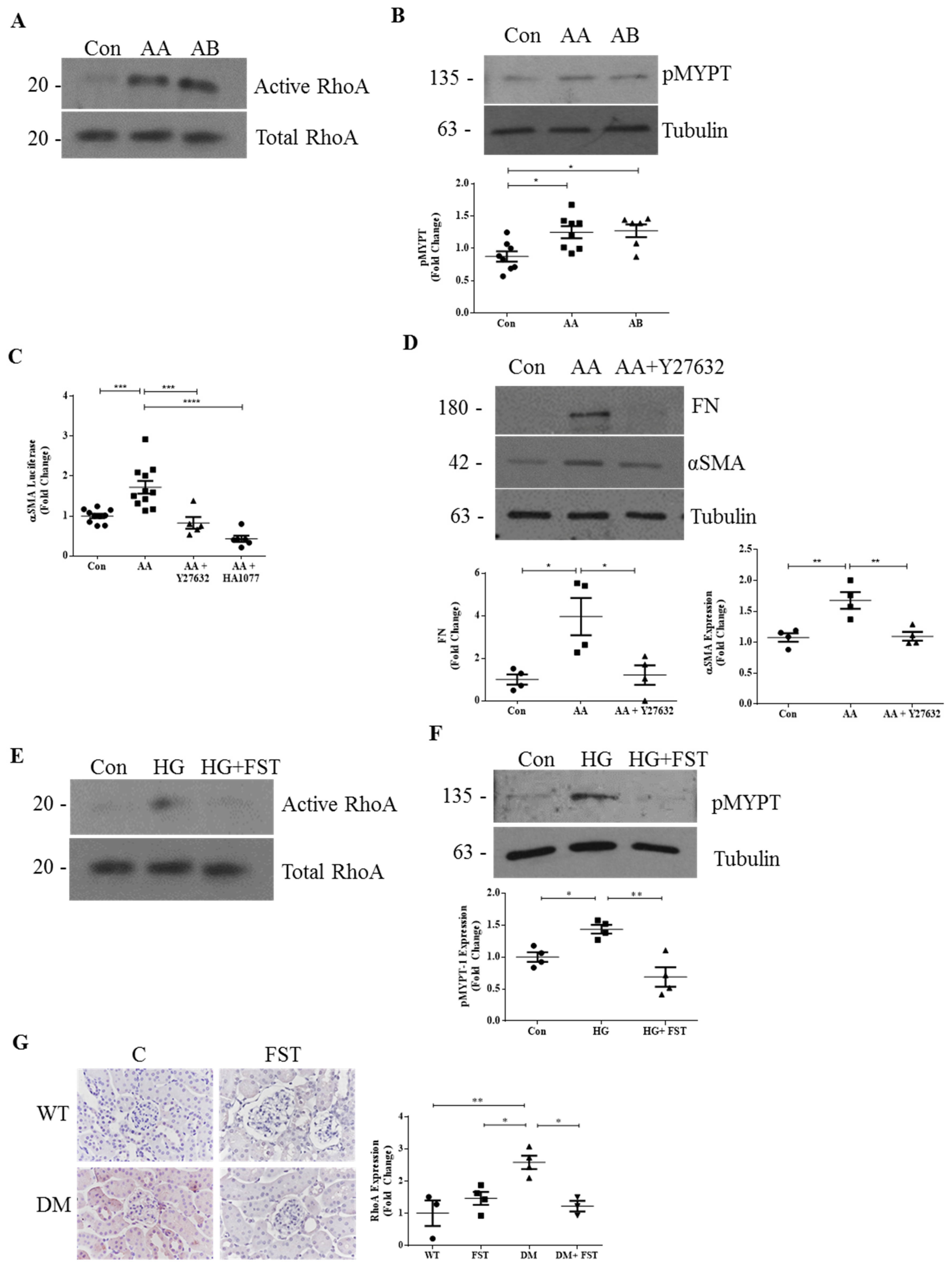
3.1. RhoA Mediates the Profibrotic Effects of Activins in DKD
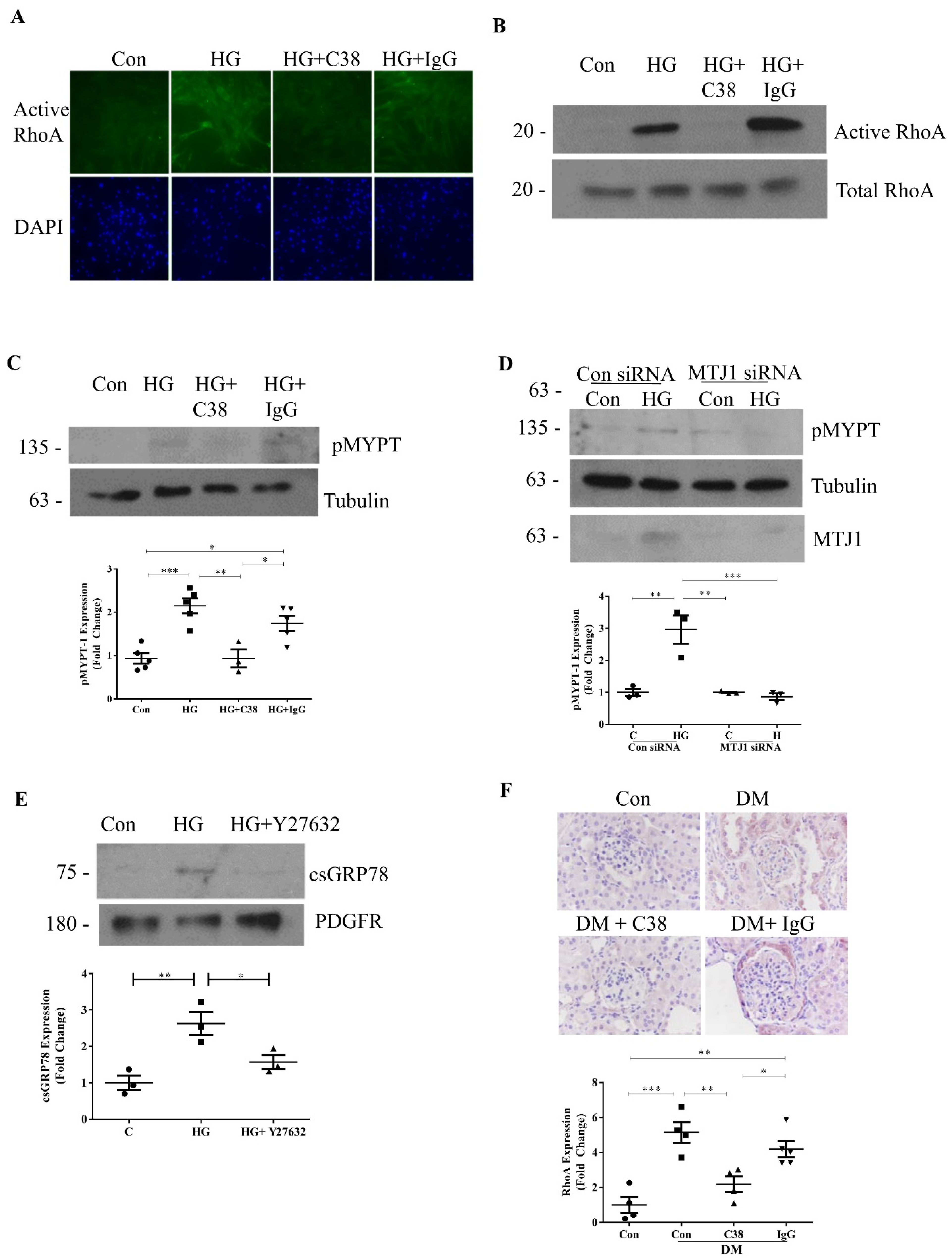
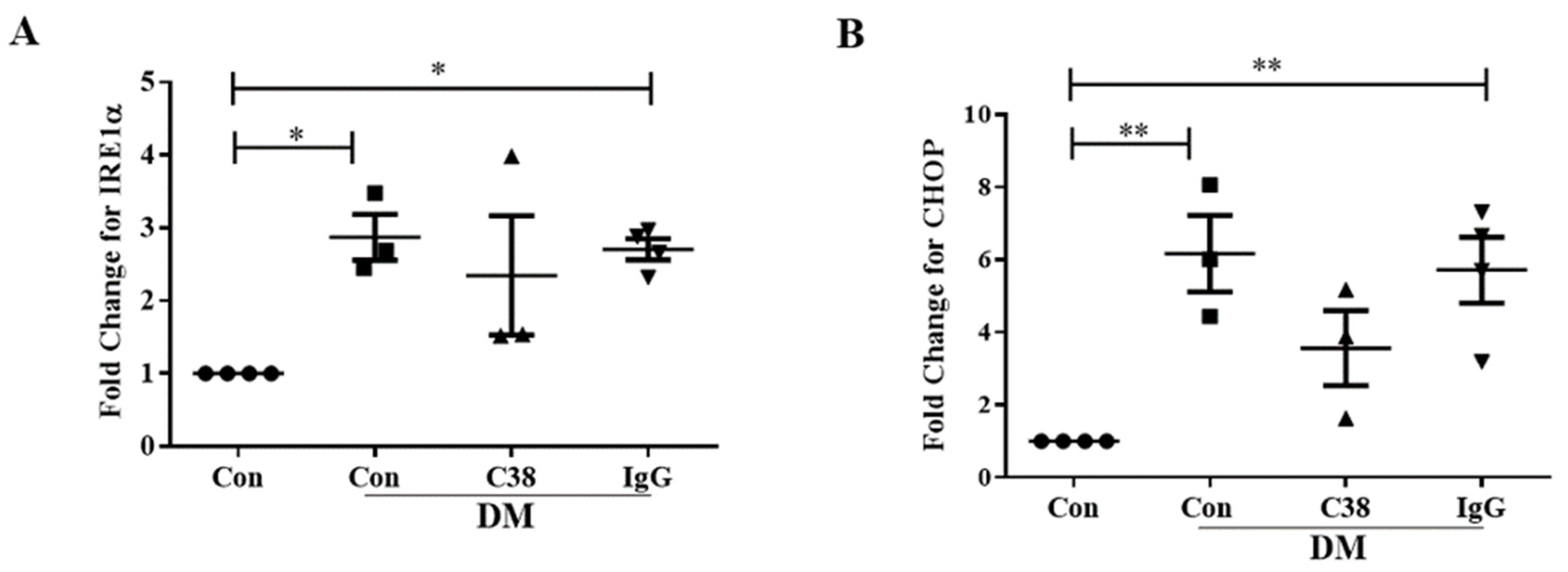
3.2. Cell-Surface GRP78 Mediates HG-Induced RhoA Activation
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C.; Cavender, M.A.; Fioretto, P.; Fowler, K.J.; Heerspink, H.J.; Manley, T.; McGuire, D.K.; Molitch, M.E.; Mottl, A.K.; et al. SGLT2 Inhibition for CKD and Cardiovascular Disease in Type 2 Diabetes: Report of a Scientific Workshop Sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 2020, 77. [Google Scholar] [CrossRef]
- Mauer, S.M.; Steffes, M.W.; Ellis, E.N.; Sutherland, D.E.; Brown, D.M.; Goetz, F.C. Structural-functional relationships in diabetic nephropathy. J. Clin. Investig. 1984, 74, 1143–1155. [Google Scholar] [CrossRef]
- Caramori, M.L.; Parks, A.; Mauer, M. Renal Lesions Predict Progression of Diabetic Nephropathy in Type 1 Diabetes. J. Am. Soc. Nephrol. 2013, 24, 1175–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Wu, D.; Gao, B.; Ingram, A.J.; Zhang, B.; Chorneyko, K.; McKenzie, R.; Krepinsky, J.C. RhoA/Rho-Kinase Contribute to the Pathogenesis of Diabetic Renal Disease. Diabetes 2008, 57, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, N.; Naruse, K.; Kobayashi, Y.; Nakamura, N.; Hamada, Y.; Nakashima, E.; Matsubara, T.; Oiso, Y.; Nakamura, J. High glucose-induced upregulation of Rho/Rho-kinase via platelet-derived growth factor receptor−β increases migration of aortic smooth muscle cells. J. Mol. Cell. Cardiol. 2008, 45, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Zhang, B.; Wu, D.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. TGFβ-induced RhoA activation and fibronectin production in mesangial cells require caveolae. Am. J. Physiol. Physiol. 2008, 295, F153–F164. [Google Scholar] [CrossRef] [Green Version]
- Krepinsky, J.C.; Ingram, A.J.; Tang, D.; Wu, D.; Liu, L.; Scholey, J.W. Nitric Oxide Inhibits Stretch-Induced MAPK Activation in Mesangial Cells Through RhoA Inactivation. J. Am. Soc. Nephrol. 2003, 14, 2790–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thumkeo, D.; Watanabe, S.; Narumiya, S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013, 92, 303–315. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Sekiguchi, K.; Yokota, T.; Utsunomiya, K.; Nishimura, R. The Physiology, Pathology, and Therapeutic Interventions for ROCK Isoforms in Diabetic Kidney Disease. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Komers, R.; Oyama, T.T.; Beard, D.R.; Anderson, S. Effects of systemic inhibition of Rho kinase on blood pressure and renal haemodynamics in diabetic rats. Br. J. Pharmacol. 2010, 162, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Krieken, R.; Mehta, N.; Wang, T.; Zheng, M.; Li, R.; Gao, B.; Ayaub, E.; Ask, K.; Paton, J.C.; Paton, A.W.; et al. Cell surface expression of 78-kDa glucose-regulated protein (GRP78) mediates diabetic nephropathy. J. Biol. Chem. 2019, 294, 7755–7768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gava, A.L.; Van Krieken, R.; Mehta, N.; Li, R.; Gao, B.; Desjardins, E.M.; Steinberg, G.R.; Hawke, T.; Krepinsky, J.C. The caveolin-1 regulated protein follistatin protects against diabetic kidney disease. Kidney Int. 2019, 96, 1134–1149. [Google Scholar] [CrossRef]
- Mehta, N.; Zhang, D.; Li, R.; Wang, T.; Gava, A.; Parthasarathy, P.; Gao, B.; Krepinsky, J.C. Caveolin-1 regulation of Sp1 controls production of the antifibrotic protein follistatin in kidney mesangial cells. Cell Commun. Signal. 2019, 17, 37. [Google Scholar] [CrossRef] [Green Version]
- De Ridder, G.G.; Ray, R.; Pizzo, S.V. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012, 22, 225–235. [Google Scholar] [CrossRef]
- Mehta, N.; Krepinsky, J.C. The emerging role of activins in renal disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 136–144. [Google Scholar] [CrossRef]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Werner, S.; Alzheimer, C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006, 17, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Wada, W.; Kuwano, H.; Hasegawa, Y.; Kojima, I. The Dependence of Transforming Growth Factor-β-Induced Collagen Production on Autocrine Factor Activin A in Hepatic Stellate Cells. Endocrinology 2004, 145, 2753–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, S.; Maeshima, A.; Kojima, I.; Nojima, Y. Activin A Is a Potent Activator of Renal Interstitial Fibroblasts. J. Am. Soc. Nephrol. 2004, 15, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Aoki, F.; Kurabayashi, M.; Hasegawa, Y.; Kojima, I. Attenuation of Bleomycin-induced Pulmonary Fibrosis by Follistatin. Am. J. Respir. Crit. Care Med. 2005, 172, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.S.; Hathaway, C.K.; Smithies, O.; Kakoki, M. Transforming growth factor-β1 and diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F689–F696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.-J.; Guan, G.-J.; Liu, G.; Zhang, T.; Liu, G.-H. Effect of activin A on tubulointerstitial fibrosis in diabetic nephropathy. Nephrology 2009, 14, 311–320. [Google Scholar] [CrossRef]
- Zhang, L.; Deng, M.; Parthasarathy, R.; Wang, L.; Mongan, M.; Molkentin, J.D.; Zheng, Y.; Xia, Y. MEKK1 Transduces Activin Signals in Keratinocytes to Induce Actin Stress Fiber Formation and Migration. Mol. Cell. Biol. 2005, 25, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Liu, N.-Y.; Wang, X.-E.; Chen, Y.-H.; Li, Q.-L.; Lu, K.-R.; Sun, L.; Jia, Q.; Zhang, L.; Zhang, L. Activin B Promotes Epithelial Wound Healing In Vivo through RhoA-JNK Signaling Pathway. PLoS ONE 2011, 6, e25143. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wu, M.; Feng, J.; Lin, X.; Gu, Z. RhoA/Rho kinase signaling regulates transforming growth factor-β1-induced chondrogenesis and actin organization of synovium-derived mesenchymal stem cells through interaction with the Smad pathway. Int. J. Mol. Med. 2012, 30, 1119–1125. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-J.; Minei, R.; Sato, T.; Taniguchi, A. The Influence of a Nanopatterned Scaffold that Mimics Abnormal Renal Mesangial Matrix on Mesangial Cell Behavior. Int. J. Mol. Sci. 2019, 20, 5349. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Sebe, A.; Péterfi, Z.; Masszi, A.; Thirone, A.C.P.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szászi, K.; Mucsi, I.; et al. Cell Contact–dependent Regulation of Epithelial–Myofibroblast Transition via the Rho-Rho Kinase-Phospho-Myosin Pathway. Mol. Biol. Cell 2007, 18, 1083–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Lee, A.S. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell. Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez–Gronow, M.; Selim, M.A.; Papalas, J.; Pizzo, S.V. GRP78: A Multifunctional Receptor on the Cell Surface. Antioxid. Redox Signal. 2009, 11, 2299–2306. [Google Scholar] [CrossRef]
- Gopal, U.; Mowery, Y.; Young, K.; Pizzo, S.V. Targeting cell surface GRP78 enhances pancreatic cancer radiosensitivity through YAP/TAZ protein signaling. J. Biol. Chem. 2019, 294, 13939–13952. [Google Scholar] [CrossRef]
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Pizzo, S.V. The Role of MTJ-1 in Cell Surface Translocation of GRP78, a Receptor for α2-Macroglobulin-Dependent Signaling. J. Immunol. 2005, 174, 2092–2097. [Google Scholar] [CrossRef] [Green Version]
- Olayioye, M.A.; Noll, B.; Hausser, A. Spatiotemporal Control of Intracellular Membrane Trafficking by Rho GTPases. Cells 2019, 8, 1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, Y.; Matoba, K.; Kawanami, D.; Takeda, Y.; Akamine, T.; Ishizawa, S.; Kanazawa, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. ROCK2 regulates TGF-β-induced expression of CTGF and profibrotic genes via NF-κB and cytoskeleton dynamics in mesangial cells. Am. J. Physiol. Physiol. 2019, 317, F839–F851. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Gojo, A.; Utsunomiya, K.; Taniguchi, K.; Yokota, T.; Ishizawa, S.; Kanazawa, Y.; Kurata, H.; Tajima, N. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2007, 568, 242–247. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Yamada, M.; Imakiire, T.; Kushiyama, T.; Higashi, K.; Hyodo, N.; Yamamoto, K.; Oda, T.; Suzuki, S.; Miura, S. A Rho-kinase inhibitor, fasudil, prevents development of diabetes and nephropathy in insulin-resistant diabetic rats. J. Endocrinol. 2007, 192, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, F.; Huang, X.R.; Liu, F.; Chen, H.-Y.; Chung, A.C.; Shi, J.; Wei, L.; Lan, H.Y.; Fu, P. Amelioration of Albuminuria in ROCK1 Knockout Mice with Streptozotocin-Induced Diabetic Kidney Disease. Am. J. Nephrol. 2011, 34, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Warren, A.M.; Knudsen, S.T.; Cooper, M.E. Diabetic nephropathy: An insight into molecular mechanisms and emerging therapies. Expert Opin. Ther. Targets 2019, 23, 579–591. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, H.; Xiang, H.; Liu, J.; Liu, Y.; Zhang, X.; Wang, J.; Tang, Y. TGF-β1 induces the dissolution of tight junctions in human renal proximal tubular cells: Role of the RhoA/ROCK signaling pathway. Int. J. Mol. Med. 2013, 32, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti–TGF-β1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tang, P.; Guo, F.; Zhang, M.; Chen, Y.; Yan, Y.; Tian, Z.; Xu, P.; Zhang, L.; Zhang, L.; et al. RhoA regulates Activin B-induced stress fiber formation and migration of bone marrow-derived mesenchymal stromal cell through distinct signaling. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-J.; Yao, F.-J.; Lu, G.-H.; Xu, C.-G.; Xu, Z.; Tang, K.; Cheng, Y.-J.; Gao, X.-R.; Wu, S.-H. The Role of the Rho/ROCK Pathway in Ang II and TGF-β1-Induced Atrial Remodeling. PLoS ONE 2016, 11, e0161625. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Peng, F.; Zhang, B.; Ingram, A.J.; Gao, B.; Krepinsky, J.C. Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signalling in mesangial cells. Diabetologia 2007, 50, 2008–2018. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, J.; Zhao, X.; Xie, W.; Wang, H.; Kong, H. Fasudil inhibits neutrophil-endothelial cell interactions by regulating the expressions of GRP78 and BMPR2. Exp. Cell Res. 2018, 365, 97–105. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soomro, A.; Trink, J.; O’Neil, K.; Li, R.; Naiel, S.; Gao, B.; Ask, K.; Krepinsky, J.C. Activin A and Cell-Surface GRP78 Are Novel Targetable RhoA Activators for Diabetic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 2839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062839
Soomro A, Trink J, O’Neil K, Li R, Naiel S, Gao B, Ask K, Krepinsky JC. Activin A and Cell-Surface GRP78 Are Novel Targetable RhoA Activators for Diabetic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(6):2839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062839
Chicago/Turabian StyleSoomro, Asfia, Jackie Trink, Kian O’Neil, Renzhong Li, Safaa Naiel, Bo Gao, Kjetil Ask, and Joan C. Krepinsky. 2021. "Activin A and Cell-Surface GRP78 Are Novel Targetable RhoA Activators for Diabetic Kidney Disease" International Journal of Molecular Sciences 22, no. 6: 2839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062839