
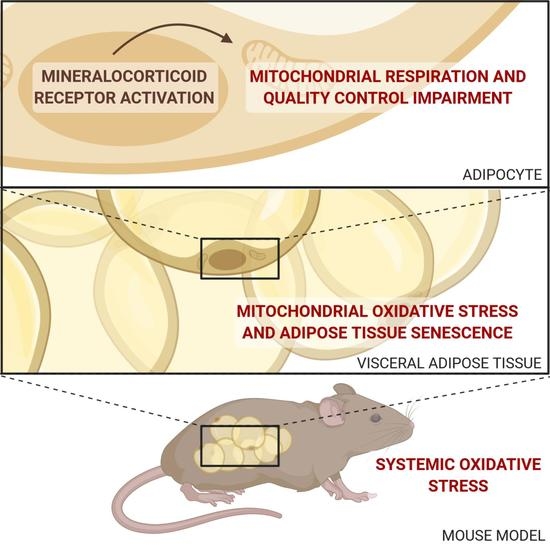
Adipocyte-Mineralocorticoid Receptor Alters Mitochondrial Quality Control Leading to Mitochondrial Dysfunction and Senescence of Visceral Adipose Tissue

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Adipocyte MR Overexpression Induces Adipocyte MR Overactivation
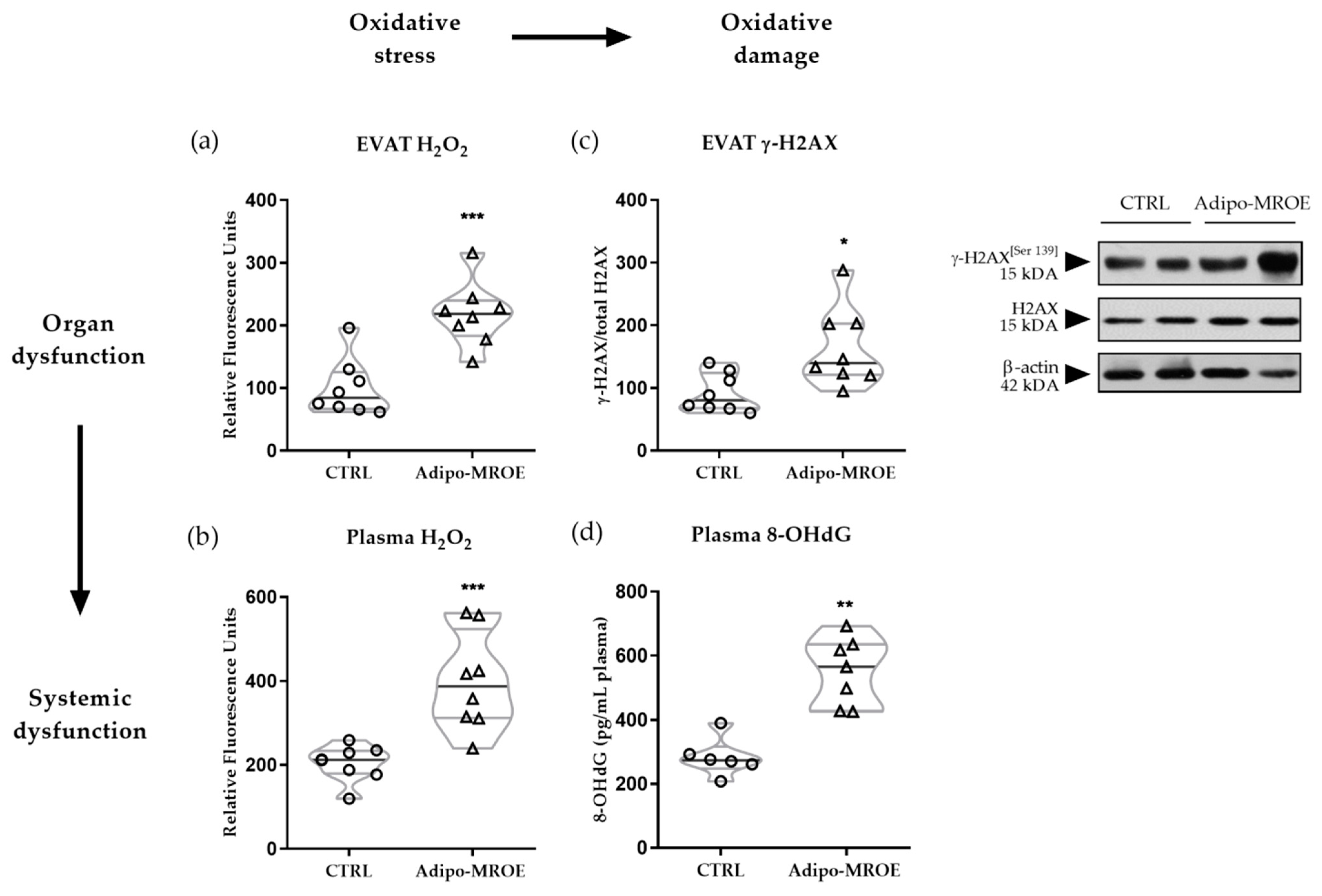
2.2. AT MR Activation Induces OS and OS-Induced DNA-Damage
2.3. Adipocyte-MR Activation Disrupts AT Mitochondrial Function
2.4. MR-Dependent Mitochondrial Dysfunction Is Due to Impaired MQC
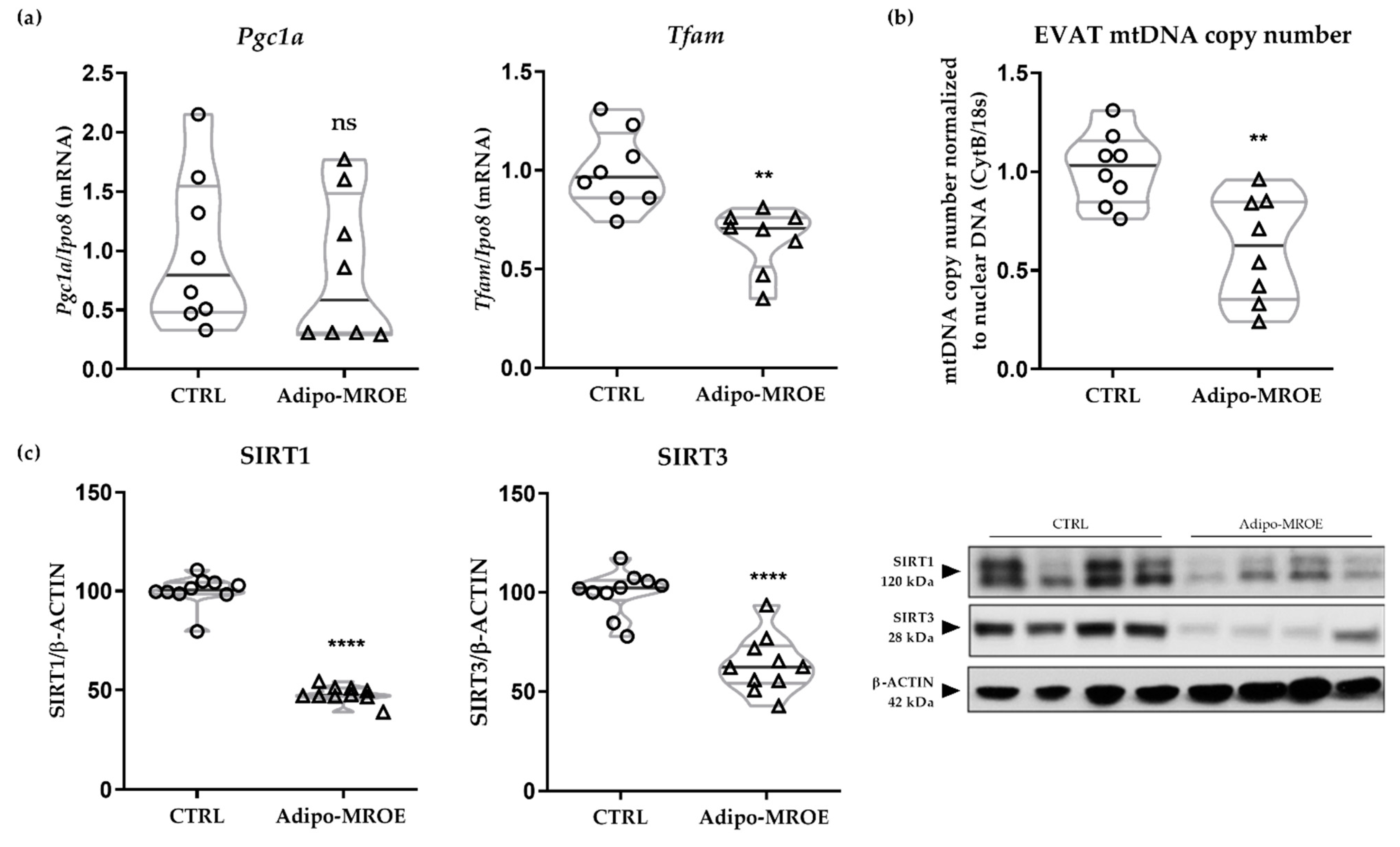
2.4.1. MR Activation Impairs Mitochondrial Biogenesis
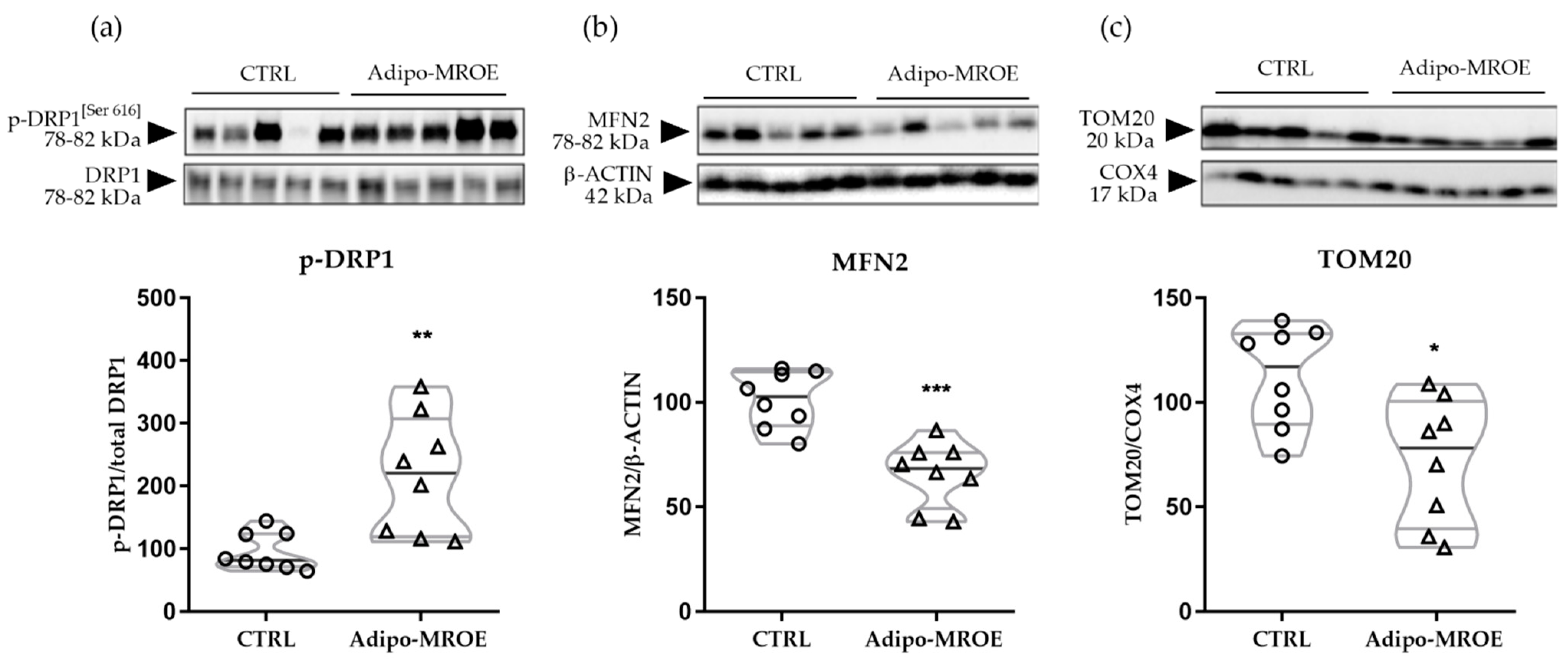
2.4.2. MR Activation Impairs Mitochondrial Dynamics
2.4.3. MR Activation Increases Mitophagy
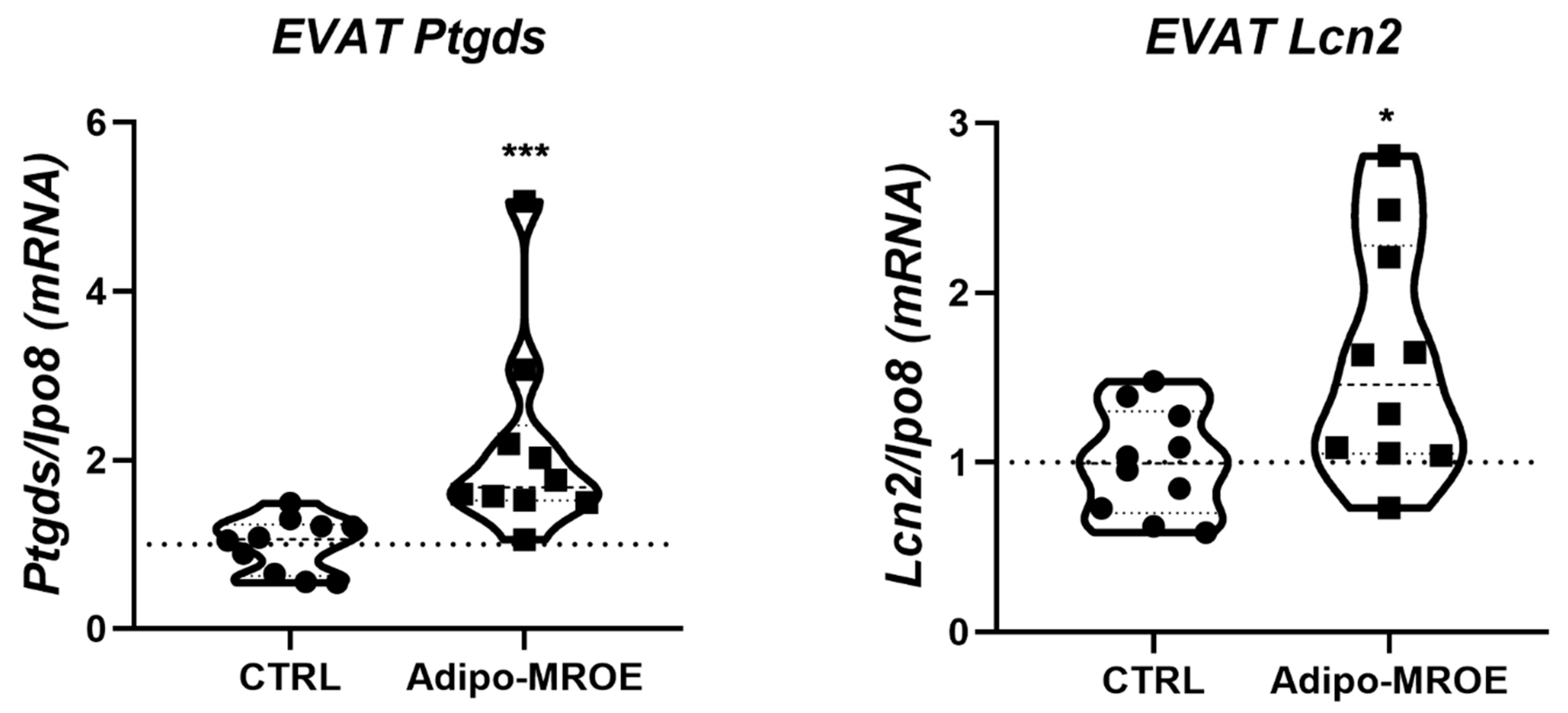
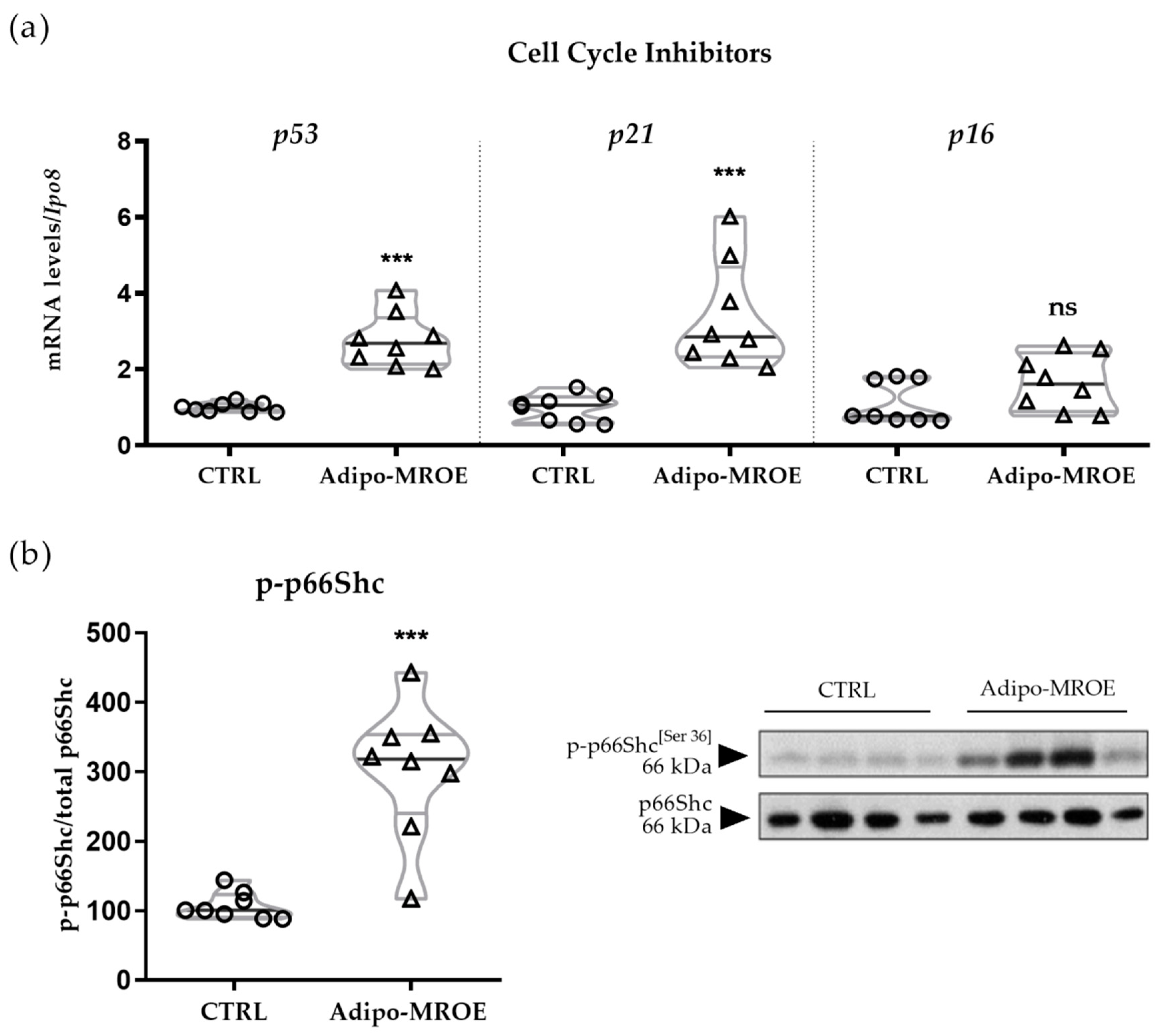
2.5. Adipocyte-MR Activation Induces Premature Senescence in AT
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.1.1. Transgenic Model
4.1.2. Animal Experiments
4.1.3. Sacrifice and Organ Collection
4.2. Mitochondrial Functional Studies
4.2.1. Mitochondria Isolation
4.2.2. Measurement of In Vitro Oxygen Consumption by High Resolution Respirometry
4.3. Molecular Studies
4.3.1. Hydrogen Peroxide Concentration Measurement
4.3.2. Superoxide Concentration Measurement
4.3.3. 8-OHdG Concentration Measurement
4.3.4. Quantitative Real-Time Polymerase Chain Reaction
4.3.5. Western Blot Analysis
4.4. Data Analysis
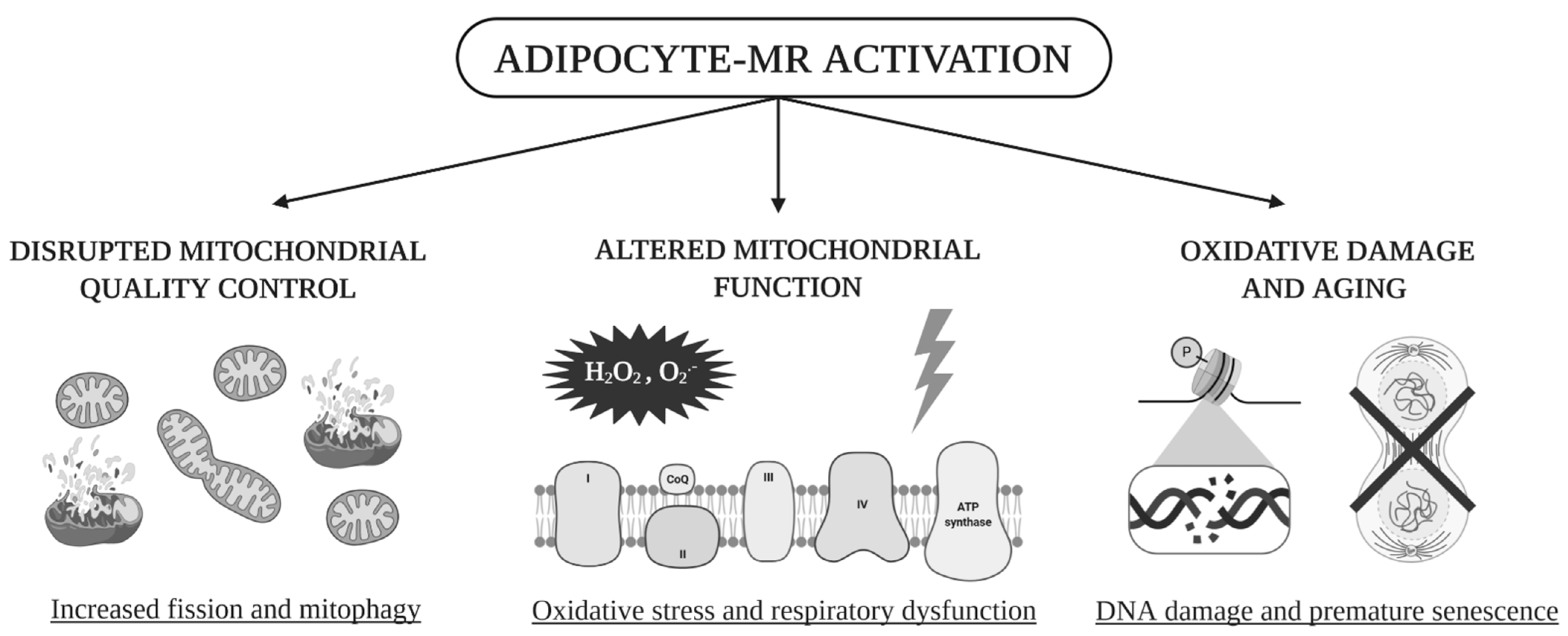
5. Conclusions
- Our data highlight the impact of visceral AT adipocyte-MR on mitochondrial function and quality.
- Our work explores mechanisms by which AT mitochondrial quality becomes compromised by MR activation, and how such perturbations alter mitochondrial function.
- Our results support promising therapeutic avenues using MR antagonism as an additive strategy to improve AT mitochondrial function in the context of MetS.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO | Obesity and Overweight. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 30 November 2017).
- Ahima, R.S.; Flier, J.S. Adipose Tissue as an Endocrine Organ. Trends Endocrinol. Metab. 2000, 11, 327–332. [Google Scholar] [CrossRef]
- Blüher, M. Adipose Tissue Dysfunction in Obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.-P.; Sechi, L.A.; Giacchetti, G.; Ronconi, V.; Strazzullo, P.; Funder, J.W. Primary Aldosteronism: Cardiovascular, Renal and Metabolic Implications. Trends Endocrinol. Metab 2008, 19, 88–90. [Google Scholar] [CrossRef]
- Fallo, F.; Pilon, C.; Urbanet, R. Primary Aldosteronism and Metabolic Syndrome. Horm. Metab. Res. 2012, 44, 208–214. [Google Scholar] [CrossRef]
- Nagase, M.; Fujita, T. Mineralocorticoid Receptor Activation in Obesity Hypertension. Hypertens Res. 2009, 32, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Adler, G.K. Role of Mineralocorticoid Receptor in Insulin Resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 168–175. [Google Scholar] [CrossRef]
- Guo, C.; Ricchiuti, V.; Lian, B.Q.; Yao, T.M.; Coutinho, P.; Romero, J.R.; Li, J.; Williams, G.H.; Adler, G.K. Mineralocorticoid Receptor Blockade Reverses Obesity-Related Changes in Expression of Adiponectin, Peroxisome Proliferator-Activated Receptor-Gamma, and Proinflammatory Adipokines. Circulation 2008, 117, 2253–2261. [Google Scholar] [CrossRef]
- Urbanet, R.; Nguyen Dinh Cat, A.; Feraco, A.; Venteclef, N.; El Mogrhabi, S.; Sierra-Ramos, C.; Alvarez de la Rosa, D.; Adler, G.K.; Quilliot, D.; Rossignol, P.; et al. Adipocyte Mineralocorticoid Receptor Activation Leads to Metabolic Syndrome and Induction of Prostaglandin D2 Synthase. Hypertension 2015, 66, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, D.; Hutson, I.; Tycksen, E.; Pietka, T.A.; Bauerle, K.; Harris, C.A. Role of Mineralocorticoid Receptor in Adipogenesis and Obesity in Male Mice. Endocrinology 2020, 161. [Google Scholar] [CrossRef] [Green Version]
- Briones, A.M.; Nguyen Dinh Cat, A.; Callera, G.E.; Yogi, A.; Burger, D.; He, Y.; Corrêa, J.W.; Gagnon, A.M.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; et al. Adipocytes Produce Aldosterone through Calcineurin-Dependent Signaling Pathways: Implications in Diabetes Mellitus-Associated Obesity and Vascular Dysfunction. Hypertension 2012, 59, 1069–1078. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-Hydroxysteroid Dehydrogenases: Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen Dinh Cat, A.; Antunes, T.T.; Callera, G.E.; Sanchez, A.; Tsiropoulou, S.; Dulak-Lis, M.G.; Anagnostopoulou, A.; He, Y.; Montezano, A.C.; Jaisser, F.; et al. Adipocyte-Specific Mineralocorticoid Receptor Overexpression in Mice Is Associated with Metabolic Syndrome and Vascular Dysfunction: Role of Redox-Sensitive PKG-1 and Rho Kinase. Diabetes 2016, 65, 2392–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, M.; Khemka, V.K.; Chatterjee, G.; Ganguly, A.; Mukhopadhyay, S.; Chakrabarti, S. Enhanced ROS Production and Oxidative Damage in Subcutaneous White Adipose Tissue Mitochondria in Obese and Type 2 Diabetes Subjects. Mol. Cell Biochem. 2015, 399, 95–103. [Google Scholar] [CrossRef] [PubMed]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial Dysfunction in Obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial Dysfunction and Type 2 Diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, C.; Friederich-Persson, M.; Braud, L.; Palacios-Ramirez, R.; Karlsson, S.; Boujardine, N.; Motterlini, R.; Jaisser, F.; Nguyen Dinh Cat, A. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension 2019, 73, 458–468. [Google Scholar] [CrossRef]
- Queisser, N.; Happ, K.; Link, S.; Jahn, D.; Zimnol, A.; Geier, A.; Schupp, N. Aldosterone Induces Fibrosis, Oxidative Stress and DNA Damage in Livers of Male Rats Independent of Blood Pressure Changes. Toxicol. Appl. Pharm. 2014, 280, 399–407. [Google Scholar] [CrossRef]
- Queisser, N.; Fazeli, G.; Schupp, N. Superoxide Anion and Hydrogen Peroxide-Induced Signaling and Damage in Angiotensin II and Aldosterone Action. Biol. Chem. 2010, 391, 1265–1279. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; Jose, C.; Rossignol, R. Relationships between Mitochondrial Dynamics and Bioenergetics. In Mitochondrial Dynamics and Neurodegeneration; Springer: Dordrecht, The Netherlands, 2011; pp. 47–68. ISBN 978-94-007-1290-4. [Google Scholar]
- Westermann, B. Bioenergetic Role of Mitochondrial Fusion and Fission. Biochim. Biophys. Acta. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.-M.; Joanisse, D.R.; Baillot, R.G.; Hood, D.A. Mitochondrial Dysregulation in the Pathogenesis of Diabetes: Potential for Mitochondrial Biogenesis-Mediated Interventions. J. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.D.; Holden, C.R.; Sansbury, B.E.; Gibb, A.A.; Shah, J.; Zafar, N.; Tang, Y.; Hellmann, J.; Rai, S.N.; Spite, M.; et al. Metabolic Remodeling of White Adipose Tissue in Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E262–E277. [Google Scholar] [CrossRef]
- Zhu, C.; Huang, S.; Yuan, Y.; Ding, G.; Chen, R.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial Dysfunction Mediates Aldosterone-Induced Podocyte Damage: A Therapeutic Target of PPARγ. Am. J. Pathol. 2011, 178, 2020–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Jia, Z.; Guo, X.; Yang, T. Aldosterone Induces Epithelial-Mesenchymal Transition via ROS of Mitochondrial Origin. Am. J. Physiol. Ren. Physiol. 2007, 293, F723–F731. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, A.; Qi, J.; Wang, H.; Liu, X.; Zhao, M.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; et al. P53/Drp1-Dependent Mitochondrial Fission Mediates Aldosterone-Induced Podocyte Injury and Mitochondrial Dysfunction. Am. J. Physiol. Ren. Physiol. 2017, 314, F798–F808. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.-P.; Murphy, M.E. The Role of the P53 Tumor Suppressor in Metabolism and Diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A Crucial Role for Adipose Tissue P53 in the Regulation of Insulin Resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Latouche, C.; El Moghrabi, S.; Messaoudi, S.; Nguyen Dinh Cat, A.; Hernandez-Diaz, I.; Alvarez de la Rosa, D.; Perret, C.; López Andrés, N.; Rossignol, P.; Zannad, F.; et al. Neutrophil Gelatinase-Associated Lipocalin Is a Novel Mineralocorticoid Target in the Cardiovascular System. Hypertension 2012, 59, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.L.; Ibarrola, J.; Fernández-Celis, A.; Schou, M.; Sonne, M.P.; Holm, M.R.; Rasmussen, J.; Dela, F.; Jaisser, F.; Faber, J.; et al. The Mineralocorticoid Receptor Antagonist Eplerenone Suppress Interstitial Fibrosis in Subcutaneous Adipose Tissue in Type 2 Diabetes Patients. Diabetes 2020. [Google Scholar] [CrossRef]
- Patni, H.; Mathew, J.T.; Luan, L.; Franki, N.; Chander, P.N.; Singhal, P.C. Aldosterone Promotes Proximal Tubular Cell Apoptosis: Role of Oxidative Stress. Am. J. Physiol. Ren. Physiol. 2007, 293, F1065–F1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Chen, Y.; Zhang, P.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial Dysfunction Accounts for Aldosterone-Induced Epithelial-to-Mesenchymal Transition of Renal Proximal Tubular Epithelial Cells. Free Radic. Biol. Med. 2012, 53, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola, J.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Peña, A.; Arrieta, V.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Cachofeiro, V.; Santamaria, E.; et al. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci. Rep. 2018, 8, 6801. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Tenorio, J.; Marin-Royo, G.; Martinez-Martinez, E.; Martin, R.; Miana, M.; Lopez-Andres, N.; Jurado-Lopez, R.; Gallardo, I.; Luaces, M.; Roman, J.A.S.; et al. The Role of Oxidative Stress in the Crosstalk between Leptin and Mineralocorticoid Receptor in the Cardiac Fibrosis Associated with Obesity. Sci. Rep. 2017, 7, 16802. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic Regulation of PGC-1α Localization and Turnover Implicates Mitochondrial Adaptation in Calorie Restriction and the Stress Response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial Dynamics and Mitochondrial Quality Control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 Deficiency-Induced Mitochondrial Dysfunction and Inflammasome Formation in the Brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Wang, F.; Yang, P.; Wang, X.; Xu, R.; Chen, J.; Yuan, Y.; Lu, Z.; Duan, J. Mitochondrial Fission Is Required for Angiotensin II-Induced Cardiomyocyte Apoptosis Mediated by a Sirt1-P53 Signaling Pathway. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladyshev, V.N. The Free Radical Theory of Aging Is Dead. Long Live the Damage Theory! Antioxid Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Fallo, F.; Veglio, F.; Bertello, C.; Sonino, N.; Della Mea, P.; Ermani, M.; Rabbia, F.; Federspil, G.; Mulatero, P. Prevalence and Characteristics of the Metabolic Syndrome in Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2006, 91, 454–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhart-Bornstein, M.; Lamounier-Zepter, V.; Schraven, A.; Langenbach, J.; Willenberg, H.S.; Barthel, A.; Hauner, H.; McCann, S.M.; Scherbaum, W.A.; Bornstein, S.R. Human Adipocytes Secrete Mineralocorticoid-Releasing Factors. Proc. Natl. Acad. Sci. USA 2003, 100, 14211–14216. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, J.L.; Bruder-Nascimento, T.; Belin de Chantemèle, E.J. The Regulation of Aldosterone Secretion by Leptin: Implications in Obesity-Related Cardiovascular Disease. Curr. Opin. Nephrol. Hypertens 2018, 27, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.-C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemèle, E.J. Adipocyte-Derived Hormone Leptin Is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bornstein, S.R.; Graessler, J.; Kopprasch, S. Very-Low-Density Lipoprotein Mediates Transcriptional Regulation of Aldosterone Synthase in Human Adrenocortical Cells through Multiple Signaling Pathways. Cell Tissue Res. 2012, 348, 71–80. [Google Scholar] [CrossRef]
- Karashima, S.; Yoneda, T.; Kometani, M.; Ohe, M.; Mori, S.; Sawamura, T.; Furukawa, K.; Seta, T.; Yamagishi, M.; Takeda, Y. Comparison of Eplerenone and Spironolactone for the Treatment of Primary Aldosteronism. Hypertens Res. 2016, 39, 133–137. [Google Scholar] [CrossRef]
- Catena, C.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.; Novello, M.; Favret, G.; Melis, A.; Cavarape, A.; Sechi, L.A. Insulin Sensitivity in Patients with Primary Aldosteronism: A Follow-up Study. J. Clin. Endocrinol. Metab. 2006, 91, 3457–3463. [Google Scholar] [CrossRef]
- Garg, R.; Kneen, L.; Williams, G.H.; Adler, G.K. Effect of Mineralocorticoid Receptor Antagonist on Insulin Resistance and Endothelial Function in Obese Subjects. Diabetes Obes. Metab. 2014, 16, 268–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, M.L.; Schou, M.; Rossignol, P.; Holm, M.R.; Rasmussen, J.; Brandt, N.; Frandsen, M.; Chabanova, E.; Dela, F.; Faber, J.; et al. Effect of the Mineralocorticoid Receptor Antagonist Eplerenone on Liver Fat and Metabolism in Patients with Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled Trial (MIRAD Trial). Diabetes Obes. Metab. 2019, 21, 2305–2314. [Google Scholar] [CrossRef]
- Urbanet, R.; Pilon, C.; Calcagno, A.; Peschechera, A.; Hubert, E.-L.; Giacchetti, G.; Gomez-Sanchez, C.; Mulatero, P.; Toffanin, M.; Sonino, N.; et al. Analysis of Insulin Sensitivity in Adipose Tissue of Patients with Primary Aldosteronism. J. Clin. Endocrinol. Metab. 2010, 95, 4037–4042. [Google Scholar] [CrossRef] [Green Version]
- Hirata, A.; Maeda, N.; Hiuge, A.; Hibuse, T.; Fujita, K.; Okada, T.; Kihara, S.; Funahashi, T.; Shimomura, I. Blockade of Mineralocorticoid Receptor Reverses Adipocyte Dysfunction and Insulin Resistance in Obese Mice. Cardiovasc. Res. 2009, 84, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Bournat, J.C.; Brown, C.W. Mitochondrial Dysfunction in Obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased Oxidative Stress in Obesity and Its Impact on Metabolic Syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased Oxidative Stress Precedes the Onset of High-Fat Diet-Induced Insulin Resistance and Obesity. Metab. Clin. Exp. 2008, 57, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative Stress and Metabolic Pathologies: From an Adipocentric Point of View. Oxid Med. Cell Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, H.-J.; Kim, J.-H.; Kwon, O.-B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.-S.; Yoon, G.; Choi, K.-M.; Ko, Y.-G. Mitochondria Are Impaired in the Adipocytes of Type 2 Diabetic Mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Schöttl, T.; Kappler, L.; Braun, K.; Fromme, T.; Klingenspor, M. Limited Mitochondrial Capacity of Visceral Versus Subcutaneous White Adipocytes in Male C57BL/6N Mice. Endocrinology 2015, 156, 923–933. [Google Scholar] [CrossRef]
- Kraunsøe, R.; Boushel, R.; Hansen, C.N.; Schjerling, P.; Qvortrup, K.; Støckel, M.; Mikines, K.J.; Dela, F. Mitochondrial Respiration in Subcutaneous and Visceral Adipose Tissue from Patients with Morbid Obesity. J. Physiol. 2010, 588, 2023–2032. [Google Scholar] [CrossRef]
- Soro-Arnaiz, I.; Li, Q.O.Y.; Torres-Capelli, M.; Meléndez-Rodríguez, F.; Veiga, S.; Veys, K.; Sebastian, D.; Elorza, A.; Tello, D.; Hernansanz-Agustín, P.; et al. Role of Mitochondrial Complex IV in Age-Dependent Obesity. Cell Rep. 2016, 16, 2991–3002. [Google Scholar] [CrossRef] [Green Version]
- Vernochet, C.; Mourier, A.; Bezy, O.; Macotela, Y.; Boucher, J.; Rardin, M.J.; An, D.; Lee, K.Y.; Ilkayeva, O.R.; Zingaretti, C.M.; et al. Adipose-Specific Deletion of TFAM Increases Mitochondrial Oxidation and Protects Mice against Obesity and Insulin Resistance. Cell Metab. 2012, 16, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.; Gottlieb, R.A. Parkin-Mediated Mitophagy Is Downregulated in Browning of White Adipose Tissue. Obesity (Silver Spring) 2017, 25, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.-Y.; Kohno, M.; Hitomi, H.; Kitada, K.; Fujisawa, Y.; Yatabe, J.; Yatabe, M.; Felder, R.A.; Ohsaki, H.; Rafiq, K.; et al. Aldosterone/Mineralocorticoid Receptor Stimulation Induces Cellular Senescence in the Kidney. Endocrinology 2011, 152, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanh, V.C.; Zulkifli, A.F.; Tokunaga, C.; Yamashita, T.; Hiramatsu, Y.; Ohneda, O. Aging Impairs Beige Adipocyte Differentiation of Mesenchymal Stem Cells via the Reduced Expression of Sirtuin 1. Biochem. Biophys. Res. Commun. 2018, 500, 682–690. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The Mitochondrial P53 Pathway. Biochim. Biophys. Acta. 2009, 1787, 414. [Google Scholar] [CrossRef] [Green Version]
- Afanas’ev, I. Reactive Oxygen Species and Age-Related Genes P66Shc, Sirtuin, FoxO3 and Klotho in Senescence. Oxid Med. Cell Longev. 2010, 3, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Nijhawan, P.; Behl, T. Role of Sirtuins in Obesity. Obes. Med. 2020, 17, 100156. [Google Scholar] [CrossRef]
- Martín-Fernández, B.; Rubio-Navarro, A.; Cortegano, I.; Ballesteros, S.; Alía, M.; Cannata-Ortiz, P.; Olivares-Álvaro, E.; Egido, J.; de Andrés, B.; Gaspar, M.L.; et al. Aldosterone Induces Renal Fibrosis and Inflammatory M1-Macrophage Subtype via Mineralocorticoid Receptor in Rats. PLoS ONE 2016, 11, e0145946. [Google Scholar] [CrossRef] [Green Version]
- Lattenist, L.; Lechner, S.M.; Messaoudi, S.; Le Mercier, A.; El Moghrabi, S.; Prince, S.; Bobadilla, N.A.; Kolkhof, P.; Jaisser, F.; Barrera-Chimal, J. Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone Protects Against Acute Kidney Injury-Mediated Chronic Kidney Disease: Role of Oxidative Stress. Hypertension 2017. [Google Scholar] [CrossRef]
- Tarjus, A.; Martínez-Martínez, E.; Amador, C.; Latouche, C.; El Moghrabi, S.; Berger, T.; Mak, T.W.; Fay, R.; Farman, N.; Rossignol, P.; et al. Neutrophil Gelatinase-Associated Lipocalin, a Novel Mineralocorticoid Biotarget, Mediates Vascular Profibrotic Effects of Mineralocorticoids. Hypertension 2015, 66, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Garg, R.; Adler, G.K. Mineralocorticoid Receptor Antagonists and the Metabolic Syndrome. Curr. Hypertens Rep. 2010, 12, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Feraco, A.; Marzolla, V.; Scuteri, A.; Armani, A.; Caprio, M. Mineralocorticoid Receptors in Metabolic Syndrome: From Physiology to Disease. Trends Endocrinol. Metab. 2020, 31, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.-C.; Caprio, M.; Fève, B. Mineralocorticoid Receptors in the Metabolic Syndrome. Trends Endocrinol. Metab. 2009, 20, 444–451. [Google Scholar] [CrossRef]
- Barrett Mueller, K.; Lu, Q.; Mohammad, N.N.; Luu, V.; McCurley, A.; Williams, G.H.; Adler, G.K.; Karas, R.H.; Jaffe, I.Z. Estrogen Receptor Inhibits Mineralocorticoid Receptor Transcriptional Regulatory Function. Endocrinology 2014, 155, 4461–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzen, S.; Gurer-Orhan, H.; Saso, L. Detection of Reactive Oxygen and Nitrogen Species by Electron Paramagnetic Resonance (EPR) Technique. Molecules 2017, 22, 181. [Google Scholar] [CrossRef] [PubMed]
- del Pozo, C.H.; Calvo, R.M.; Vesperinas-García, G.; Gómez-Ambrosi, J.; Frühbeck, G.; Corripio-Sánchez, R.; Rubio, M.A.; Obregon, M.-J. IPO8 and FBXL10: New Reference Genes for Gene Expression Studies in Human Adipose Tissue. Obes. (Silver Spring) 2010, 18, 897–903. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefranc, C.; Friederich-Persson, M.; Foufelle, F.; Nguyen Dinh Cat, A.; Jaisser, F. Adipocyte-Mineralocorticoid Receptor Alters Mitochondrial Quality Control Leading to Mitochondrial Dysfunction and Senescence of Visceral Adipose Tissue. Int. J. Mol. Sci. 2021, 22, 2881. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062881
Lefranc C, Friederich-Persson M, Foufelle F, Nguyen Dinh Cat A, Jaisser F. Adipocyte-Mineralocorticoid Receptor Alters Mitochondrial Quality Control Leading to Mitochondrial Dysfunction and Senescence of Visceral Adipose Tissue. International Journal of Molecular Sciences. 2021; 22(6):2881. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062881
Chicago/Turabian StyleLefranc, Clara, Malou Friederich-Persson, Fabienne Foufelle, Aurélie Nguyen Dinh Cat, and Frédéric Jaisser. 2021. "Adipocyte-Mineralocorticoid Receptor Alters Mitochondrial Quality Control Leading to Mitochondrial Dysfunction and Senescence of Visceral Adipose Tissue" International Journal of Molecular Sciences 22, no. 6: 2881. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062881