
Mycobacterial Epoxide Hydrolase EphD Is Inhibited by Urea and Thiourea Derivatives


1
Department of Biochemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Mlynská Dolina, Ilkovičova 6, 842 15 Bratislava, Slovakia
2
Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO 80523-1682, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(6), 2884; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062884
Submission received: 29 January 2021
/
Revised: 1 March 2021
/
Accepted: 10 March 2021
/
Published: 12 March 2021
(This article belongs to the Special Issue The Role of Hydrolases in Biology and Xenobiotics Metabolism)
Abstract
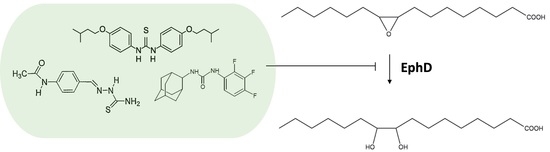
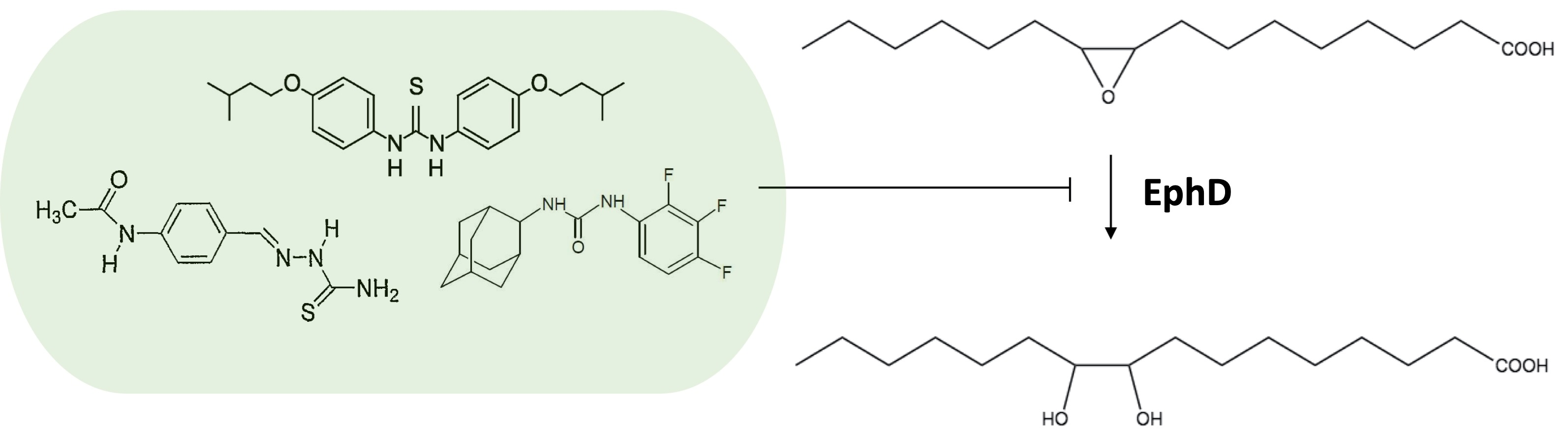
:The genome of the human intracellular pathogen Mycobacterium tuberculosis encodes an unusually large number of epoxide hydrolases, which are thought to be involved in lipid metabolism and detoxification reactions needed to endure the hostile environment of host macrophages. These enzymes therefore represent suitable targets for compounds such as urea derivatives, which are known inhibitors of soluble epoxide hydrolases. In this work, we studied in vitro the effect of the thiourea drug isoxyl on six epoxide hydrolases of M. tuberculosis using a fatty acid substrate. We show that one of the proteins inhibited by isoxyl is EphD, an enzyme involved in the metabolism of mycolic acids, key components of the mycobacterial cell wall. By analyzing mycolic acid profiles, we demonstrate the inhibition of EphD epoxide hydrolase activity by isoxyl and two other urea-based inhibitors, thiacetazone and AU1235, inside the mycobacterial cell.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
With about 10 million new cases and 1.4 million deaths in 2019, tuberculosis (TB), a disease caused by pathogenic bacterium Mycobacterium tuberculosis, remains a serious global health problem. The End TB Strategy adopted by the World Health Organization (WHO) envisioned a 20% decline in global incidence of TB and a 35% reduction in TB deaths between 2015 and 2020; however, despite some progress, these milestones have not been reached [1]. Accelerating this decline and putting a stop to the TB epidemic requires intensifying the search for a new vaccine and new chemotherapeutics, tasks which face several challenges [2,3,4]. Currently, a six-month regimen is routinely implemented in treatment of drug susceptible TB, consisting of a combination of isoniazid (INH), rifampicin (RIF), ethambutol (EMB) and pyrazinamide (PZA). In the case of multiple-drug-resistant TB (MDR-TB), the treatment can last up to 20 months and involves administration of second-line TB drugs, which are associated with more severe side effects and toxicity [5].
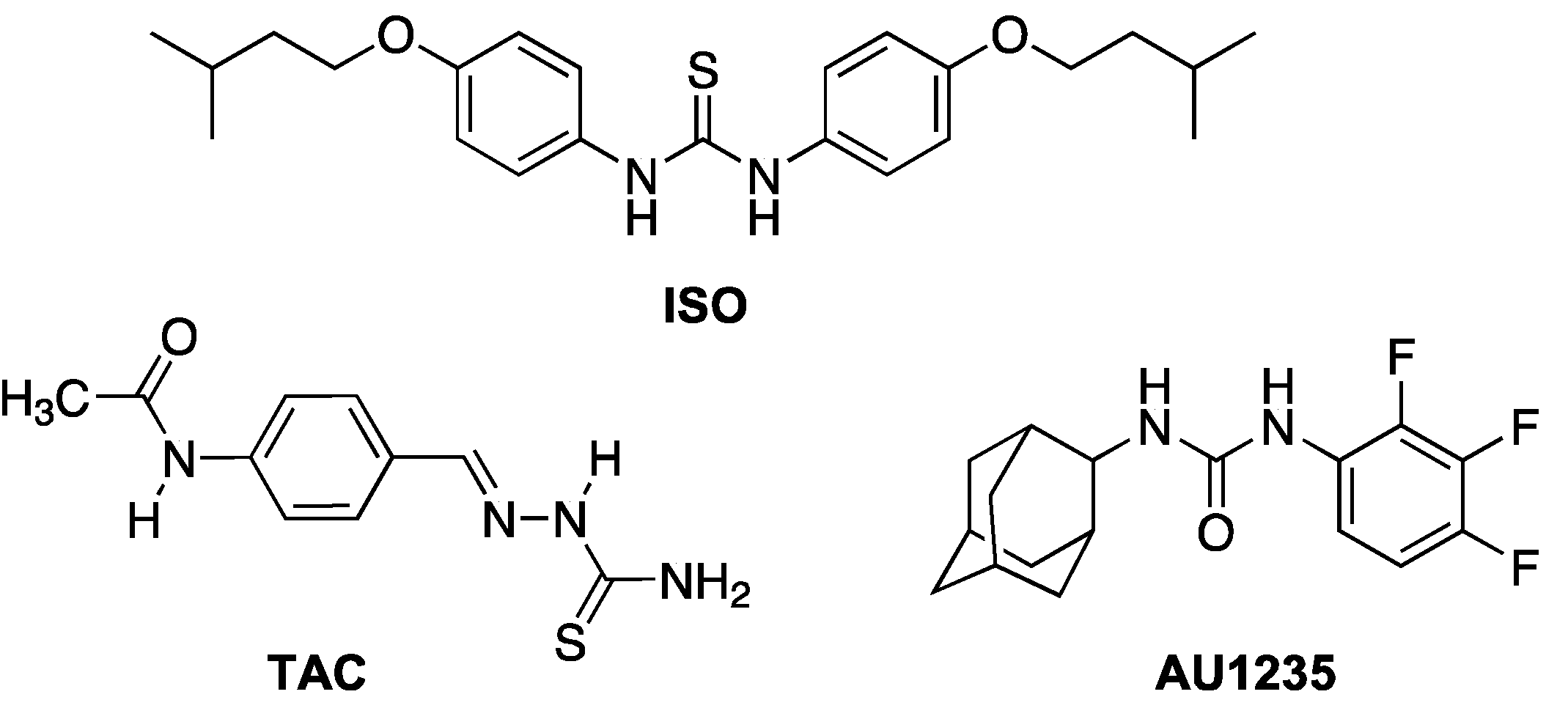
Isoxyl (thiocarlide; 4,4′-diisoamyloxydiphenylthiourea) (ISO) (Figure 1) is a thiourea compound that was used for treatment of TB in the 1960s, but its use was discontinued due to poor bioavailability [6]. Nevertheless, ISO shows potent antimycobacterial activity in vitro with low minimal inhibitory concentrations (MICs) on M. tuberculosis, including strains resistant to RIF and INH [7]. Initial studies found that ISO inhibits the stearoyl-CoA Δ9-desaturase DesA3. However, this did not explain the inhibition of mycolic acids seen in ISO treated M. tuberculosis [8]. Mycolic acids are very long chain (C60–C90) α-alkyl β-hydroxy fatty acids and are key components of the mycobacterial outer membrane (mycomembrane), their presence being essential for the viability of the bacterium [9,10]. Mycobacteria produce several types of mycolates differing in the presence of different functional groups in their hydrocarbon chain, such as double bonds, cyclopropane rings and oxygenated functions [11]. In subsequent studies, it was shown that ISO and thiacetazone (TAC), a related thiosemicarbazone drug (Figure 1), both target HadAB, the β-hydroxyacyl-ACP dehydratase component of the fatty acid synthase II (FASII) system, which is responsible for the synthesis of mycolic acids [12]. It is well established that ISO and TAC are prodrugs and require modification by the mycobacterial monooxygenase EthA for activity [13,14,15].
Mycobacteria have an extremely hydrophobic cell envelope that consists of a conventional plasma membrane; a layer of peptidoglycan; and a polysaccharide, arabinogalactan, which is esterified by mycolic acids, forming the inner leaflet of the mycomembrane [16]. The outer leaflet of the mycomembrane is composed of various complex lipids such as trehalose dimycolate (TDM), di- and polyacyl trehaloses, phthiocerol dimycocerosates and sulfolipids [17,18]. The mycobacterial cell envelope is an important target for antimycobacterial compounds, with the most notable examples being first-line antituberculotics INH, which inhibits the FASII enoyl-ACP reductase InhA [19,20], and EMB, which interferes with the synthesis of arabinogalactan [21]. In recent years, several new inhibitors were found to target the cell envelope, for example, by affecting the transporter MmpL3, which transfers trehalose monomycolate (TMM) across the plasma membrane [22]. Once transferred to the periplasmic space, TMM is a substrate for the mycolyltransferases FbpA, FbpB and FbpC (also known collectively as the antigen 85 complex) that catalyze the transfer of mycolic acyl chains onto cell wall arabinogalactan or the synthesis of trehalose dimycolate (TDM), thus incorporating mycolates into the mycomembrane [23]. One of the MmpL3 inhibitors is an adamantyl urea AU1235 (Figure 1), which inhibits a range of mycobacterial strains, including MDR isolates of M. tuberculosis [22].
Initial analysis of the genome of virulent strain M. tuberculosis H37Rv discovered five genes encoding putative epoxide hydrolases (EHs) homologous to eukaryotic α/β hydrolase fold EHs, each about 300 amino acids long (ephA, ephB, ephC, ephE and ephF), and another one which appeared to be an N-terminal part of a fusion protein with a short-chain dehydrogenase (SDR) at its C-terminus, named ephD [24,25]. Another EH from M. tuberculosis (EphG) homologous to the limonene 1,2-epoxide hydrolase from Rhodococcus erythropolis was described [26]. Recently, we were able to produce these seven mycobacterial EHs in soluble form and we have shown, by using 9,10-cis-epoxystearic acid as substrate, that, with the exception of EphC, all of them display epoxide hydrolase activity in vitro. At the same time, we demonstrated that the N-terminal EH domain of EphD is responsible for its EH activity, which, in turn, results in the ability of this protein to modify epoxymycolates in non-pathogenic Mycobacterium smegmatis [27]. To date, structures of several EHs from M. tuberculosis or their orthologues from Mycobacterium thermoresistibile were solved [26,28,29], and although their physiological substrates still remain to be discovered, it was proposed that they might be important in detoxification reactions [25].
It is known that human-soluble epoxide hydrolase (sEH) can be inhibited by different urea derivatives [30,31], which was also shown for mycobacterial EHs EphB and EphE [32,33]. In this work we tested the activity of EphA, EphB, EphD, EphE, EphF and EphG in vitro on 9,10-cis-epoxystearic acid in the presence of ISO. We show that the activity of EphD is inhibited inside mycobacterial cells by ISO, TAC and AU1235, and that this enzyme is thus an additional target for these antimycobacterial compounds.
2. Results
2.1. Monitoring the Activity of Mycobacterial EHs in the Presence of ISO
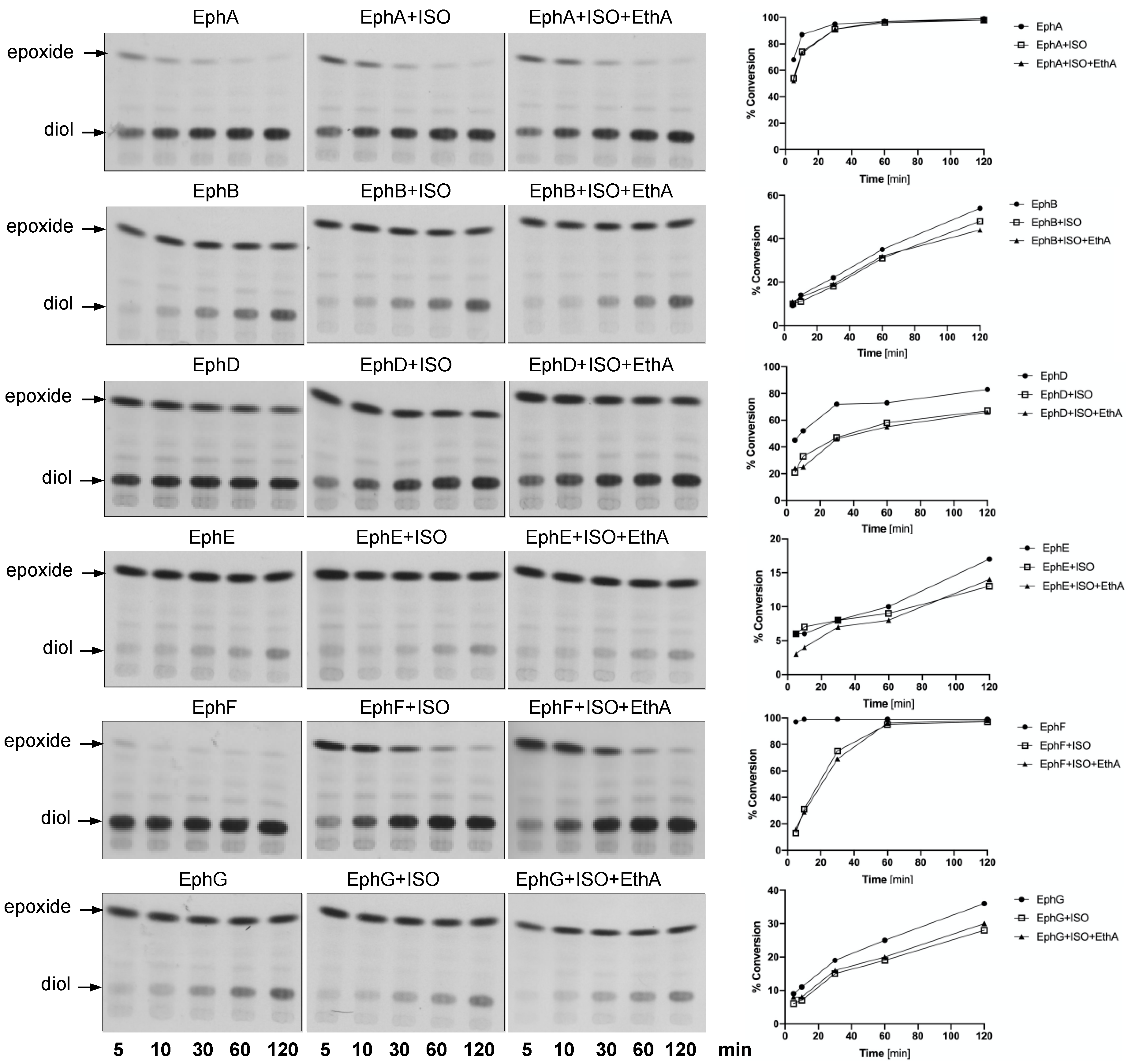
In order to monitor the EH activity of mycobacterial EHs in vitro, we used a radioactively labelled substrate, [14C]-9,10-cis-epoxystearic acid [34,35] and 10,000× g supernatants of the lysates of E. coli strains individually producing each EH—E. coli BL21(DE3) pET29a-ephA, E. coli BL21-AI pDEST17-ephB, E. coli BL21(DE3) pET29a-ephD, E. coli BL21-AI pET28a-ephE, E. coli BL21(DE3) pET28a-ephF and E. coli BL21(DE3) pET29a-ephG—as enzyme sources. ISO was added to the reactions at a final concentration of 10 µg/mL and left incubating for 5, 10, 30, 60 or 120 min. Partially purified monooxygenase EthA was added to some reaction mixtures together with ISO. Extracted lipids were separated by thin-layer chromatography (Figure 2, left), and the bands corresponding to the substrate, [14C]-9,10-cis-epoxystearic acid or the corresponding diol product were isolated by preparative thin-layer chromatography and quantified by scintillation counting. The EH activity in each reaction was expressed as a substrate conversion percentage (Figure 2, right; Table S1). With the exception of EphD and EphF, the activities of the studied EHs were not significantly affected by ISO. Moreover, in the case of all the EHs tested, we did not observe any significant effect of the addition of EthA to the reactions and, as expected, the proteins of the E. coli BL21(DE3) pET28a lysate did not catalyze the conversion of [14C]-9,10-cis-epoxystearic acid to the corresponding diol (Figure S1).
2.2. EphD and EphF Interact Differently with ISO
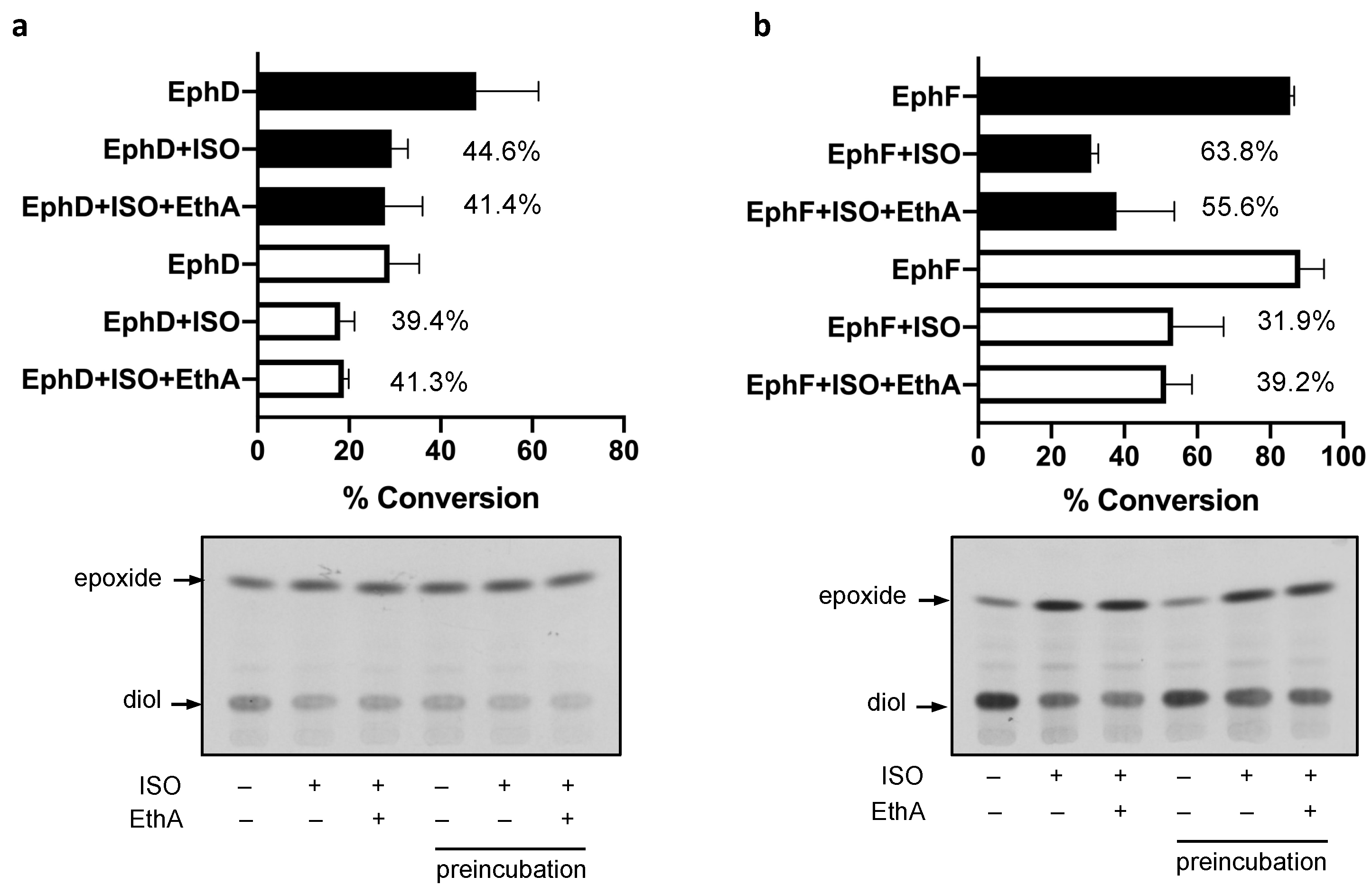
The activities of EphD and EphF are inhibited by ISO; however, there seemed to be a marked difference in the kinetics of this inhibition—in contrast to EphD, the activity of EphF was inhibited only at the beginning of the incubation and the conversion was comparable to that of the untreated control at later time points (Figure 2). We therefore included a 15 min preincubation step with ISO before adding the substrate and allowing the reaction to run for 15 min. We observed that in the case of EphD, preincubation does not alter the inhibitory effect of ISO, as the percentage of inhibition remains at the same level when compared to the corresponding control reaction (Figure 3a). On the other hand, the preincubation step with EphF resulted in lower inhibitory activity of ISO (Figure 3b), clearly suggesting a different type of interaction between the enzyme and the inhibitor. Again, the presence of monooxygenase EthA in the reaction did not affect the inhibitory activity of ISO.
2.3. EphD Is Inhibited by Urea and Thiourea Derivatives inside the Cell
We next sought to determine whether the inhibition of EphD by ISO observed in vitro translated into EphD inhibition inside the bacteria. In our previous work, we showed that protein EphD is involved in the modification of oxygenated mycolic acids in mycobacteria [27]. The overproduction of EphD and its orthologue from M. smegmatis mc2155, MSMEG_4280, in M. smegmatis led to a decrease in the amounts of epoxymycolic acids, which was accompanied by the accumulation of dicarboxymycolates in mycolic acid extracts. Dicarboxymycolates are formed during isolation of mycolic acids and are products of saponification of wax-ester mycolates, which probably arise upon opening of the oxirane ring by EphD. We reasoned that if ISO inhibits EphD inside the mycobacterial cells, cultivation of M. smegmatis EphD and MSMEG_4280 overproducing strains in the presence of ISO might lead to a decrease in dicarboxymycolates in these strains. In parallel, we also tested the inhibitory activity of thiosemicarbazone TAC and adamantyl urea AU1235, while INH was used as a control drug.
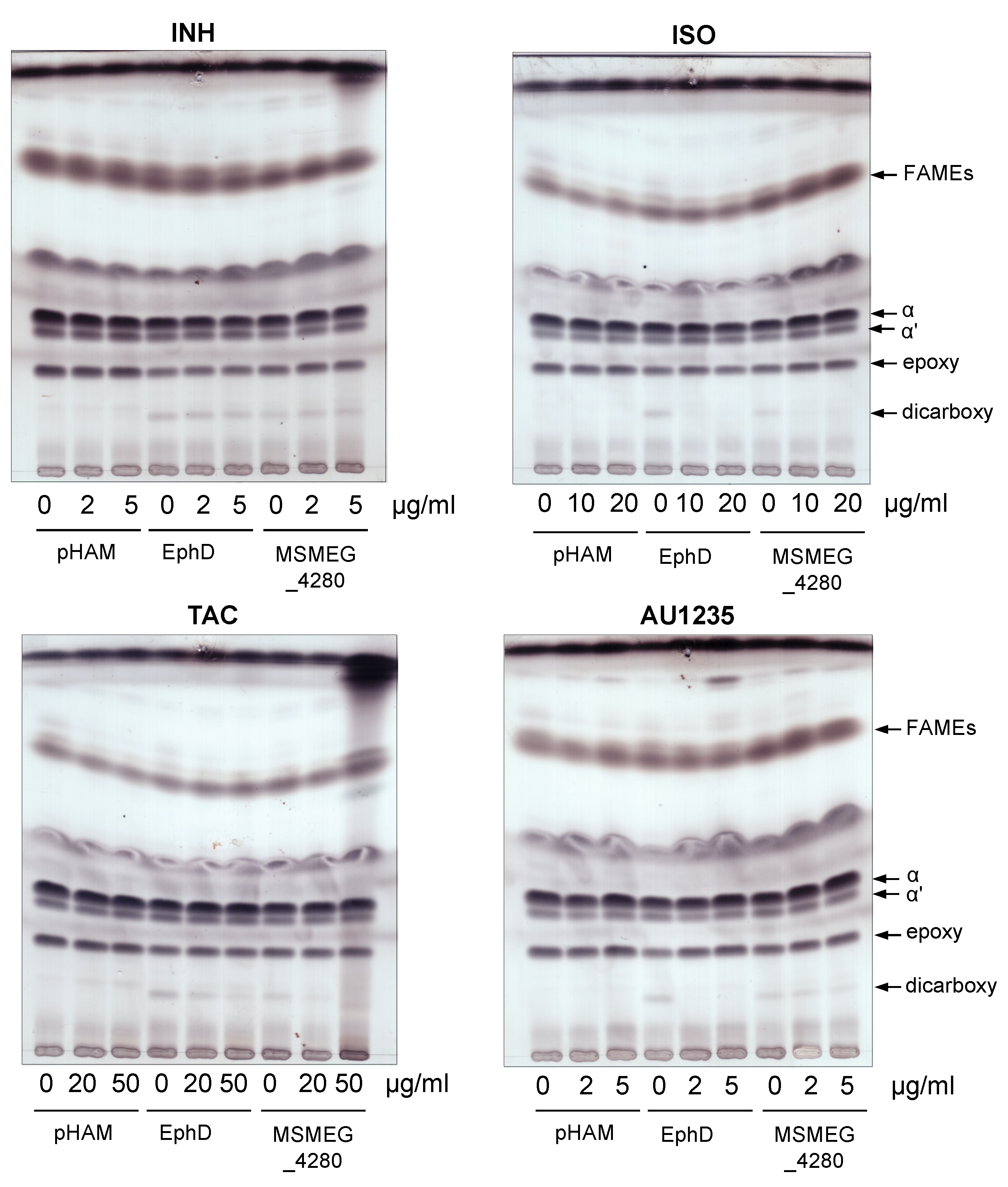
M. smegmatis is intrinsically resistant to ISO and TAC, probably due to the presence of an additional dehydratase, which is able to substitute for the loss of HadAB activity [36]. Higher concentrations of ISO (10 and 20 µg/mL) and TAC (20 and 50 µg/mL) than the MICs of these drugs for M. tuberculosis were therefore chosen for our analysis. The concentrations of AU1235 used were lower (2 µg/mL) and higher (5 µg/mL) than the reported MIC of 3.2 µg/mL for M. smegmatis [22]. For overproduction of EphD and MSMEG_4280 in M. smegmatis, we used an acetamide inducible expression system pHAM [37], and INH, ISO, TAC or AU1235 were added at the same time as the inducer. After 24 h of incubation, the cells were harvested and mycolic acids were extracted and methylated and the resulting mycolic acid methyl esters (MAMEs) were analyzed by thin-layer chromatography (TLC). As observed previously, the overproduction of EphD and MSMEG_4280, compared to control strain, led to lower amounts of epoxymycolates and concomitant accumulation of dicarboxymycolates, while the non-oxygenated α and α’ forms were not affected. In the presence of sub-inhibitory concentrations of INH, the profiles of control strain M. smegmatis pHAM as well as overproducing strains remained unchanged (Figure 4, upper left). However, addition of ISO, TAC and AU1235 to the overproducing strains caused a decrease of dicarboxymycolates compared to their untreated controls (Figure 4 and Figure S2), suggesting that the EH activity of EphD is targeted by these drugs inside the cells.
3. Discussion
Mycobacterium tuberculosis produces an unusually large number of EHs, which probably arose from lateral gene transfer events [38]. As the primary function of this class of enzymes is elimination of toxic epoxides by converting them to less reactive diols, it is tempting to speculate that in M. tuberculosis their function lies in suppressing the oxidative damage caused by reactive oxygen species produced by host macrophages. To date, however, data about the native substrates of most mycobacterial EHs are still lacking. Our previous work has shown that EphD, a protein with an N-terminal α/β hydrolase domain and a C-terminal short-chain dehydrogenase domain, is involved in the metabolism of mycolic acids, essential cell-wall lipids of mycobacteria. In the same work we have demonstrated that mycobacterial proteins EphA, EphB, EphD, EphE, EphF and EphG, but not EphC, can cleave 9,10-cis-epoxystearic acid into a corresponding diol. In our current study, we used the same substrate to monitor the activities of these EHs in the presence of ISO and we show that, in our experimental setup, only the activities of EphD and EphF are significantly inhibited by this thiourea drug. Brown et al. have observed no inhibition of EH activity by ISO when using purified EphB and a relatively low inhibition when using a crude extract of an E. coli strain producing EphE, both on a fluorescent substrate [33]. Our results confirm that ISO does not inhibit EphB, regardless of the enzyme source or substrate used. However, due to low activity of EphE monitored in our assay, we cannot make any conclusion regarding the inhibition of this epoxide hydrolase. Interestingly, while the EH activity of EphD is clearly inhibited by ISO throughout the experiment, in case of EphF, after strong initial inhibition, the substrate conversion returns to control levels over time. This may reflect different binding affinity of the inhibitor, as seen in the case of murine and mammalian sEH [39], or it may indicate a modification of ISO by EphF, which recognizes ISO as a substrate and inactivates this drug. Further efforts are needed to elucidate the interaction between EphF and ISO, the most crucial being the isolation of the enzyme and subsequent structural analysis of possible ISO metabolites produced by catalytic activity of this protein.
Our in vitro assays show that the mycobacterial monooxygenase EthA, which is responsible for the activation of ISO does not affect the inhibition of mycobacterial EHs. It was shown that for exerting their effect on mycobacterial dehydratase HadAB, ISO and TAC need to be metabolized to their sulfenic acid derivatives and as such they covalently bind to the Cys166 residue of HadA, blocking the binding of its physiological substrate [14]. Inhibition of EHs by urea compounds, on the other hand, functions by mimicking the transition state of the substrate upon oxirane ring opening, without prior activation [40,41]. The difference in the mode of action of these inhibitors on the two classes of enzymes might therefore explain that no activation by EthA is needed for the inhibition of mycobacterial EHs.
The clear inhibition of EphD EH activity by ISO in in vitro conditions prompted us to test this inhibition in mycobacterial cells. We took advantage of a direct consequence of the EH activity of this enzyme in the occurrence of dicarboxymycolates in MAME extracts of overproducing strains of M. smegmatis. We confirmed that ISO, as well as structurally related drug TAC and adamantyl urea AU1235, inhibit EphD inside the mycobacterial cells, showing that (thio)urea-based inhibitors of different structures can inhibit this enzyme. Although a non-essential gene for in vitro growth of M. tuberculosis [27,42], ephD is one of the 126 genes shown to be essential for its survival in macrophages [43]. We have observed that deletion of ephD in M. tuberculosis causes a defect in synthesis of ketomycolates, which appear to be important for TB pathogenesis [44,45]. However, it remains to be determined what is the precise role of EphD in the biosynthesis of this mycolate class and how exactly are its EH and SDR domains involved in this process.
Altogether, our results support the notion that an approach consisting of refurbishing known drugs with strong antimycobacterial activity, such as urea and thiourea derivatives, and improving their pharmacokinetic properties is worth considering in combating M. tuberculosis. Several works [33,46,47,48] indicate that structural optimization of these compounds might indeed lead to promising results.
4. Materials and Methods
4.1. Cloning, Bacterial Strains, Growth Conditions and Recombinant Protein Production
For cloning purposes, strain E. coli DH5α (Stratagene, San Diego, CA, USA) was used and cells were grown in Luria-Bertani (LB) medium (Invitrogen, Carlsbad, CA, USA) with shaking or on LB agar plates at 37 °C. The coding sequence of gene ethA was excised from the construct pVV16-ethA [15] using restriction endonucleases NdeI and HindIII and cloned into pET29a(+) (Novagen, Piacenza, Italy). The resulting construct pET29a-ethA was transformed into E. coli BL21(DE3) (Stratagene).
Strains E. coli BL21(DE3) pET29a-ephA, E. coli BL21-AI pDEST17-ephB, E. coli BL21(DE3) pET29a-ephD, E. coli BL21-AI pET28a-ephE, E. coli BL21(DE3) pET28a-ephF, E. coli BL21(DE3) pET29a-ephG [27] and E. coli BL21(DE3) pET29a-ethA were grown in Luria-Bertani (LB) medium (Invitrogen) at 37 °C with shaking, until optical density at 600 nm (OD600) reached 0.4–0.6, when recombinant protein production was induced by adding 0.4 mM isopropyl-β-D-1-thiogalactopyranoside. Strains were further incubated for 16 h at 16 °C, except for E. coli BL21(DE3) pET29a-ephD, which was incubated at 30 °C for 4 h.
Strains M. smegmatis mc2155 pHAM, M. smegmatis mc2155 pHAM-ephD and pHAM-msmeg_4280 [27] were grown in liquid modified M63 medium (7.6 × 10−2 M (NH4)2SO4, 0.5 M KH2PO4, 1 mM MgSO4, 0.2% succinate, 0.025% tyloxapol, pH 7) at 30 °C with shaking, to an OD600 of 0.6–0.9, when acetamide was added to a concentration of 0.2%. At the same time, INH (0, 2 and 5 µg/mL), ISO (0, 10 and 20 µg/mL), TAC (0, 20 and 50 µg/mL) or AU1235 (0, 2 and 5 µg/mL) were added with the final concentration of dimethyl sulfoxide (DMSO) of 1% and cultures were further grown for 24 h at 30 °C.
When needed, 20 µg/mL hygromycin and 20 µg/mL kanamycin were added to the medium.
4.2. Purification of EthA
The recombinant His-tagged protein EthA was isolated from E. coli BL21(DE3) pET29a-ethA. One gram (wet weight) of cells was resuspended in 4 mL of buffer A (25 mM Tris-HCl, pH = 7.5, 10% glycerol, 300 mM NaCl) with addition of DNase (1.5 µg/mL). The suspension was subjected to probe sonication (10 times 20 s pulses with 60 s cooling intervals), and the sonicate was centrifuged for 10 min, 10,000× g at 4 °C. The resulting supernatant was loaded onto 2 mL of TALON metal affinity resin (Clontech, Mountain View, CA, USA), and the unbound protein was removed by washing with buffer A. His-tagged EthA was gradually eluted with buffer A containing 10 mM, 50 mM and 300 mM imidazole, and fractions containing the partially purified protein were combined, desalted using a PD-10 column (GE Healthcare, Chicago, IL, USA) and concentrated using Amicon Ultra-4 (Merck, Kenilworth, NJ, USA) centrifugal filter.
4.3. Epoxide Hydrolase Assays
Cell lysates of E. coli BL21(DE3) pET29a-ephA, E. coli BL21-AI pDEST17-ephB, E. coli BL21(DE3) pET29a-ephD, E. coli BL21-AI pET28a-ephE, E. coli BL21(DE3) pET28a-ephF and E. coli BL21(DE3) pET29a-ephG were prepared as described earlier [27], and these were centrifuged for 10 min at 10,000× g. EH assays [34] were performed with equivalents of 130 μg of proteins from 10,000× g supernatants prepared from the above mentioned strains, with the exception of BL21(DE3) pET29a-ephA and BL21-AI pDEST17-ephB 10,000× g supernatants, where 2 μg and 500 μg of proteins were used, respectively. The reaction mixture contained 22,000 dpm of [14C]-9,10-cis-epoxystearic acid (56 mCi/mmol) as the substrate, ISO in a final concentration 10 µg/mL (2% DMSO) and, alternatively, 10 μg of partially purified monooxygenase EthA. Adding higher concentrations of ISO was also attempted; however, this caused precipitation of the compound. Reaction volume was adjusted with 50 mM Tris-HCl pH = 7.5 to 250 μL, and the reactions were incubated at 37 °C for 5, 10, 30, 60 or 120 min. For preincubation experiments, all reaction components except the substrate were incubated for 15 min at 37 °C with ISO, after which the substrate was added, and the mixture was incubated for additional 15 min at 37 °C. Reactions were stopped by adding 500 μL of ethyl acetate and vortexing. The samples were further briefly centrifuged, and the upper organic phase was transferred to a clean tube, dried under a stream of nitrogen and resuspended in 20 μL of CHCl3:CH3OH (2:1). Half of the suspension was analyzed by thin-layer chromatography (TLC) on Silica gel 60 F254 plates (Merck) in n-hexane:diethyl ether:formic acid (70:30:2), and the radiolabeled compounds were visualized on Kodak BioMax MR film.
For quantification, spots corresponding to the substrate and the product from each reaction were carefully scraped off silica plates, 5 mL of EcoLite(+) liquid scintillation cocktail (MP Biomedicals, Irvine, CA, USA) was added and samples were analyzed on TriCarb 2900TR (PerkinElmer, Waltham, MA, USA).
4.4. Analysis of Mycolic Acids
Mycolic acids were isolated as described earlier [7]. Briefly, 1 mL of 15% tetrabutylammonium hydroxide (TBAH) was added to M. smegmatis mc2155 pHAM, M. smegmatis mc2155 pHAM-ephD and pHAM-msmeg_4280 cell pellets (0.1–0.5 g wet weight) and the samples were incubated for 100 °C overnight. One milliliter of water, 1.5 mL dichloromethane and 150 µL of iodomethane were then added to the saponified mycolic acids, and they were left shaking for 4 h at room temperature. The organic phase was then washed twice with water and dried under nitrogen flow, and fatty acid and mycolic acid methyl esters (FAMEs, MAMEs) were extracted from the dry residue with 2 mL of diethyl ether. Extracts were transferred to clean tubes, dried, and resuspended in CHCl3:CH3OH (2:1) according to OD600 of cultures at time of harvesting. Samples were analyzed by TLC on Silica gel 60 F254 plates (Merck) in n-hexane:ethyl acetate (95:5, three runs), and different MAME populations were visualized with 10% cupric sulfate in 8% phosphoric acid and charring.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/1422-0067/22/6/2884/s1. Reference [49] are cited in the supplementary materials.
Author Contributions
Conceptualization, J.K. and M.J.; Funding acquisition, J.K. and M.J.; Investigation, J.M. and M.K.; Methodology, J.K. and M.J.; Writing—original draft, J.M.; Writing—review and editing, J.K. and M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Slovak Research and Development Agency (grant n. APVV-19-0189) (to J.K.) and by grants from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (AI153477 and AI116525) (to M.J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or Supplementary Material.
Acknowledgments
We are grateful to Michael Arand for the kind gift of [14C]-9,10-cis-epoxystearic acid.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- WHO. WHO Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; Licence: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- Huszár, S.; Chibale, K.; Singh, V. The quest for the holy grail: New antitubercular chemical entities, targets and strategies. Drug Discov. Today 2020, 25, 772–780. [Google Scholar] [CrossRef]
- Kroesen, V.M.; Madacki, J.; Frigui, W.; Sayes, F.; Brosch, R. Mycobacterial virulence: Impact on immunogenicity and vaccine research. F1000Research 2019, 8, 2025. [Google Scholar] [CrossRef]
- McShane, H. Insights and challenges in tuberculosis vaccine development. Lancet Respir. Med. 2019, 7, 810–819. [Google Scholar] [CrossRef]
- WHO. WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2019; Licence: CC BY-NC-SA 3.0 IGO. [Google Scholar]
- Tousek, J. On the Clinical effectiveness of isoxyl. Antibiot. Chemother. 1970, 16, 149–155. [Google Scholar] [PubMed]
- Phetsuksiri, B.; Baulard, A.R.; Cooper, A.M.; Minnikin, D.E.; Douglas, J.D.; Besra, G.S.; Brennan, P.J. Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 1999, 43, 1042–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phetsuksiri, B.; Jackson, M.; Scherman, H.; McNeil, M.; Besra, G.S.; Baulard, A.R.; Slayden, R.A.; DeBarber, A.E.; Barry, C.E.; Baird, M.S.; et al. Unique mechanism of action of the thiourea drug isoxyl on mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 53123–53130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portevin, D.; de Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffé, M.; Guilhot, C. A Polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchèze, C.; Morbidoni, H.R.; Weisbrod, T.R.; Iwamoto, H.; Kuo, M.; Sacchettini, J.C.; Jacobs, W.R. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. [Google Scholar] [CrossRef] [Green Version]
- Barry, C.E., III; Lee, R.E.; Mdluli, K.; Sampson, A.E.; Schroeder, B.G.; Slayden, R.A.; Yuan, Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog. Lipid Res. 1998, 37, 143–179. [Google Scholar] [CrossRef] [Green Version]
- Grzegorzewicz, A.E.; Korduláková, J.; Jones, V.; Born, S.E.M.; Belardinelli, J.M.; Vaquié, A.; Gundi, V.A.K.B.; Madacki, J.; Slama, N.; Laval, F.; et al. A common mechanism of inhibition of the mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef] [Green Version]
- Dover, L.G.; Alahari, A.; Gratraud, P.; Gomes, J.M.; Bhowruth, V.; Reynolds, R.C.; Besra, G.S.; Kremer, L. EthA, a common activator of thiocarbamide-containing drugs acting on different mycobacterial targets. Antimicrob. Agents Chemother. 2007, 51, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzegorzewicz, A.E.; Eynard, N.; Quémard, A.; North, E.J.; Margolis, A.; Lindenberger, J.J.; Jones, V.; Korduláková, J.; Brennan, P.J.; Lee, R.E.; et al. Covalent modification of the mycobacterium tuberculosis FAS-II dehydratase by isoxyl and thiacetazone. ACS Infect. Dis. 2015, 1, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Korduláková, J.; Janin, Y.L.; Liav, A.; Barilone, N.; Vultos, T.D.; Rauzier, J.; Brennan, P.J.; Gicquel, B.; Jackson, M. Isoxyl activation is required for bacteriostatic activity against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 3824–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daffé, M.; Marrakchi, H. Unraveling the structure of the mycobacterial envelope. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leis, A.; Niederweis, M.; Plitzko, J.M.; Engelhardt, H. Disclosure of the mycobacterial outer membrane: Cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. USA 2008, 105, 3963–3967. [Google Scholar] [CrossRef] [Green Version]
- Zuber, B.; Chami, M.; Houssin, C.; Dubochet, J.; Griffiths, G.; Daffé, M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 2008, 190, 5672–5680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R. InhA, a gene encoding a target for isoniazid and ethionamide in mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Wang, L.; David, H.L. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1972, 2, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Belanger, A.E.; Besra, G.S.; Ford, M.E.; Mikusová, K.; Belisle, J.T.; Brennan, P.J.; Inamine, J.M. The EmbAB genes of mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. USA 1996, 93, 11919–11924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.K.B.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.M.; Korduláková, J.; et al. Inhibition of mycolic acid transport across the mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belisle, J.T.; Vissa, V.D.; Sievert, T.; Takayama, K.; Brennan, P.J.; Besra, G.S. Role of the major antigen of mycobacterium tuberculosis in cell wall biogenesis. Science 1997, 276, 1420–1422. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Tekaia, F.; Gordon, S.V.; Garnier, T.; Brosch, R.; Barrell, B.G.; Cole, S.T. Analysis of the proteome of mycobacterium tuberculosis in silico. Tuber. Lung Dis. 1999, 79, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Unge, T.; Cronin, A.; Arand, M.; Bergfors, T.; Jones, T.A.; Mowbray, S.L. Structure of an Atypical epoxide hydrolase from mycobacterium tuberculosis gives insights into its function. J. Mol. Biol. 2005, 351, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Madacki, J.; Laval, F.; Grzegorzewicz, A.; Lemassu, A.; Záhorszká, M.; Arand, M.; McNeil, M.; Daffé, M.; Jackson, M.; Lanéelle, M.-A.; et al. Impact of the epoxide hydrolase EphD on the metabolism of mycolic acids in mycobacteria. J. Biol. Chem. 2018, 293, 5172–5184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswal, B.K.; Garen, G.; Cherney, M.M.; Garen, C.; James, M.N.G. Cloning, expression, purification, crystallization and preliminary X-ray studies of epoxide hydrolases A and B from mycobacterium tuberculosis. Acta Cryst. F 2006, 62, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.C.; Henderson, S.R.; Illarionov, B.; Crosskey, T.; Southall, S.M.; Krichel, B.; Uetrecht, C.; Fischer, M.; Wilmanns, M. The crystal structure of mycobacterial epoxide hydrolase A. Sci. Rep. 2020, 10, 16539. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morisseau, C.; Goodrow, M.H.; Dowdy, D.; Zheng, J.; Greene, J.F.; Sanborn, J.R.; Hammock, B.D. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 8849–8854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswal, B.K.; Morisseau, C.; Garen, G.; Cherney, M.M.; Garen, C.; Niu, C.; Hammock, B.D.; James, M.N.G. The molecular structure of epoxide hydrolase b from mycobacterium tuberculosis and its complex with a urea-based inhibitor. J. Mol. Biol. 2008, 381, 897–912. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; North, E.J.; Hurdle, J.G.; Morisseau, C.; Scarborough, J.S.; Sun, D.; Korduláková, J.; Scherman, M.S.; Jones, V.; Grzegorzewicz, A.; et al. The structure–activity relationship of urea derivatives as anti-tuberculosis agents. Bioorganic Med. Chem. 2011, 19, 5585–5595. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.; Arand, M.; Frank, H.; Seidel, A.; Hinz, W.; Winkler, L.; Hänel, K.; Blée, E.; Beetham, J.K.; Hammock, B.D.; et al. Visualization of a covalent intermediate between microsomal epoxide hydrolase, but not cholesterol epoxide hydrolase, and their substrates. Eur. J. Biochem. 1997, 245, 490–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arand, M.; Cronin, A.; Adamska, M.; Oesch, F. Epoxide hydrolases: Structure, function, mechanism, and assay. In Phase II Conjugation Enzymes and Transport Systems; Sies, H., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2005; Volume 400, pp. 569–588. ISBN 978-0-12-182805-9. [Google Scholar]
- Carrère-Kremer, S.; Blaise, M.; Singh, V.K.; Alibaud, L.; Tuaillon, E.; Halloum, I.; van de Weerd, R.; Guérardel, Y.; Drancourt, M.; Takiff, H.; et al. A New dehydratase conferring innate resistance to thiacetazone and intra-amoebal survival of mycobacterium smegmatis. Mol. Microbiol. 2015, 96, 1085–1102. [Google Scholar] [CrossRef] [PubMed]
- Triccas, J.A.; Parish, T.; Britton, W.J.; Gicquel, B. An inducible expression system permitting the efficient purification of a recombinant antigen from mycobacterium smegmatis. FEMS Microbiol. Lett. 1998, 167, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, B.; Kingma, J.; Arand, M.; Wubbolts, M.G.; Janssen, D.B. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl. Environ. Microbiol. 2006, 72, 2905–2917. [Google Scholar] [CrossRef] [Green Version]
- Gomez, G.A.; Morisseau, C.; Hammock, B.D.; Christianson, D.W. Human soluble epoxide hydrolase: Structural basis of inhibition by 4-(3-Cyclohexylureido)-carboxylic acids. Protein Sci. 2006, 15, 58–64. [Google Scholar] [CrossRef]
- Argiriadi, M.A.; Morisseau, C.; Hammock, B.D.; Christianson, D.W. Detoxification of environmental mutagens and carcinogens: Structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. USA 1999, 96, 10637–10642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argiriadi, M.A.; Morisseau, C.; Goodrow, M.H.; Dowdy, D.L.; Hammock, B.D.; Christianson, D.W. Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J. Biol. Chem. 2000, 275, 15265–15270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive essentiality analysis of the mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 2017, 8, e02133-16. [Google Scholar] [CrossRef] [Green Version]
- Rengarajan, J.; Bloom, B.R.; Rubin, E.J. Genome-wide requirements for mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 8327–8332. [Google Scholar] [CrossRef] [Green Version]
- Dao, D.N.; Sweeney, K.; Hsu, T.; Gurcha, S.S.; Nascimento, I.P.; Roshevsky, D.; Besra, G.S.; Chan, J.; Porcelli, S.A.; Jacobs, W.R. Mycolic acid modification by the MmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog. 2008, 4, e1000081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubnau, E.; Chan, J.; Raynaud, C.; Mohan, V.P.; Lanéelle, M.-A.; Yu, K.; Quémard, A.; Smith, I.; Daffé, M. Oxygenated mycolic acids are necessary for virulence of mycobacterium tuberculosis in mice. Mol. Microbiol. 2000, 36, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Bhowruth, V.; Brown, A.K.; Reynolds, R.C.; Coxon, G.D.; Mackay, S.P.; Minnikin, D.E.; Besra, G.S. Symmetrical and unsymmetrical analogues of isoxyl; active agents against mycobacterium tuberculosis. Bioorganic Med. Chem. Lett. 2006, 16, 4743–4747. [Google Scholar] [CrossRef] [PubMed]
- Coxon, G.D.; Craig, D.; Corrales, R.M.; Vialla, E.; Gannoun-Zaki, L.; Kremer, L. Synthesis, antitubercular activity and mechanism of resistance of highly effective thiacetazone analogues. PLoS ONE 2013, 8, e53162. [Google Scholar] [CrossRef] [Green Version]
- North, E.J.; Scherman, M.S.; Bruhn, D.F.; Scarborough, J.S.; Maddox, M.M.; Jones, V.; Grzegorzewicz, A.; Yang, L.; Hess, T.; Morisseau, C.; et al. Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorganic Med. Chem. 2013, 21, 2587–2599. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structures of isoxyl (ISO), thiacetazone (TAC) and AU1235.

Figure 2.
Epoxide hydrolase activity of EphA, EphB, EphD, EphE, EphF and EphG proteins in vitro in the presence of 10 µg/mL of ISO. Left: Thin-layer chromatography of in vitro reactions with 10,000× g supernatants of lysates of E. coli strains producing EHs from M. tuberculosis using [14C]-9,10-cis-epoxystearic acid as the substrate. Reactions ran for 5, 10, 30, 60 or 120 min at 37 °C. Plates were developed in n-hexane:diethyl ether:formic acid (70:30:2) and visualized by autoradiography. Right: Quantification of results of thin-layer chromatography (TLC) analysis. For each condition, the EH activity was expressed as percentage of substrate conversion.
Figure 2.
Epoxide hydrolase activity of EphA, EphB, EphD, EphE, EphF and EphG proteins in vitro in the presence of 10 µg/mL of ISO. Left: Thin-layer chromatography of in vitro reactions with 10,000× g supernatants of lysates of E. coli strains producing EHs from M. tuberculosis using [14C]-9,10-cis-epoxystearic acid as the substrate. Reactions ran for 5, 10, 30, 60 or 120 min at 37 °C. Plates were developed in n-hexane:diethyl ether:formic acid (70:30:2) and visualized by autoradiography. Right: Quantification of results of thin-layer chromatography (TLC) analysis. For each condition, the EH activity was expressed as percentage of substrate conversion.

Figure 3.
In vitro EH activity of EphD (a) and EphF (b) in the case of preincubation with 10 µg/mL of isoxyl (thiocarlide; 4,4′-diisoamyloxydiphenylthiourea) (ISO). Bar graph representing average percentages of substrate conversion from two experiments (error bars showing standard deviation). Filled bars represent reactions where ISO (or DMSO in control reactions) was added directly with other reaction components, empty bars represent reactions where the enzyme was incubated with ISO for 15 min prior to adding the substrate. Numbers next to bars represent average percentage of inhibition per reaction. The lower part of the figure shows one representative chromatogram from the TLC analysis of these reaction products. The presence (+) or absence (–) of ISO and EthA is indicated for each reaction.
Figure 3.
In vitro EH activity of EphD (a) and EphF (b) in the case of preincubation with 10 µg/mL of isoxyl (thiocarlide; 4,4′-diisoamyloxydiphenylthiourea) (ISO). Bar graph representing average percentages of substrate conversion from two experiments (error bars showing standard deviation). Filled bars represent reactions where ISO (or DMSO in control reactions) was added directly with other reaction components, empty bars represent reactions where the enzyme was incubated with ISO for 15 min prior to adding the substrate. Numbers next to bars represent average percentage of inhibition per reaction. The lower part of the figure shows one representative chromatogram from the TLC analysis of these reaction products. The presence (+) or absence (–) of ISO and EthA is indicated for each reaction.

Figure 4.
Thin-layer chromatography of fatty and mycolic acid methyl esters from strains M. smegmatis mc2155 pHAM, pHAM-ephD and pHAM-msmeg_4280 treated with isoniazid (INH), ISO, thiacetazone (TAC) or AU1235. Plates were developed in n-hexane:ethyl acetate (95:5, three runs) and visualized with 10% cupric sulfate in 8% phosphoric acid and charring. FAMEs—fatty acid methyl esters; α, αʹ, epoxy and dicarboxy represent different forms of mycolic acid methyl esters (MAMEs).
Figure 4.
Thin-layer chromatography of fatty and mycolic acid methyl esters from strains M. smegmatis mc2155 pHAM, pHAM-ephD and pHAM-msmeg_4280 treated with isoniazid (INH), ISO, thiacetazone (TAC) or AU1235. Plates were developed in n-hexane:ethyl acetate (95:5, three runs) and visualized with 10% cupric sulfate in 8% phosphoric acid and charring. FAMEs—fatty acid methyl esters; α, αʹ, epoxy and dicarboxy represent different forms of mycolic acid methyl esters (MAMEs).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Madacki, J.; Kopál, M.; Jackson, M.; Korduláková, J. Mycobacterial Epoxide Hydrolase EphD Is Inhibited by Urea and Thiourea Derivatives. Int. J. Mol. Sci. 2021, 22, 2884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062884
AMA Style
Madacki J, Kopál M, Jackson M, Korduláková J. Mycobacterial Epoxide Hydrolase EphD Is Inhibited by Urea and Thiourea Derivatives. International Journal of Molecular Sciences. 2021; 22(6):2884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062884
Chicago/Turabian StyleMadacki, Jan, Martin Kopál, Mary Jackson, and Jana Korduláková. 2021. "Mycobacterial Epoxide Hydrolase EphD Is Inhibited by Urea and Thiourea Derivatives" International Journal of Molecular Sciences 22, no. 6: 2884. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062884
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.