
Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence

 ,
, 
 and
and
Abstract
:1. Introduction
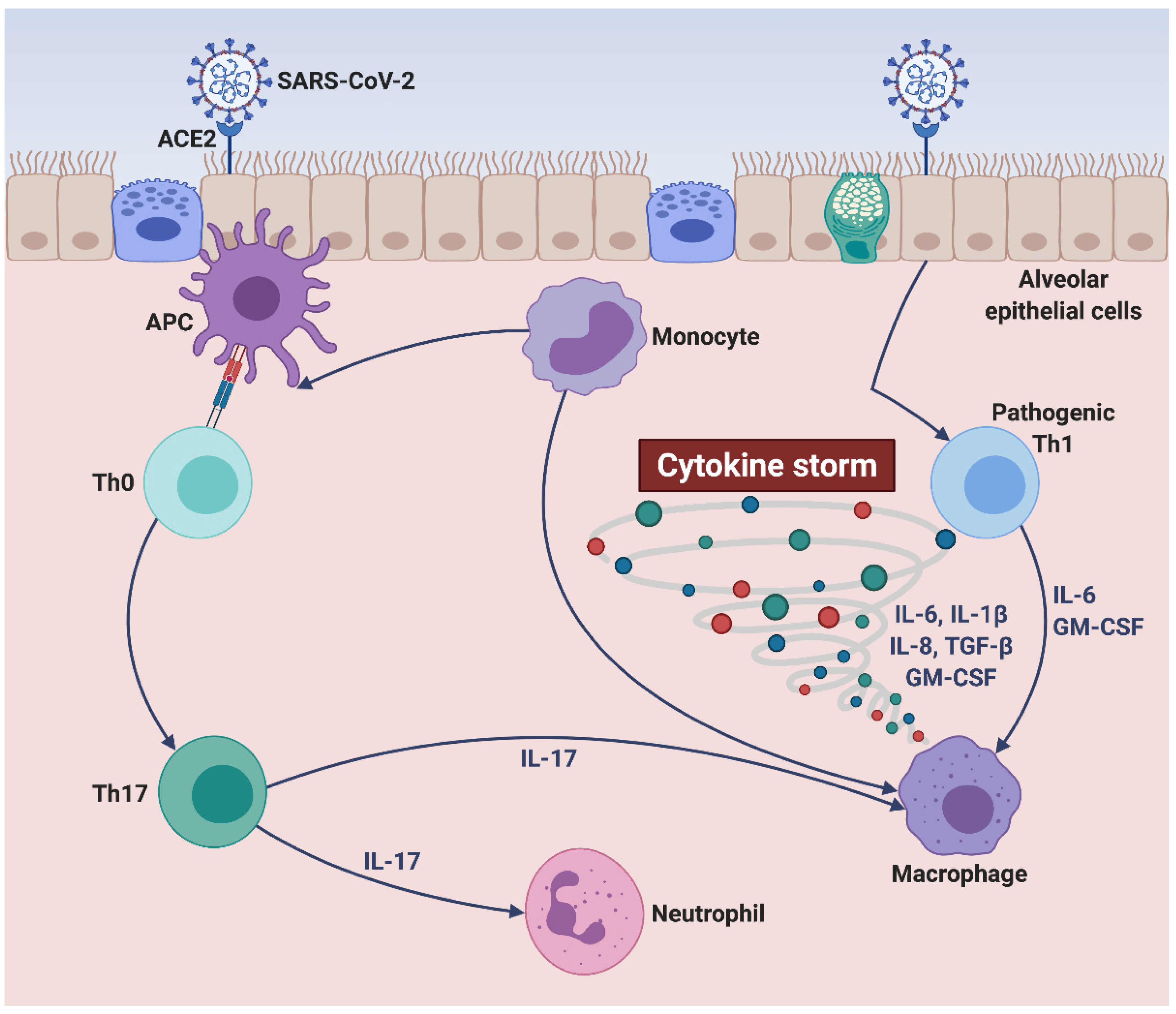
2. Cellular and Molecular Pathophysiology of SARS-CoV-2-Driven Cytokine Storm
2.1. Pathogenic Mechanisms Underlying SARS-CoV-2 Infection
2.2. Innate and Adaptive Immune Responses Triggered by SARS-CoV-2
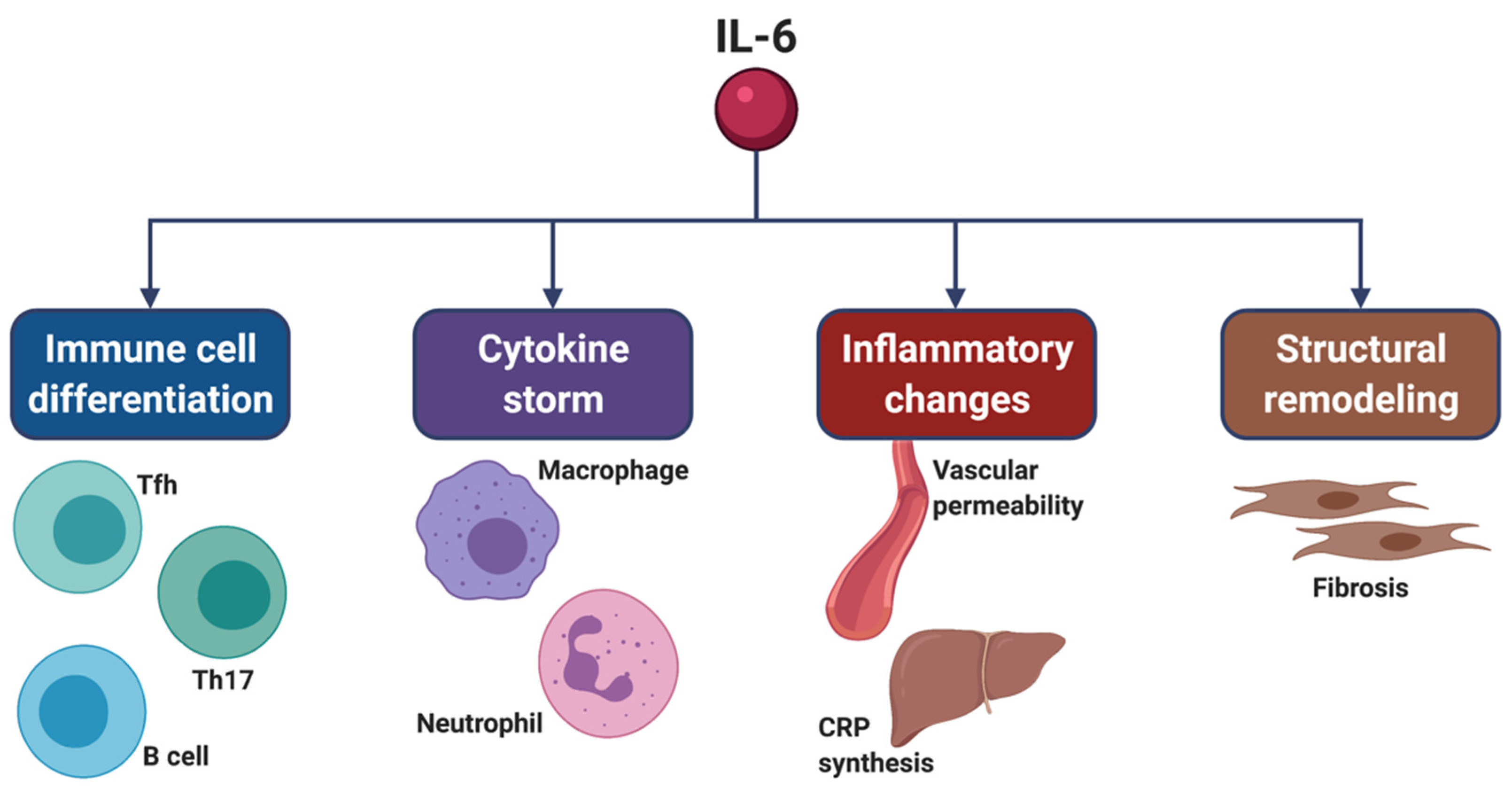
2.3. Role of IL-6 in SARS-CoV-2-Induced Cytokine Storm
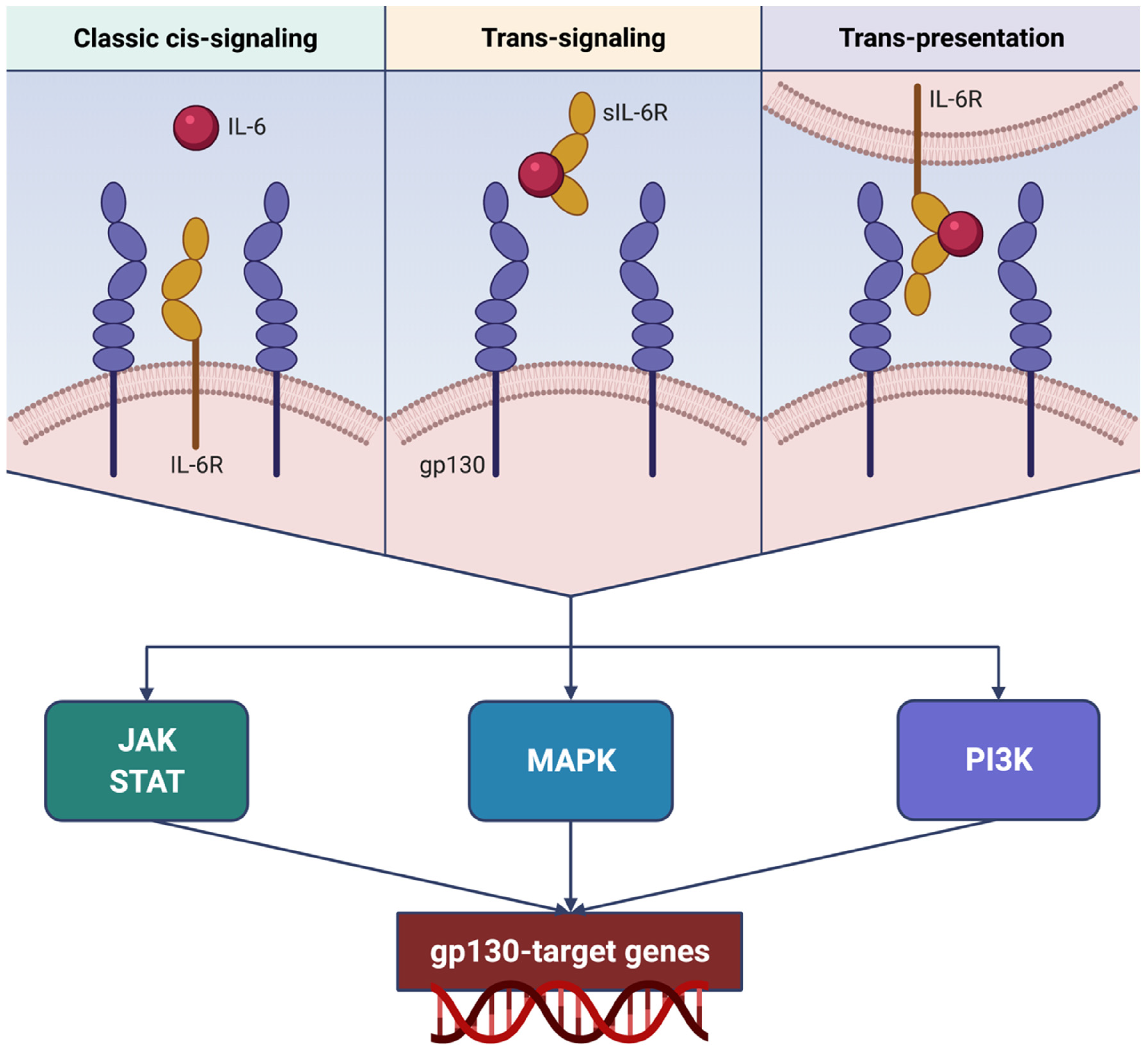
3. Molecular Mechanisms Underlying IL-6 Bioactivities
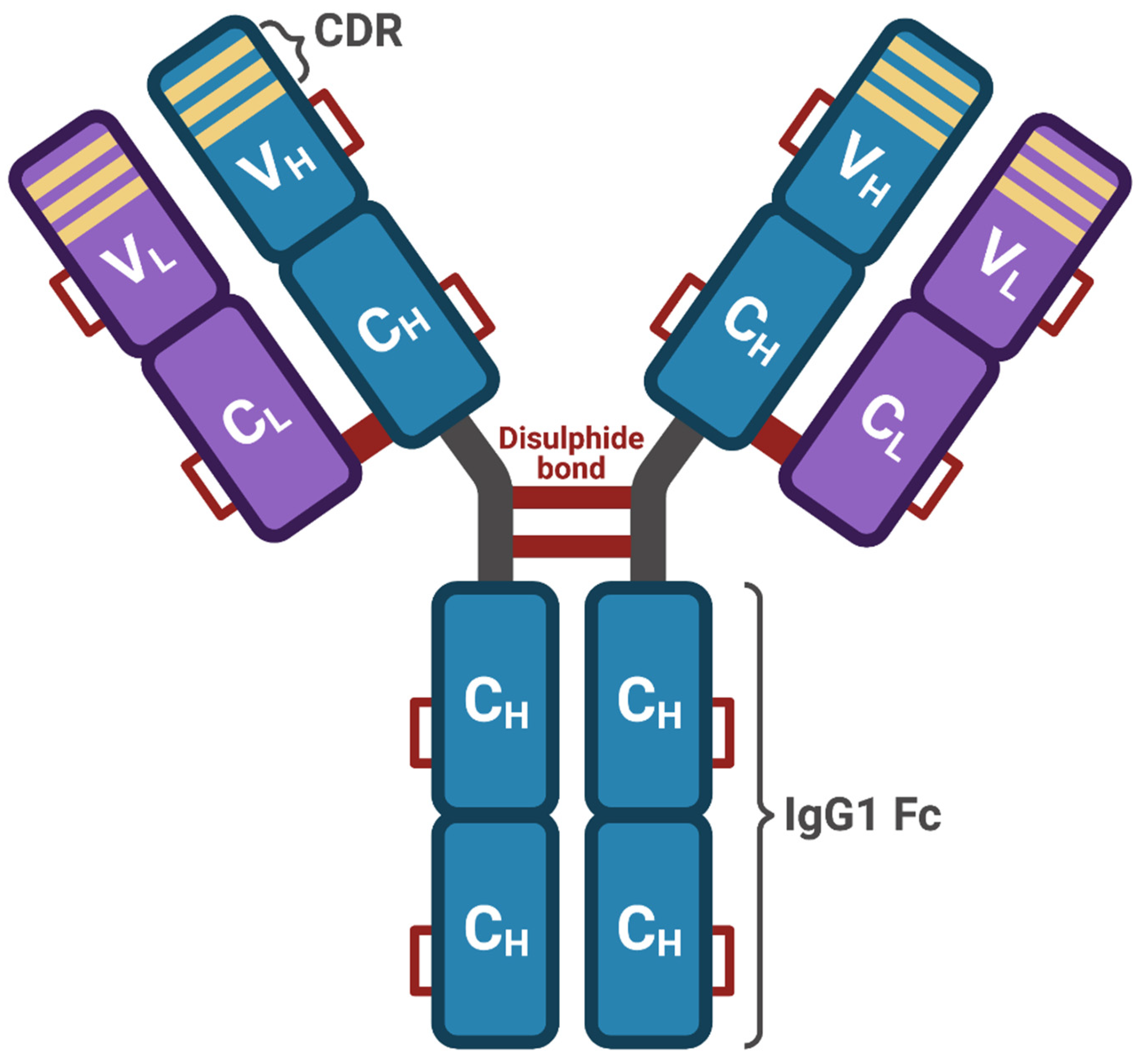
4. Tocilizumab: Mechanism of Action and COVID-19 Clinical Trials
4.1. Clinical Studies Supporting the Use of Tocilizumab in COVID-19 Patients
4.2. Clinical Studies Questioning the Use of Tocilizumab in COVID-19 Patients
4.3. Most Recent Studies Evaluating the Use of Tocilizumab in COVID-19 Patients
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| ADAM17 | a disintegrin and metalloproteinase domain-containing protein 17 |
| ARDS | acute respiratory distress syndrome |
| COVID-19 | coronavirus disease 2019 |
| CRP | C-reactive protein |
| CYP | cytochrome P450 enzyme |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| gp130 | glycoprotein 130 |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IL-6R | interleukin-6 receptor |
| IL-8 | interleukin-8 |
| IL-17 | interleukin-17 |
| IP-10 | interferon γ-induced protein 10 |
| JAK | Janus kinase |
| MAPK | mitogen-activated protein kinase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MIP-1α | macrophage inflammatory protein-1α |
| MERS | Middle East respiratory syndrome |
| MHC | major histocompatibility complex |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor family, pyrin do- main containing 3 activation |
| PI3K | phosphoinositide 3-kinase |
| RdRp | RNA-dependent RNA polymerase |
| SARS | severe acute respiratory syndrome |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| S protein | spike glycoprotein |
| STAT | signal transducer and activator of transcription |
| Th1 | T helper 1 |
| Th17 | T helper 17 |
| Tfh | T follicular helper |
| TGF-β | transforming growth factor-β |
| TLR7 | toll-like receptor-7 |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Pelaia, C.; Tinello, C.; Vatrella, A.; De Sarro, G.; Pelaia, G. Lung under attack by COVID-19-induced cytokine storm: Pathogenic mechanisms and therapeutic implications. Ther. Adv. Respir. Dis. 2020, 14, 1753466620933508. [Google Scholar] [CrossRef] [PubMed]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Lagniton, P.N.; Ye, S.; Li, E.; Xu, R.H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Ahmadpoor, P.; Rostaing, L. Why the immune system fails to mount an adaptive immune response to a Covid-19 infection. Transpl. Int. 2020, 33, 824–825. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef]
- Procopio, G.; Cancelliere, A.; Trecarichi, E.M.; Mazzitelli, M.; Arrighi, E.; Perri, G.; Serapide, F.; Pelaia, C.; Lio, E.; Busceti, M.T.; et al. Oxygen therapy via high flow nasal cannula in severe respiratory failure caused by SARS-CoV-2 infection: A real-life observational study. Ther. Adv. Respir. Dis. 2020, 14, 1753466620963016. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 cytokine storm; what we know so far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Hill, T.; Li, K.; Peters, C.J.; Tseng, C.T. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J. Virol. 2009, 83, 3039–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The role of IL-6 during viral infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, J.; Zhan, Y.; Wu, L.; Yu, X.; Zhang, W.; Ye, L.; Xu, S.; Sun, R.; Wang, Y.; et al. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect. Immun. 2004, 72, 4410–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, V.; Dhillon, S.; Plosker, G.L. Tocilizumab: A review of its use in the management of rheumatoid arthritis. Drugs 2009, 69, 609–632. [Google Scholar] [CrossRef]
- Lan, S.H.; Lai, C.C.; Huang, H.T.; Chang, S.P.; Lu, L.C.; Hsueh, P.R. Tocilizumab for severe COVID-19: A systematic review. Int. J. Antimicrob. Agents 2020, 56, 106103. [Google Scholar] [CrossRef] [PubMed]
- Okoh, A.; Bishburg, E.; Grinberg, S.; Nagarakanti, S. Tocilizumab use in COVID-19-associated pneumonia. J. Med. Virol. 2021, 93, 1023–1028. [Google Scholar] [CrossRef]
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak–an update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically-proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. BioRxiv 2020, 202, 756–759. [Google Scholar]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.Y.; Yuen, K.S.; Ye, Z.W.; Chan, C.P.; Jin, D.Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defense: Lessons from other pathogenic viruses. Emerg. Microbes. Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Hossain, M.F.; Abdulhakim, J.A.; Alam, M.A.; Ashraf, G.M.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Aleya, L. nCOVID-19 pandemic: From molecular pathogenesis to potential investigational therapeutics. Front. Cell Dev. Biol. 2020, 8, 616. [Google Scholar] [CrossRef]
- Chen, R.F.; Chang, J.C.; Yeh, W.T.; Lee, C.H.; Liu, J.W.; Eng, H.L.; Yang, K.D. Role of vascular cell adhesion molecules and leukocyte apoptosis in the lymphopenia and thrombocytopenia of patients with severe acute respiratory syndrome (SARS). Microbes Infect. 2006, 8, 122–127. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Subbarao, K. The immunobiology of SARS. Annu. Rev. Immunol. 2007, 25, 443–472. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Khaitan, A.; Unutmaz, D. Revisiting immune exhaustion during HIV infection. Curr. HIV/AIDS Rep. 2011, 8, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Toor, S.M.; Saleh, R.; Nair, V.S.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Tartaro, D.L.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- Orlov, M.; Wander, P.L.; Morrell, E.D.; Mikacenic, C.; Wurfel, M.M. A case for targeting Th17 cells and IL-17A in SARS-CoV-2 infections. J. Immunol. 2020, 205, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Chen, L.; Liu, H.G.; Liu, W.; Liu, J.; Liu, K.; Shang, J.; Deng, Y.; Wei, S. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, E005. [Google Scholar]
- Okabayashi, T.; Kariwa, H.; Yokota, S.I.; Iki, S.; Indoh, T.; Yokosawa, N.; Takashima, I.; Tsutsumi, H.; Fujii, N. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 2006, 78, 417–424. [Google Scholar] [CrossRef]
- Wu, D.; Yang, X.O. TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor fedratinib. J. Microbiol. Immunol. Infect. 2020, 53, 368–370. [Google Scholar] [CrossRef]
- Kishimoto, T. IL6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6 designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepathol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-J.; Zhang, L.-N.; Hou, H.; Xu, L.; Ji, K. Interleukin-6 signaling blockade treatment for cytokine release syndrome in COVID-19. Exp. Ther. Med. 2021, 21, 24. [Google Scholar] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Hermanns, H.M.; Schaper, F. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Düsterhöft, S.; Zhu, Y.; Grötzinger, J.; et al. Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, P.; Hansen, S.; Waetzig, G.H.; Akbarzadeh, M.; Lamertz, L.; Huber, H.J.; Ahmadian, M.R.; Moll, J.M.; Scheller, J. The balance of interleukin (IL)-6, IL-6.soluble IL-6 receptor (sIL-6R), and IL-6.sIL-6R.sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 2018, 293, 6762–6775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, J.; Zhou, C.; Wu, Z.; Zhong, S.; Liu, J.; Luo, W.; Chen, T.; Qin, Q.; Deng, P. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2005, 171, 850–857. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. RespiR. Res. 2020, 21, 56. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Therapeutic targeting of the interleukin-6 receptor. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 199–219. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.W.; Zhao, H.; Wang, G.Q. Cytokine release syndrome in severe COVID-19: Interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef]
- Navarro, G.; Taroumian, S.; Barroso, N.; Duan, L.; Furst, D. Tocilizumab in rheumatoid arthritis: A meta-analysis of efficacy and selected clinical conundrums. Semin. Arthritis Rheum. 2014, 43, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, M.; Laskou, F.; Stapleton, P.P.; Hadavi, S.; Dasgupta, B. Tocilizumab (Actemra). Hum. Vaccin. Immunother. 2017, 13, 1972–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J. Tocilizumab: A review in rheumatoid arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Rios-Fernández, R.; Callejas-Rubio, J.L.; García-Rodríguez, S.; Sancho, J.; Zubiaur, M.; Ortego-Centeno, N. Tocilizumab as an adjuvant therapy for hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. Am. J. Ther. 2016, 23, e1193–e1196. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Perrone, F.; Piccirillo, M.C.; Ascierto, P.A.; Salvarani, C.; Parrella, R.; Marata, A.M.; Popoli, P.; Ferraris, L.; Marrocco-Trischitta, M.M.; Ripamonti, D.; et al. Tocilizumab for patients with COVID-19 pneumonia. The single-arm TOCIVID-19 prospective trial. J. Transl. Med. 2020, 18, 405. [Google Scholar] [CrossRef]
- Price, C.C.; Altice, F.L.; Shyr, Y.; Koff, A.; Pischel, L.; Goshua, G.; Azar, M.M.; Mcmanus, D.; Chen, S.C.; Gleeson, S.E.; et al. Tocilizumab treatment for cytokine release syndrome in hospitalized patients with coronavirus disease 2019: Survival and clinical outcomes. Chest 2020, 158, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Toniati, P.; Piva, S.; Cattalini, M.; Garrafa, E.; Regola, F.; Castelli, F.; Franceschini, F.; Focà, E.; Andreoli, L.; Latronico, N.; et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun. Rev. 2020, 19, 102568. [Google Scholar] [CrossRef]
- Eimer, J.; Vesterbacka, J.; Svensson, A.K.; Stojanovic, B.; Wagrell, C.; Sönnerborg, A.; Nowak, P. Tocilizumab shortens time on mechanical ventilation and length of hospital stay in patients with severe COVID-19: A retrospective cohort study. J. Intern. Med. 2020, in press. [Google Scholar] [CrossRef]
- Potere, N.; Di Nisio, M.; Cibelli, D.; Scurti, R.; Frattari, A.; Porreca, E.; Abbate, A.; Parruti, G. Interleukin-6 receptor blockade with subcutaneous tocilizumab in severe COVID-19 pneumonia and hyperinflammation: A case-control study. Ann. Rheum. Dis. 2021, 80, 271–272. [Google Scholar] [CrossRef]
- Berardicurti, O.; Ruscitti, P.; Ursini, F.; D’Andrea, S.; Ciaffi, J.; Meliconi, R.; Iagnocco, A.; Cipriani, P.; Giacomelli, R. Mortality in tocilizumab-treated patients with COVID-19: A systematic review and meta-analysis. Clin. Exp. Rheumatol. 2020, 38, 1247–1254. [Google Scholar]
- Gupta, S.; Wang, W.; Hayek, S.S.; Chan, L.; Mathews, K.S.; Melamed, M.L.; Brenner, S.K.; Leonberg-Yoo, A.; Schenck, E.J.; Radbel, J.; et al. Association between early treatment with tocilizumab and mortality among critically ill patients with COVID-19. JAMA Intern. Med. 2021, 181, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hermine, O.; Mariette, X.; Tharaux, P.L.; Resche-Rigon, M.; Porcher, R.; Ravaud, P.; Bureau, S.; Dougados, M.; Tibi, A.; Azoulay, E.; et al. Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: A randomized clinical trial. JAMA Intern. Med. 2021, 181, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, C.; Della-Torre, E.; Cavalli, G.; De Luca, G.; Ripa, M.; Boffini, N.; Tomelleri, A.; Baldissera, E.; Rovere-Querini, P.; Ruggeri, A.; et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: A single-centre retrospective cohort study. Eur. J. Intern. Med. 2020, 76, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Ippolito, M.; Greco, M.; Granone, V.; Protti, A.; Gregoretti, C.; Giarratano, A.; Einav, S.; Cecconi, M. Rationale and evidence on the use of tocilizumab in COVID-19: A systematic review. Pulmonology 2021, 27, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N. Engl. J. Med. 2021, 383, 2333–2344. [Google Scholar] [CrossRef]
- Salama, C.; Han, J.; Yau, L.; Reiss, W.G.; Kramer, B.; Neidhart, J.D.; Criner, G.J.; Kaplan-Lewis, E.; Baden, R.; Pandit, L.; et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N. Engl. J. Med. 2021, 384, 20–30. [Google Scholar] [CrossRef]
- The REMAP-CAP Investigators. Interleukin-6 receptor antagonists in critically ill patients with Covid-19 pneumonia. N. Engl. J. Med. 2021, in press. [Google Scholar]
- Rosas, I.O.; Bräu, N.; Waters, M.; Go, R.C.; Hunter, B.D.; Bhagani, S.; Skiest, D.; Aziz, M.S.; Cooper, N.; Douglas, I.S.; et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N. Engl. J. Med. 2021, in press. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Shen, A.; Chen, Y.; Qi, Z. Rational use of tocilizumab in the treatment of novel coronavirus pneumonia. Clin. Drug Invest. 2020, 40, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Bennardo, F.; Buffone, C.; Giudice, A. New therapeutic opportunities for COVID-19 patients with tocilizumab: Possible correlation of interleukin-6 receptor inhibitors with osteonecrosis of the jaws. Oral Oncol. 2020, 106, 104659. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Rivas, M.; Ronda, M.; Padulles, A.; Mitjavila, F.; Riera-Mestre, A.; García-Forero, C.; Iriarte, A.; Mora, J.M.; Padulles, N.; Gonzalez, M.; et al. Beneficial effect of corticosteroids in preventing mortality in patients receiving tocilizumab to treat severe COVID-19 illness. Int. J. Infect. Dis. 2020, 101, 290–297. [Google Scholar] [CrossRef]
- Pelaia, G.; Vatrella, A.; Cuda, G.; Maselli, R.; Marsico, S.A. Molecular mechanisms of corticosteroid actions in chronic inflammatory airway diseases. Life Sci. 2003, 72, 1549–1561. [Google Scholar] [CrossRef]
- Rubin, E.J.; Longo, D.L.; Baden, L.R. Interleukin-6 receptor inhibition in Covid-19–Cooling the inflammatory soup. N. Engl. J. Med. 2021, in press. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Dosage | No. Pt. | Main Results |
|---|---|---|---|
| Xu et al. [66] | 4–8 mg/kg | 21 | Improvements of clinical, laboratory, and radiologic parameters |
| Perrone et al. [TOCIVID-19] [67] | 8 mg/kg | 301 | Decrease of 30-day mortality |
| Price et al. [68] | 8 mg/kg | 239 | Decrease of inflammatory biomarkers and survival prolongation |
| Toniati et al. [69] | 8 mg/kg | 100 | Quick and prolonged clinical improvement |
| Eimer et al. [70] | 8 mg/kg | 29 | Shortening of hospital stay |
| Potere et al. [71] | 324 mg | / | Decrease of mortality |
| Gupta et al. [73] | 8 mg/kg | 3924 | Decrease of death risk |
| Hermine et al. [74] | 8 mg/kg | 64 | No change in 28-day mortality |
| Okoh et al. [75] | 8 mg/kg | 77 | No change in overall survival |
| Campochiaro et al. [76] | 400 mg | 65 | No change in 28-day mortality |
| Stone et al. [79] | 8 mg/kg | 243 | No change in intubation risk and day 14 mortality |
| Salama et al. [80] | 8 mg/kg | 389 | No change in overall survival |
| Gordon et al. [REMAP-CAP] [81] | 8 mg/kg | 353 | Prolongation of 90-day survival |
| Rosas et al. [COVACTA] [82] | 8 mg/kg | 294 | No change in day 28 mortality |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelaia, C.; Calabrese, C.; Garofalo, E.; Bruni, A.; Vatrella, A.; Pelaia, G. Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence. Int. J. Mol. Sci. 2021, 22, 3059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063059
Pelaia C, Calabrese C, Garofalo E, Bruni A, Vatrella A, Pelaia G. Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence. International Journal of Molecular Sciences. 2021; 22(6):3059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063059
Chicago/Turabian StylePelaia, Corrado, Cecilia Calabrese, Eugenio Garofalo, Andrea Bruni, Alessandro Vatrella, and Girolamo Pelaia. 2021. "Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence" International Journal of Molecular Sciences 22, no. 6: 3059. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063059