
Evaluation of a Novel Plasmid for Simultaneous Gene Electrotransfer-Mediated Silencing of CD105 and CD146 in Combination with Irradiation

 ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
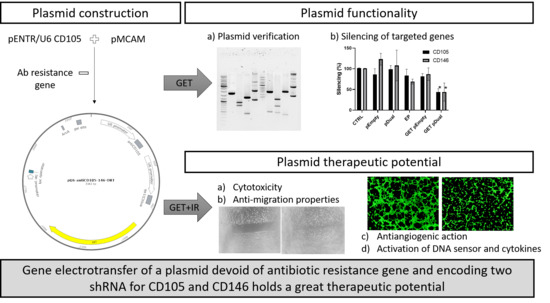
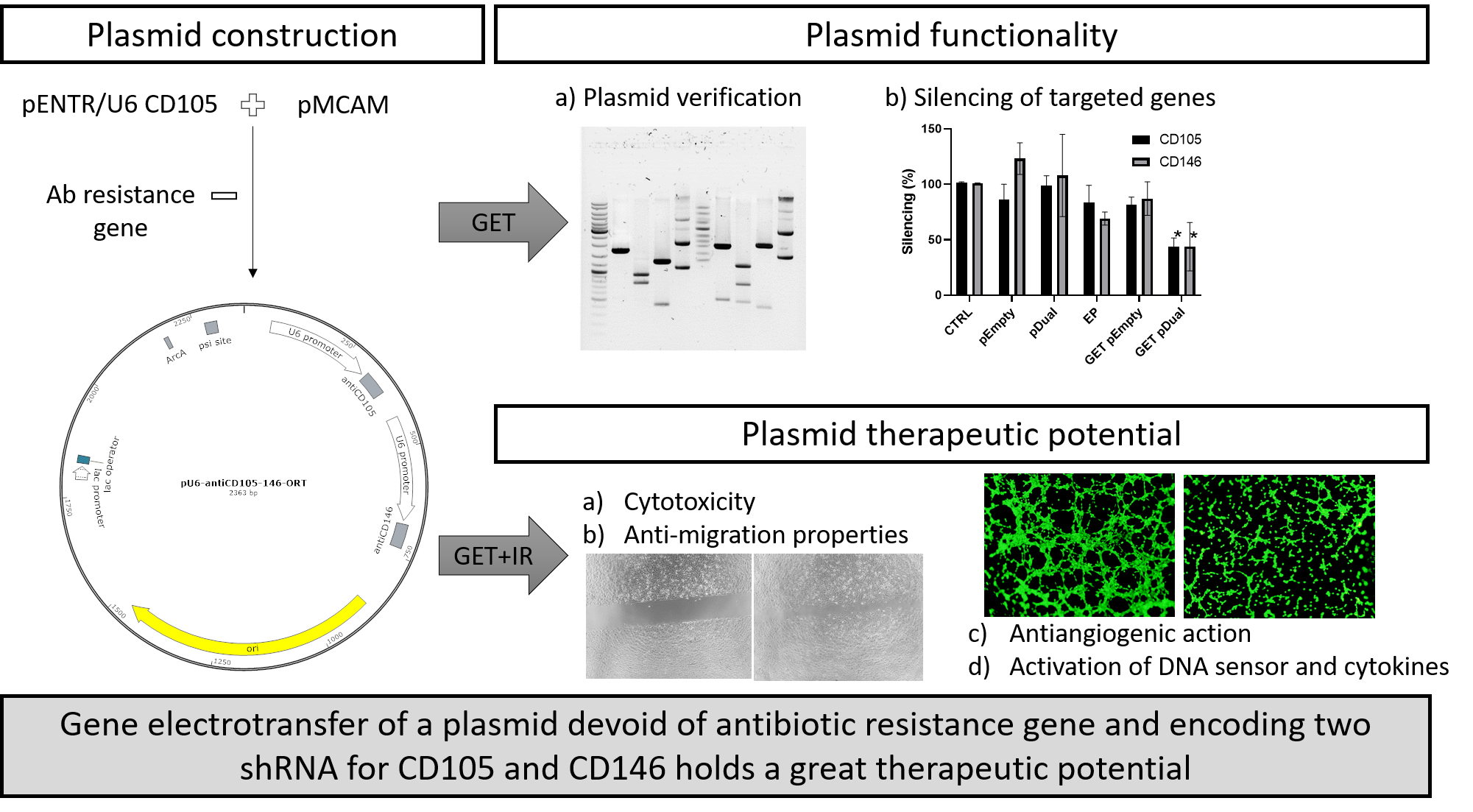
2. Results
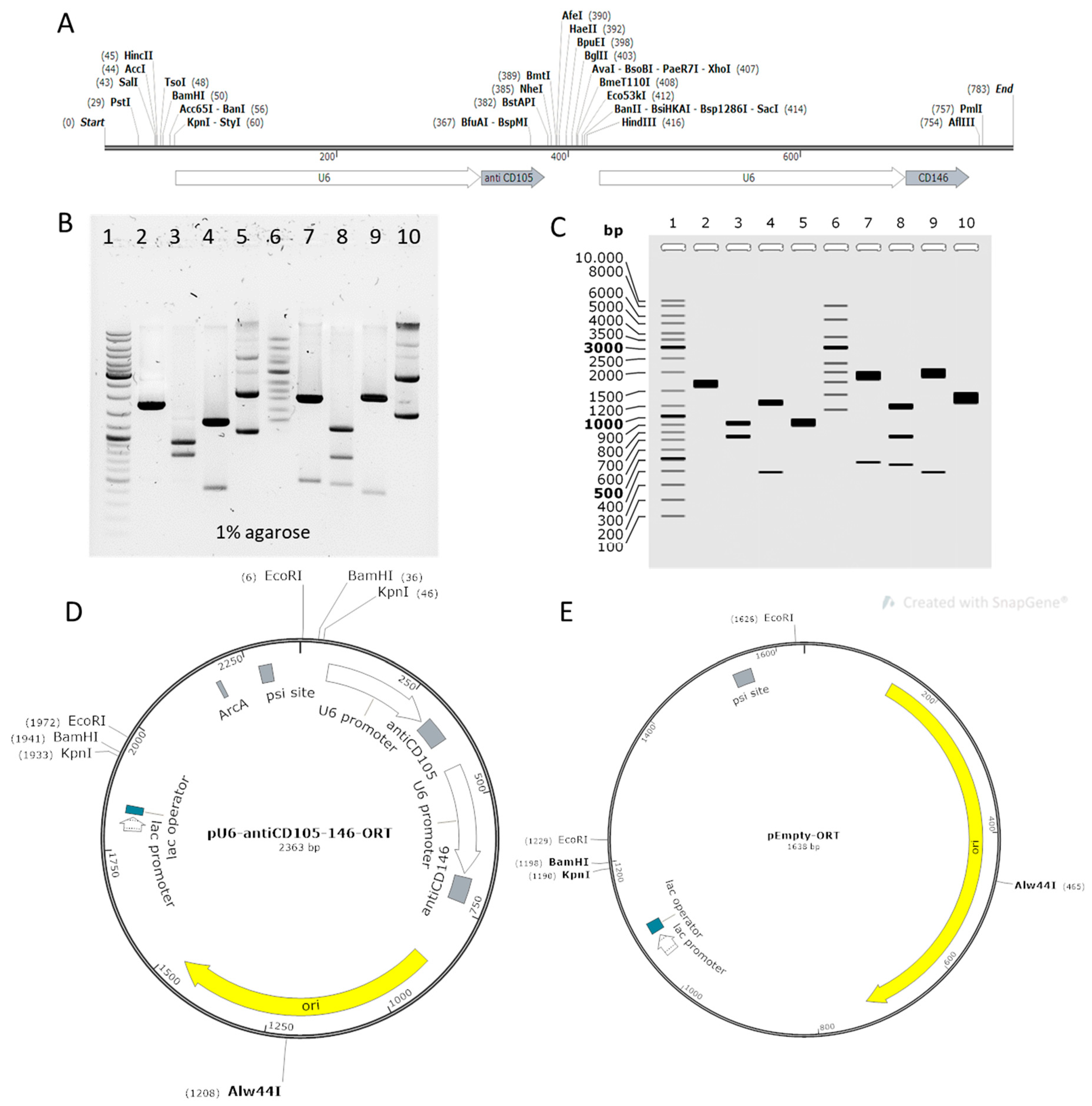
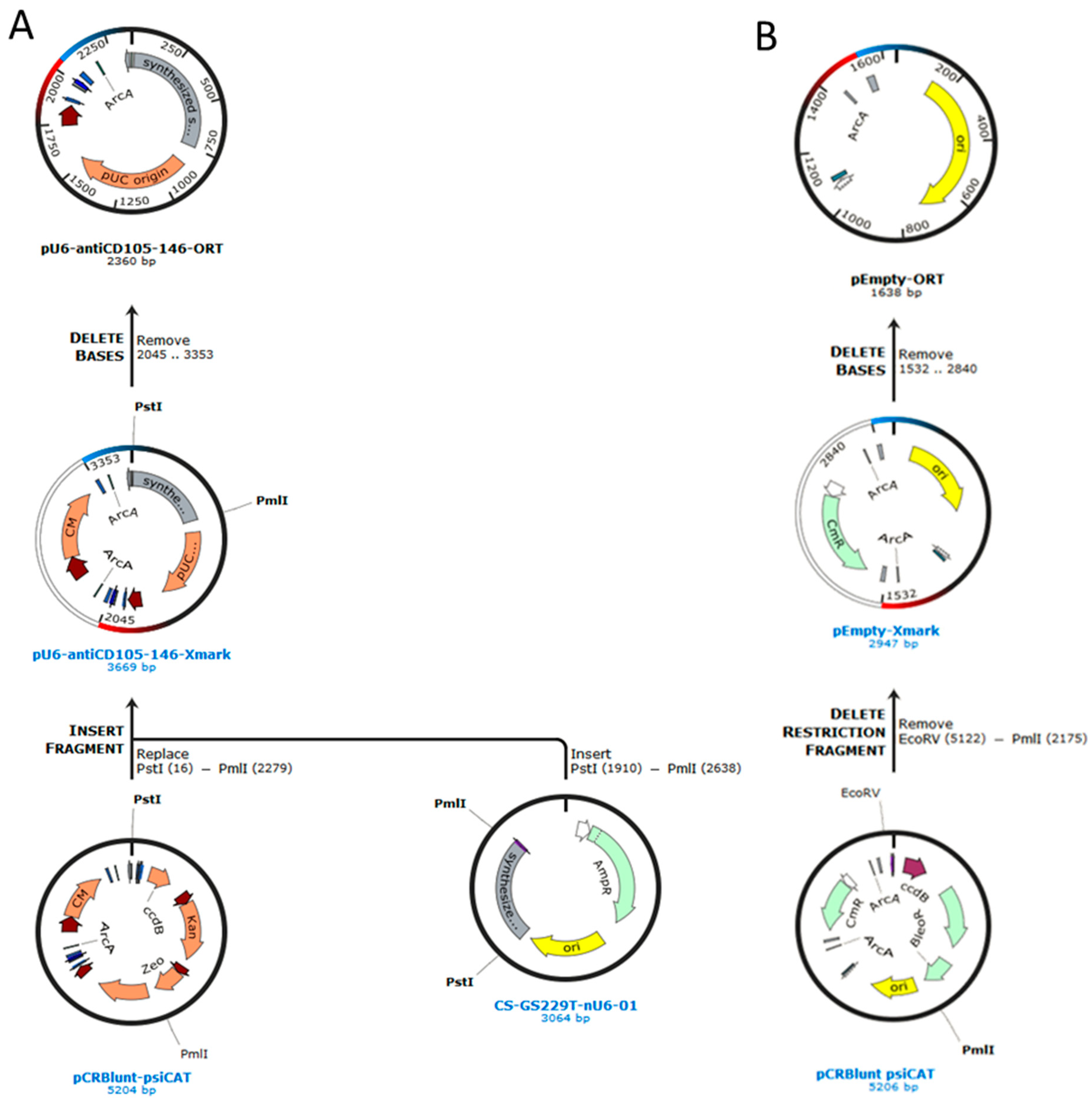
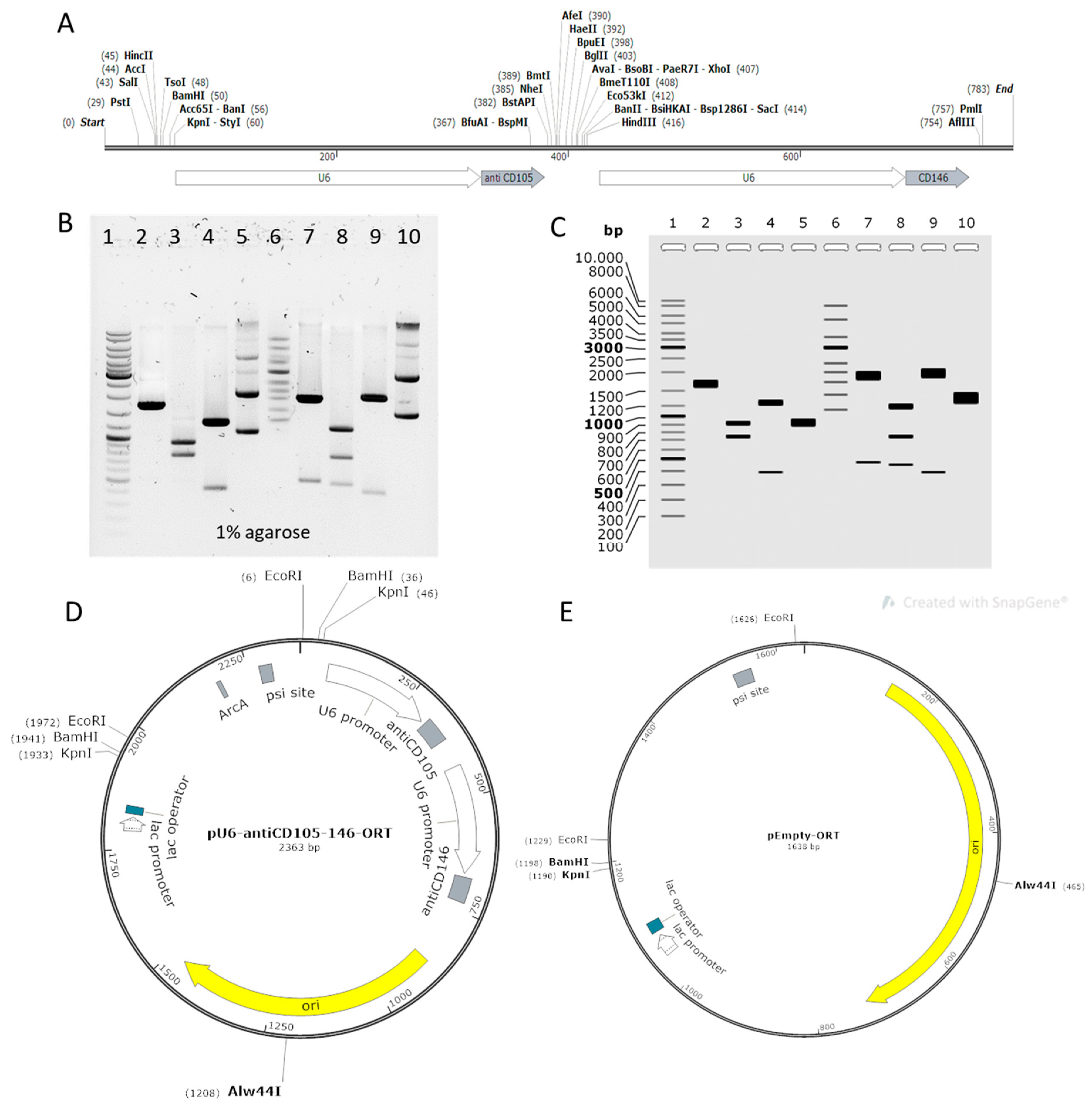
2.1. Construction of Plasmids
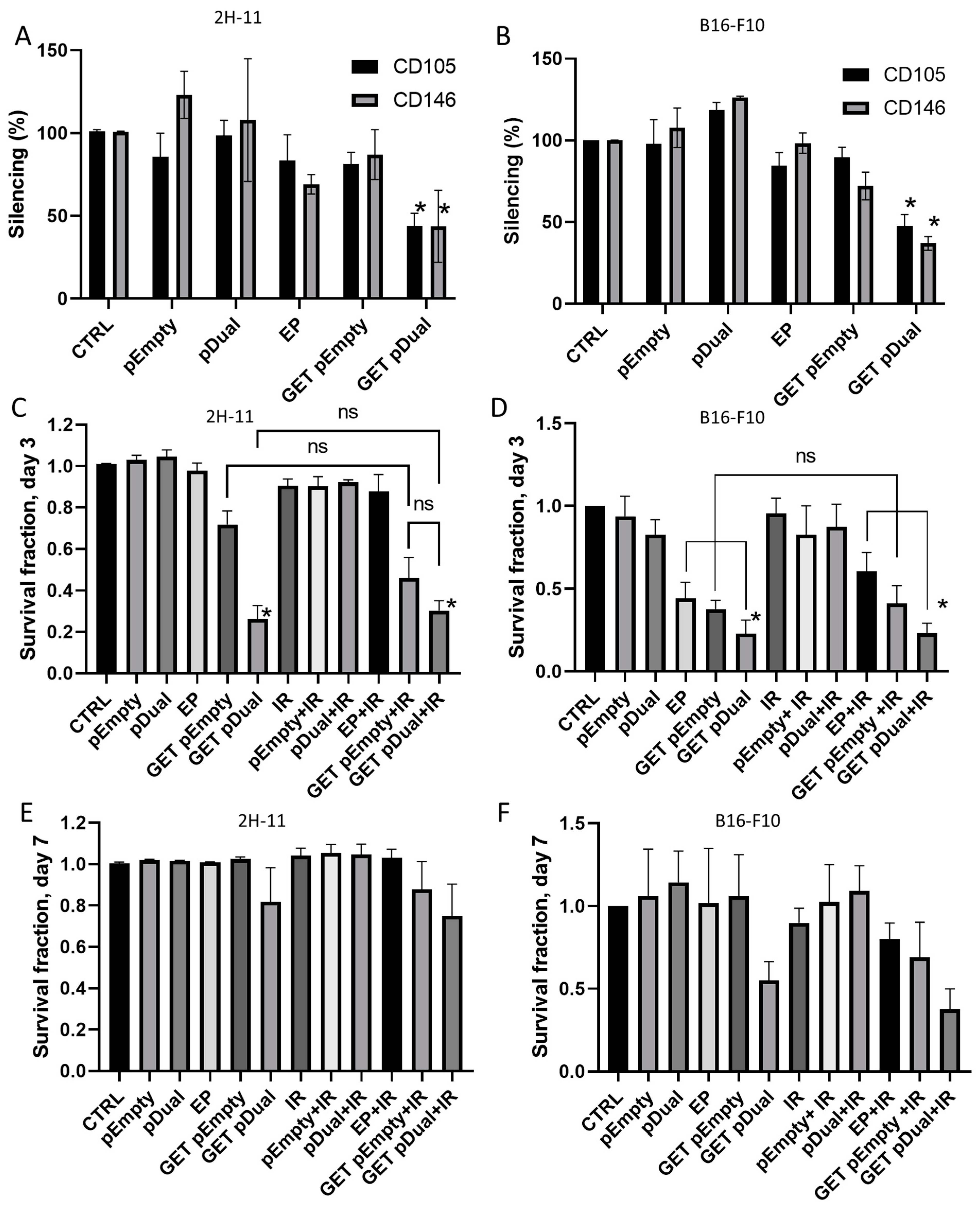
2.2. CD105 and CD146 Silencing
2.3. Cytotoxicity
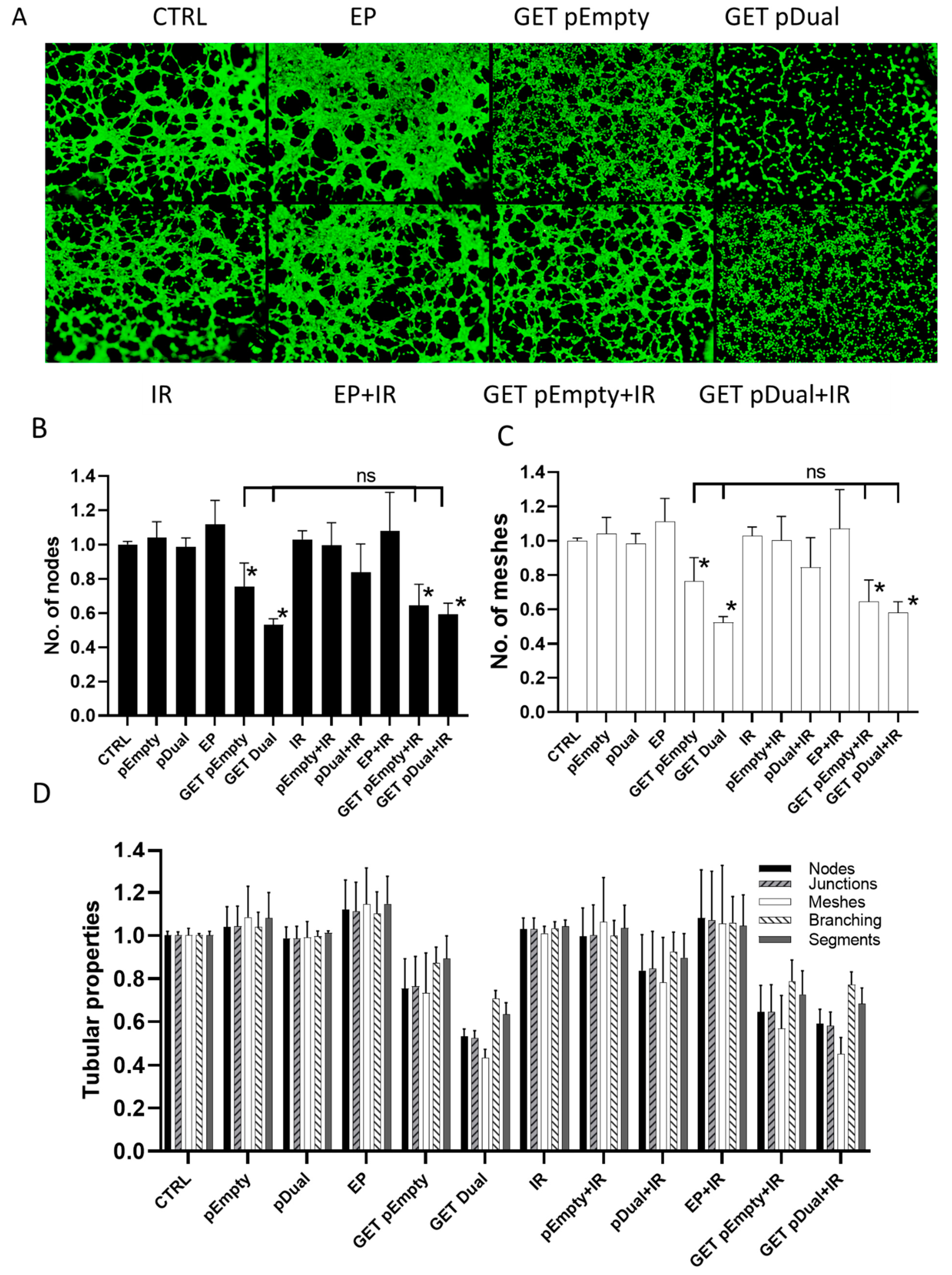
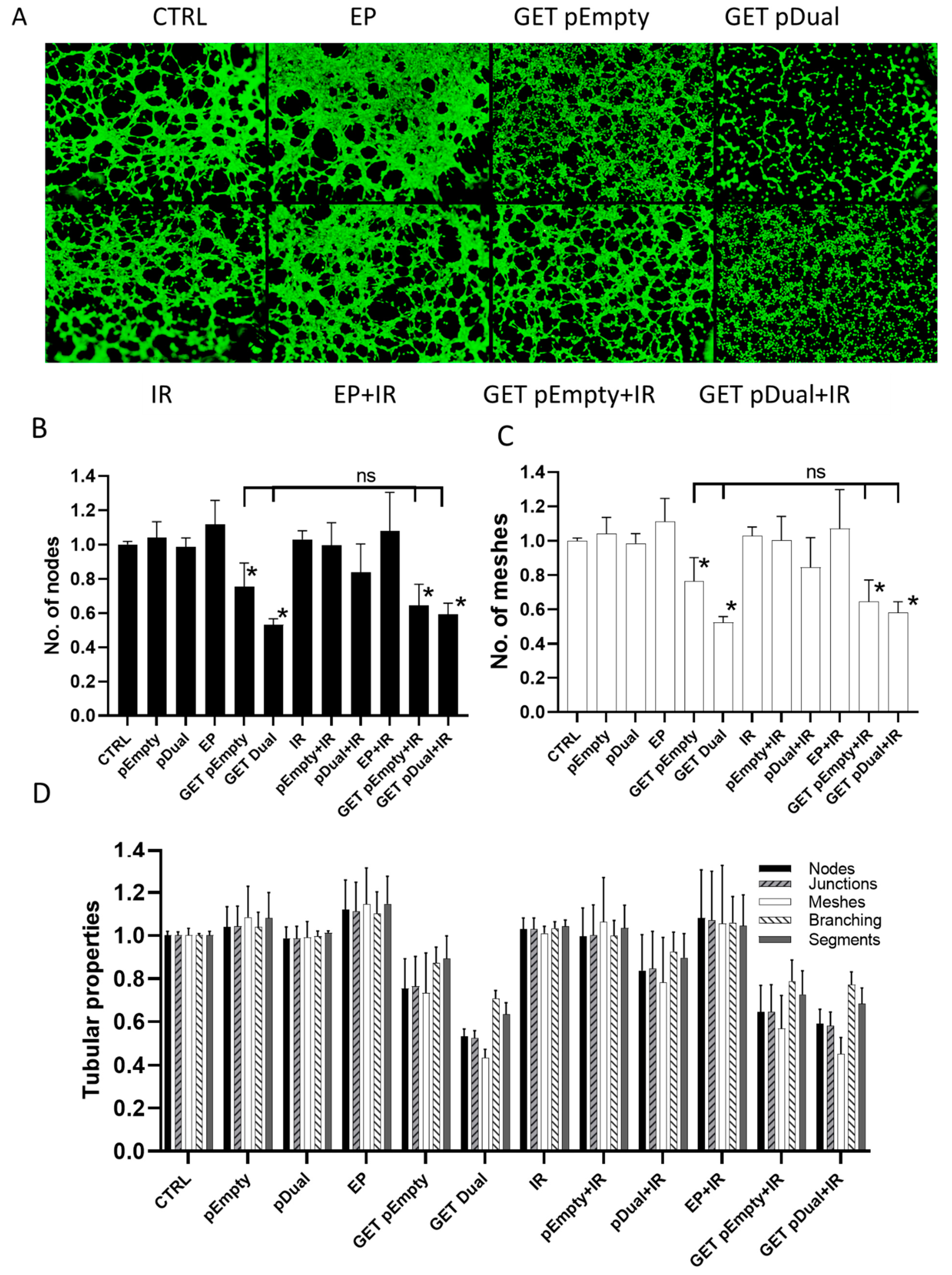
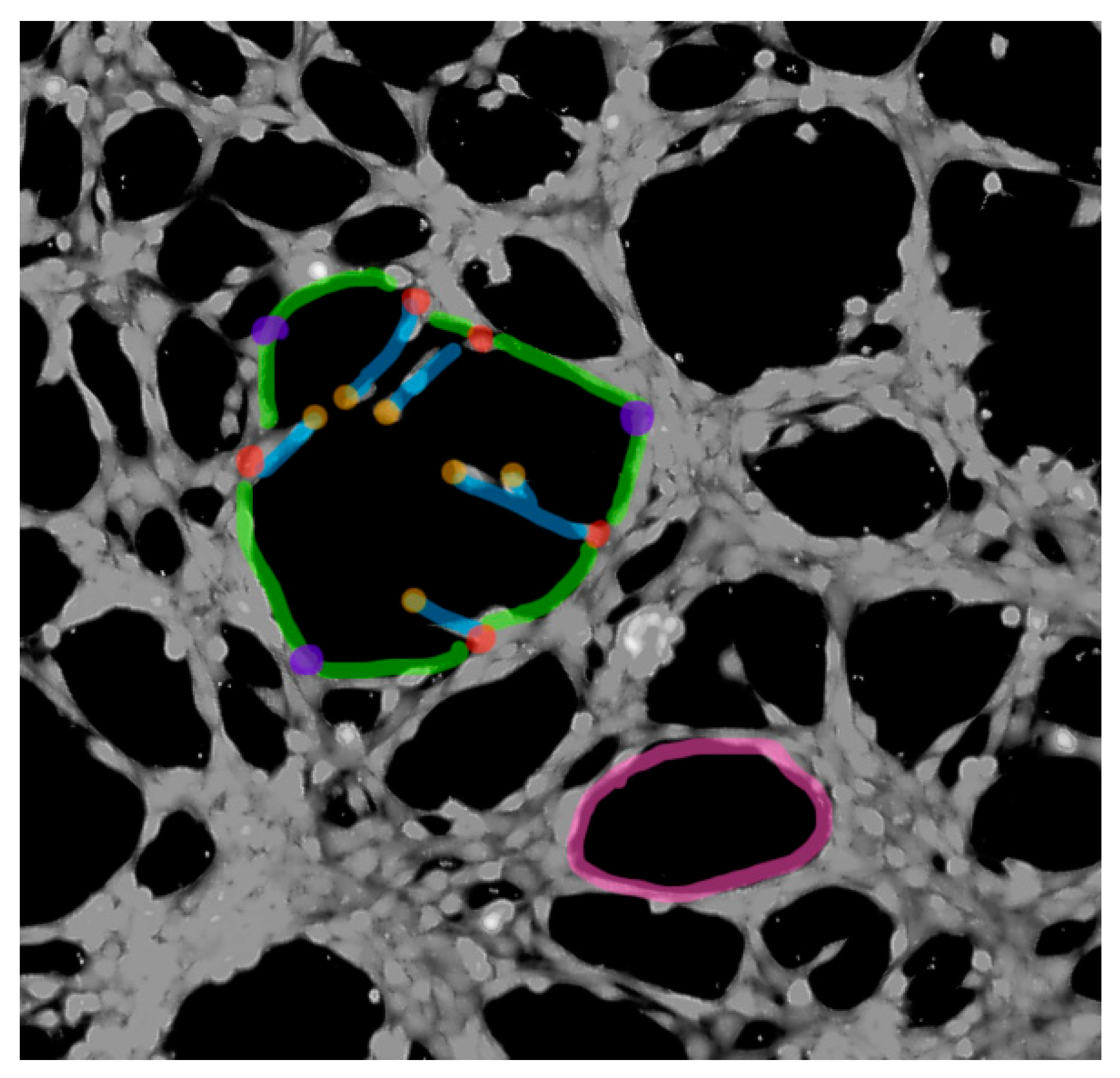
2.4. Inhibition of Capillary Tube Formation
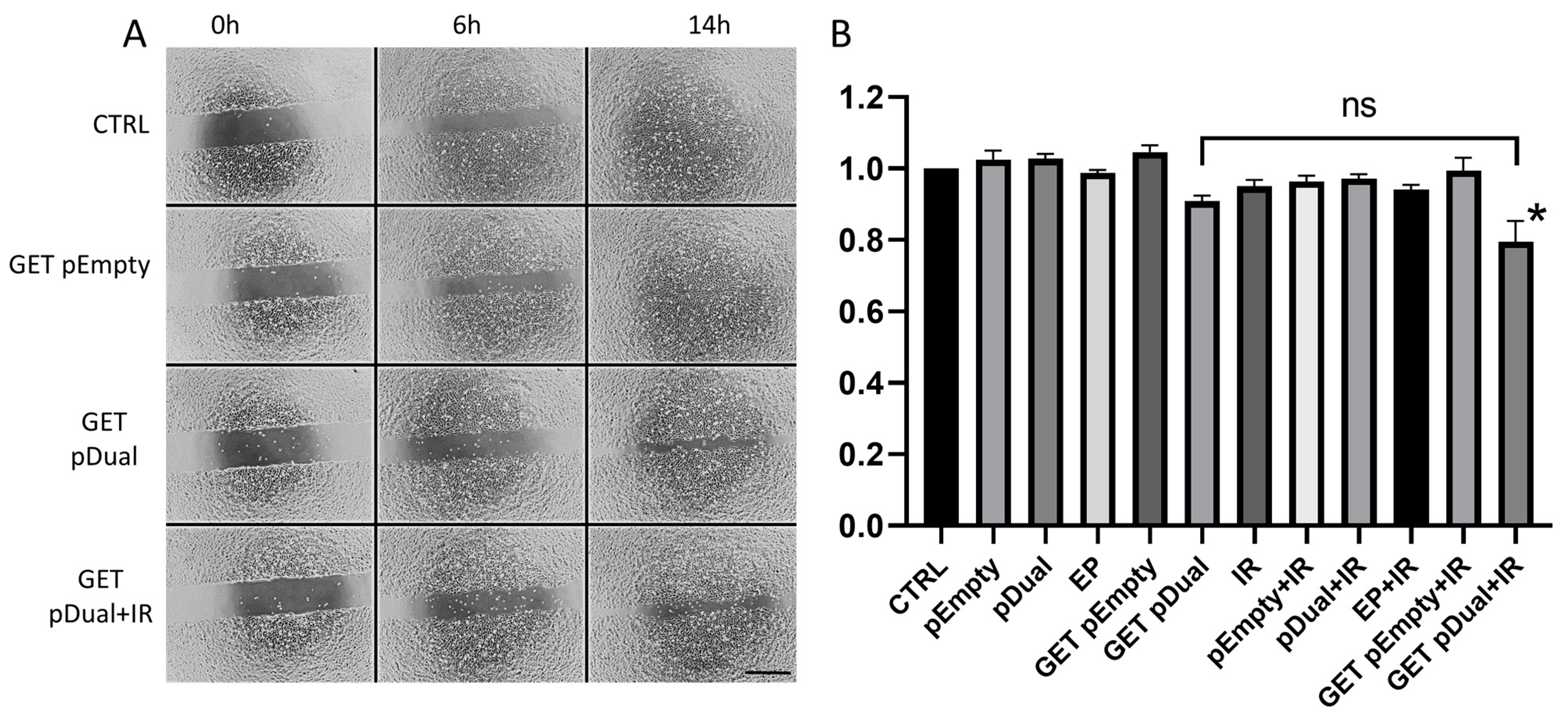
2.5. Inhibition of Endothelial Cell Migration
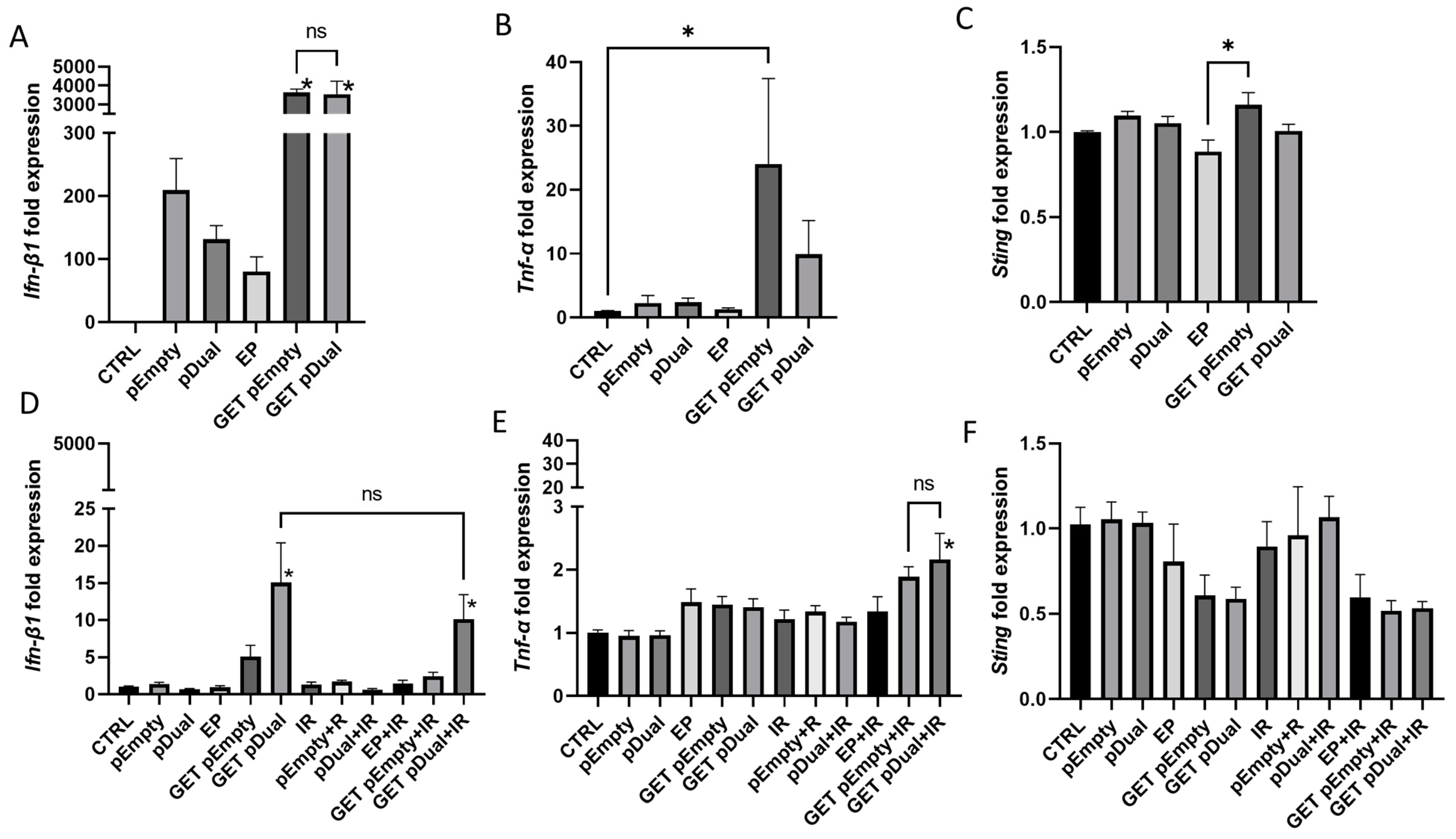
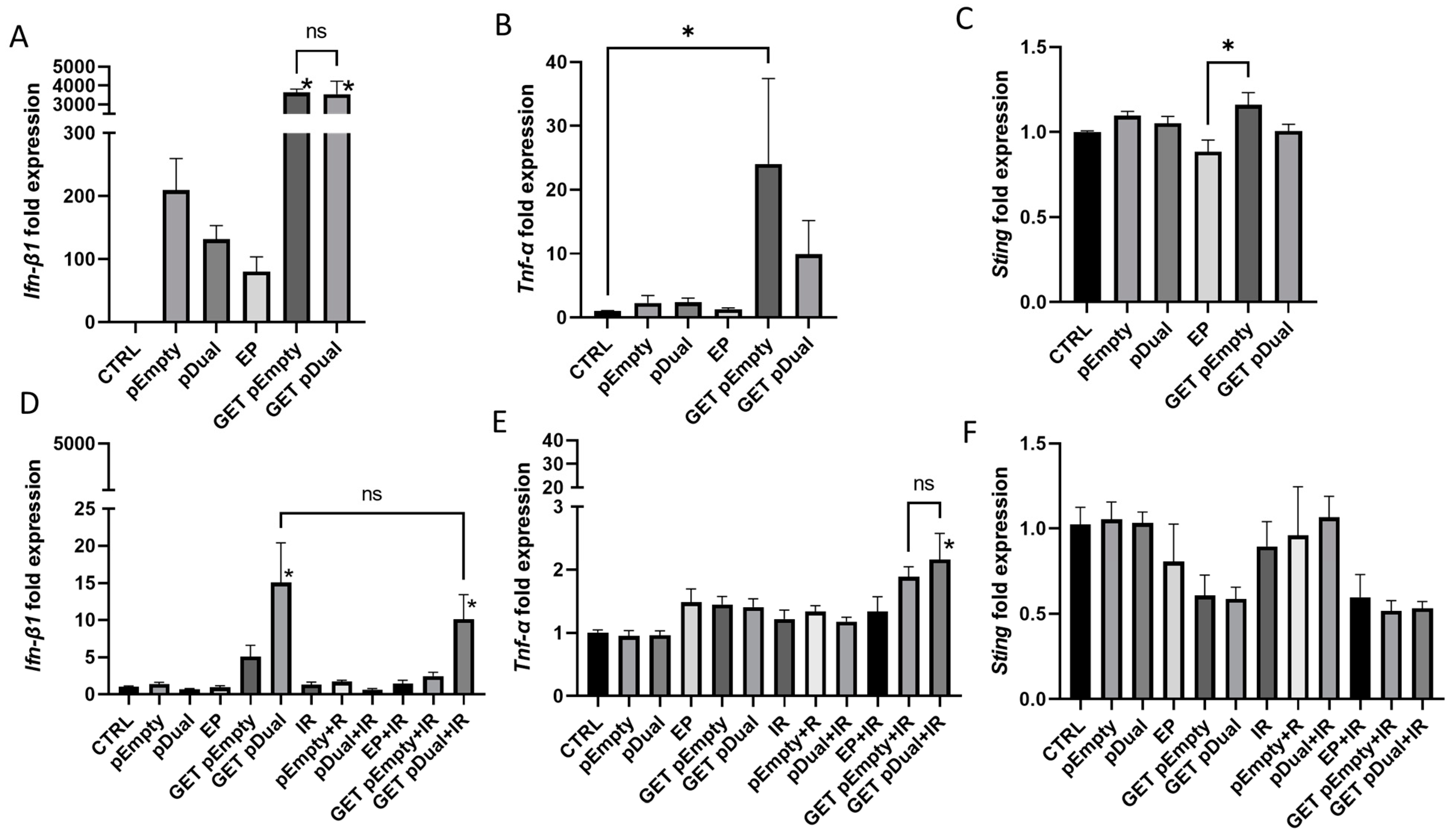
2.6. Induction of Cytokine and STING Expression
3. Discussion
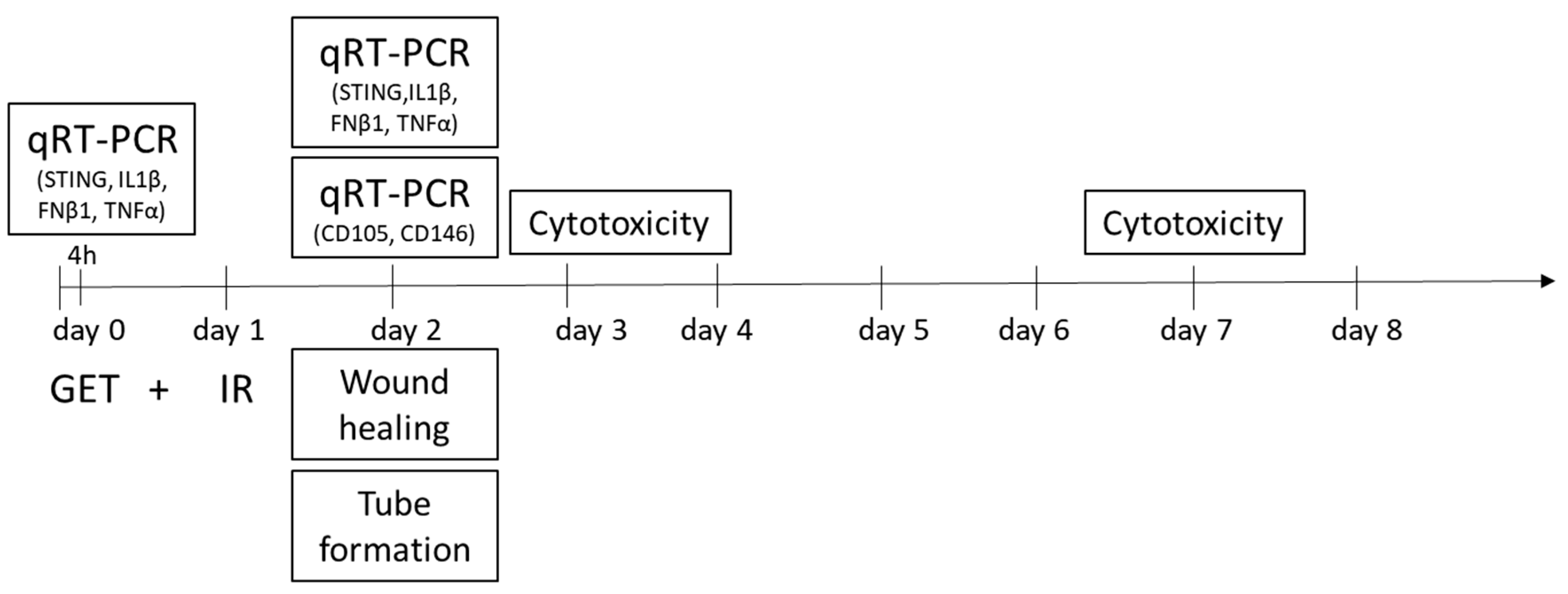
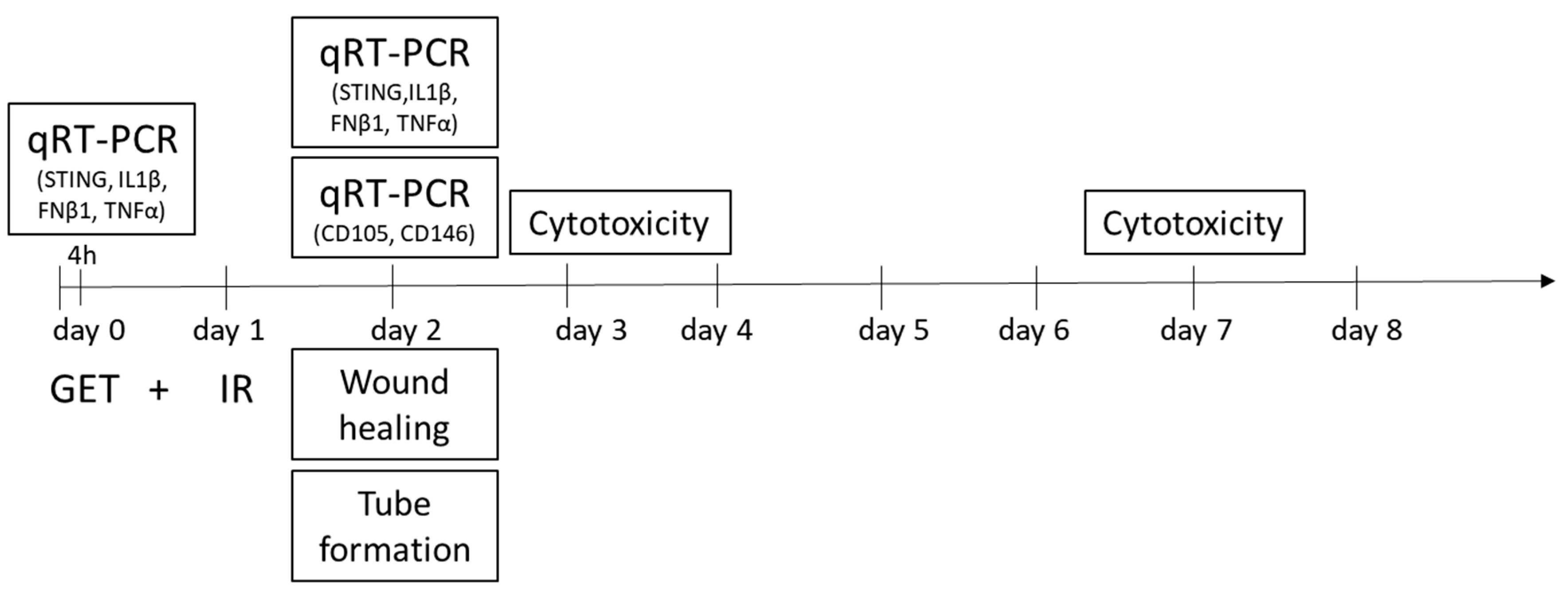
4. Materials and Methods
4.1. Construction of Plasmids
4.2. Purification of Transfection-Grade Plasmids
4.3. Cells
4.4. Gene Electrotransfer
4.5. Irradiation
4.6. Cytotoxicity Assay
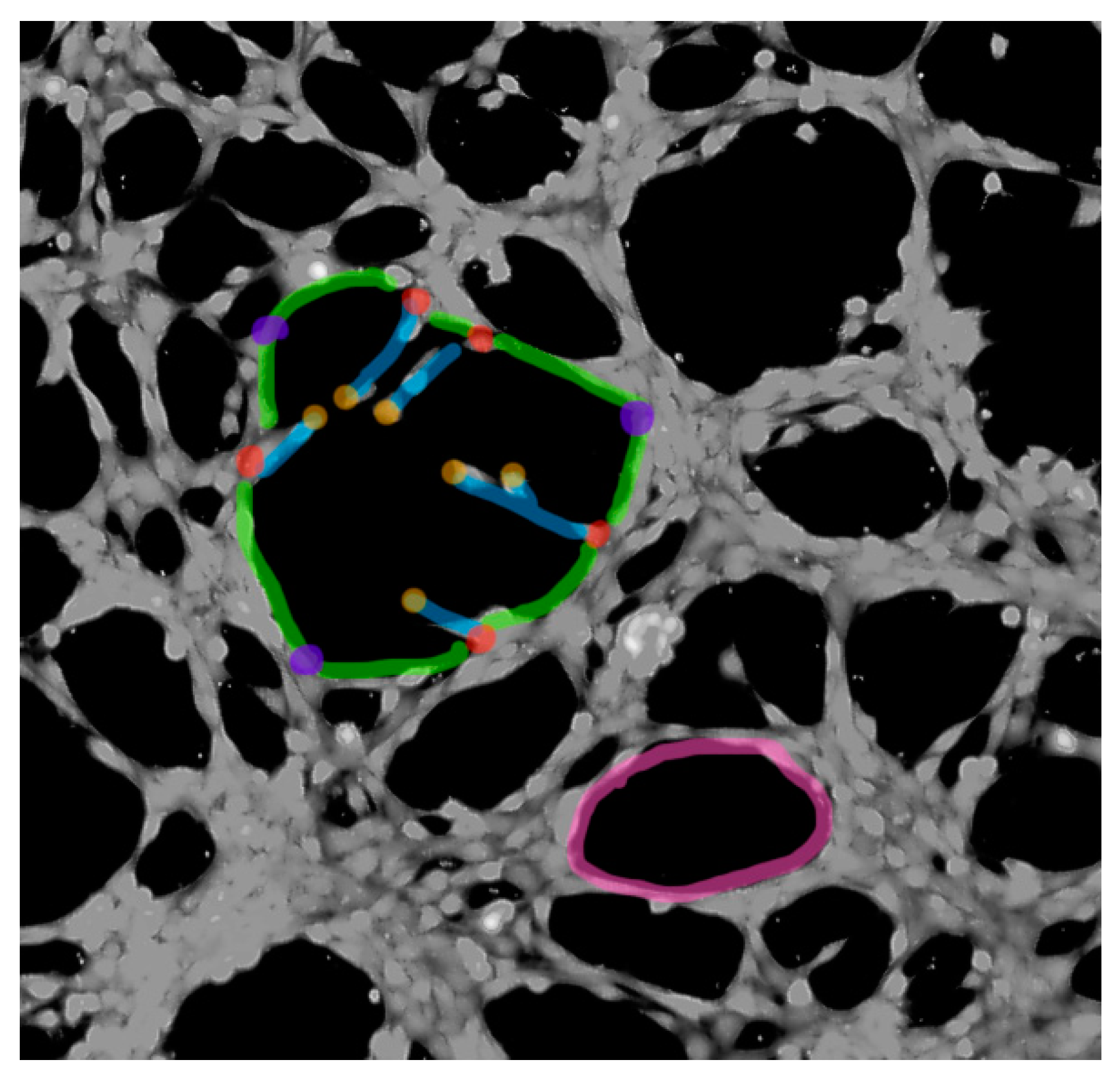
4.7. Tube Formation Assay
4.8. Wound-Healing Assay
4.9. Quantitative Real-Time PCR
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CD105 | Endoglin |
| CD146 | Mcam |
| DAMP | Danger Associated Molecular Pattern |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GET | Gene electrotransfer |
| IFN-β1 | Interferon β1 |
| IL1β | Interleukin 1-β |
| IR | Irradiation |
| ORT | Operator repressor titration |
| PAMP | Pathogen Associated Molecular Pattern |
| qRT-PCR | Quantitative reverse transcription polymerase chain reaction |
| shRNA | Short hairpin RNA |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor Necrosis Factor alpha |
| VEGF | Vascular endothelial growth factor |
References
- Denekamp, J. Angiogenesis, Neovascular proliferation and vascular pathophysiology as targets for cancer-therapy. Br. J. Radiol. 1993, 66, 181–196. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Rosen, L.S.; Gordon, M.S.; Robert, F.; Matei, D.E. Endoglin for targeted cancer treatment. Curr. Oncol. Rep. 2014, 16, 365. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A.; Adamek, A. Role of Endoglin (CD105) in the Progression of Hepatocellular Carcinoma and Anti-Angiogenic Therapy. Int. J. Mol. Sci. 2018, 19, 3887. [Google Scholar] [CrossRef] [Green Version]
- Dallas, N.A.; Samuel, S.; Xia, L.; Fan, F.; Gray, M.J.; Lim, S.J.; Ellis, L.M. Endoglin (CD105): A marker of tumor vasculature and potential target for therapy. Clin. Cancer Res. 2008, 14, 1931–1937. [Google Scholar] [CrossRef] [Green Version]
- Fonsatti, E.; Altomonte, M.; Arslan, P.; Maio, M. Endoglin (CD105): A Target for anti-angiogenetic cancer therapy. Curr. Drug Targets 2005, 4, 291–296. [Google Scholar] [CrossRef]
- Zheng, C.; Qiu, Y.; Zeng, Q.; Zhang, Y.; Lu, D.; Yang, D.; Feng, J.; Yan, X. Endothelial CD146 is required for in vitro tumor-induced angiogenesis: The role of a disulfide bond in signaling and dimerization. Int. J. Biochem. Cell Biol. 2009, 41, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhuang, J.; Duan, H.; Luo, Y.; Zeng, Q.; Fan, K.; Yan, H.; Lu, D.; Ye, Z.; Hao, J.; et al. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 2012, 120, 2330–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, M.C.; Zhao, Y.J.; Lachman, L.B.; Hwu, P.; Bar-Eli, M. Immunization against MUC18/MCAM, a novel antigen that drives melanoma invasion and metastasis. Gene Ther. 2007, 14, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Dolinsek, T.; Markelc, B.; Sersa, G.; Coer, A.; Stimac, M.; Lavrencak, J.; Brozic, A.; Kranjc, S.; Cemazar, M. Multiple Delivery of siRNA against Endoglin into Murine Mammary Adenocarcinoma Prevents Angiogenesis and Delays Tumor Growth. PLoS ONE 2013, 8, e0058723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsek, T.; Markelc, B.; Bosnjak, M.; Blagus, T.; Prosen, L.; Kranjc, S.; Stimac, M.; Lampreht, U.; Sersa, G.; Cemazar, M. Endoglin Silencing has Significant Antitumor Effect on Murine Mammary Adenocarcinoma Mediated by Vascular Targeted Effect. Curr. Gene Ther. 2015, 15, 228–244. [Google Scholar] [CrossRef]
- Dolinsek, T.; Sersa, G.; Prosen, L.; Bosnjak, M.; Stimac, M.; Razborsek, U.; Cemazar, M. Electrotransfer of plasmid DNA encoding an anti-mouse endoglin (CD105) shRNA to B16 melanoma tumors with low and high metastatic potential results in pronounced anti-tumor effects. Cancers 2015, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesic, N.; Kamensek, U.; Sersa, G.; Kranjc, S.; Stimac, M.; Lampreht, U.; Preat, V.; Vandermeulen, G.; Butinar, M.; Turk, B. Endoglin (CD105) Silencing Mediated by shRNA Under the Control of Endothelin-1 Promoter for Targeted Gene Therapy of Melanoma. Mol. Ther. Acids 2015, 4, e239. [Google Scholar] [CrossRef] [PubMed]
- Stimac, M.; Kamensek, U.; Cemazar, M.; Kranjc, S.; Coer, A.; Sersa, G. Tumor radiosensitization by gene therapy against endoglin. Cancer Gene Ther. 2016, 23. [Google Scholar] [CrossRef] [PubMed]
- Savarin, M.; Kamensek, U.; Cemazar, M.; Heller, R.; Sersa, G. Electrotransfer of plasmid DNA radiosensitizes B16F10 tumors through activation of immune response. Radiol. Oncol. 2017, 51, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosen, L.; Markelc, B.; Dolinsek, T.; Music, B.; Cemazar, M.; Sersa, G. Mcam silencing with RNA interference using magnetofection has antitumor effect in murine melanoma. Mol. Ther. Nucleic Acids 2014, 3, e205. [Google Scholar] [CrossRef]
- Costello, B.; Li, C.; Duff, S.; Butterworth, D.; Khan, A.; Perkins, M.; Owens, S.; Al-Mowallad, A.F.; O’Dwyer, S.; Kumar, S. Perfusion of99Tcm-labeled CD105 Mab into kidneys from patients with renal carcinoma suggests that CD105 is a promising vascular target. Int. J. Cancer 2004, 109, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.; Wälchli, S.; Sioud, M. Transcriptional targeting of small interfering RNAs into cancer cells. Biochem. Biophys. Res. Commun. 2006, 350, 854–859. [Google Scholar] [CrossRef]
- Rols, M.-P.; Golzio, M.; Kolosnjaj-Tabi, J. Electric Field Based Therapies in Cancer Treatment. Cancers 2020, 12, 3420. [Google Scholar] [CrossRef]
- Maglietti, F.; Tellado, M.; De Robertis, M.; Michinski, S.; Fernández, J.; Signori, E.; Marshall, G. Electroporation as the immunotherapy strategy for cancer in veterinary medicine: State of the art in Latin America. Vaccines 2020, 8, 537. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Jungel, A.; Kurowska-Stolarska, M.; Michel, B.A.; Gay, R.E.; Gay, S.; Distler, O. Nucleofection: A new, highly efficient transfection method for primary human keratinocytes *. Exp. Dermatol. 2005, 14, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Yarmush, M.L.; Golberg, A.; Serša, G.; Kotnik, T.; Miklavčič, D. Electroporation-based technologies for medicine: Principles, applications, and challenges. Annu. Rev. Biomed. Eng. 2014, 16, 295–320. [Google Scholar] [CrossRef] [Green Version]
- Faurie, C.; Rebersek, M.; Golzio, M.; Kanduser, M.; Escoffre, J.-M.; Pavlin, M.; Teissie, J.; Miklavcic, D.; Rols, M.-P. Electro-mediated gene transfer and expression are controlled by the life-time of DNA/membrane complex formation. J. Gene Med. 2010, 12, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Pavlin, M.; Pucihar, G.; Kandušer, M. The role of electrically stimulated endocytosis in gene electrotransfer. Bioelectrochemistry 2012, 83, 38–45. [Google Scholar] [CrossRef]
- Rosazza, C.; Deschout, H.; Buntz, A.; Braeckmans, K.; Rols, M.P.; Zumbusch, A. Endocytosis and endosomal trafficking of DNA after gene electrotransfer in vitro. Mol. Ther. Nucleic Acids 2016, 5, e286. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Wang, L.; Chang, C.C.; Rothenberg, K.E.; Huang, J.; Wang, Y.; Hoffman, B.D.; Liton, P.B.; Yuan, F. Involvement of a Rac1-dependent macropinocytosis pathway in plasmid DNA delivery by electrotransfection. Mol. Ther. 2017, 25, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Cervia, L.D.; Chang, C.-C.; Wang, L.; Mao, M.; Yuan, F. Enhancing electrotransfection efficiency through improvement in nuclear entry of plasmid DNA. Mol. Ther. Nucleic Acids 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chang, C.C.; Sylvers, J.; Yuan, F. A statistical framework for determination of minimal plasmid copy number required for transgene expression in mammalian cells. Bioelectrochemistry 2021, 138, 107731. [Google Scholar] [CrossRef]
- Wu, M.; Yuan, F. Membrane binding of plasmid DNA and endocytic pathways are involved in electrotransfection of mammalian cells. PLoS ONE 2011, 6, e20923. [Google Scholar] [CrossRef]
- Golzio, M.; Teissié, J.; Rols, M.P. Direct visualization at the single-cell level of electrically mediated gene delivery. Proc. Natl. Acad. Sci. USA 2002, 99, 1292–1297. [Google Scholar] [CrossRef] [Green Version]
- Heller, R.; Heller, L.C. Gene Electrotransfer Clinical Trials. Adv. Genet. 2015, 89, 235–262. [Google Scholar] [CrossRef] [PubMed]
- Gehl, J. Gene electrotransfer in clinical trials. Methods Mol. Biol. 2014, 1121, 241–246. [Google Scholar] [CrossRef]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef] [Green Version]
- Low, L.; Mander, A.; McCann, K.; Dearnaley, D.; Tjelle, T.; Mathiesen, I.; Stevenson, F.; Ottensmeier, C.H. DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum. Gene Ther. 2009, 20, 1269–1278. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Chiappori, A.; Aurisicchio, L.; Bagchi, A.; Clark, J.; Dubey, S.; Fridman, A.; Fabregas, J.C.; Marshall, J.; Scarselli, E.; et al. Phase 1 studies of the safety and immunogenicity of electroporated HER2/CEA DNA vaccine followed by adenoviral boost immunization in patients with solid tumors. J. Transl. Med. 2013, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.R.; Pringle, I.A.; Hyde, S.C. Progress and prospects: The design and production of plasmid vectors. Gene Ther 2009, 16, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, M.; Chen, C.; Yao, Q. Small interfering RNA therapy in cancer: Mechanism, potential targets, and clinical applications. Expert Opin. Ther. Targets 2008, 12, 637–645. [Google Scholar] [CrossRef]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef]
- McAnuff, M.A.; Rettig, G.R.; Rice, K.G. Potency of siRNA versus shRNA mediated knockdown in vivo. J. Pharm. Sci. 2007, 96, 2922–2930. [Google Scholar] [CrossRef]
- Lambricht, L.; Lopes, A.; Kos, S.; Sersa, G.; Préat, V.; Vandermeulen, G. Erratum: Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin. Drug Deliv. 2016, 13, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Wagner, E. Therapeutic plasmid DNA versus siRNA delivery: Common and different tasks for synthetic carriers. J. Control. Release 2012, 161, 554–565. [Google Scholar] [CrossRef]
- Sodoyer, R.; Courtois, V.; Peubez, I.; Mignon, C. Antibiotic-free selection for bio-production: Moving towards a new „gold standard“. Antibiot. Resist. Bact. 2012, 531–550. [Google Scholar] [CrossRef] [Green Version]
- Vandermeulen, G.; Marie, C.; Scherman, D.; Preat, V. New generation of plasmid backbones devoid of antibiotic resistance marker for gene therapy trials. Mol. Ther. 2011, 19, 1942–1949. [Google Scholar] [CrossRef] [Green Version]
- Mairhofer, J.; Pfaffenzeller, I.; Merz, D.; Grabherr, R. A novel antibiotic free plasmid selection system: Advances in safe and efficient DNA therapy. Biotechnol. J. 2008, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Carnes, A.; Williams, J. Plasmid DNA Manufacturing technology. Recent Pat. Biotechnol. 2007, 1, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Garmory, H.S.; Leckenby, M.W.; Griffin, K.F.; Elvin, S.J.; Taylor, R.R.; Hartley, M.G.; Hanak, J.A.J.; Williamson, E.D.; Cranenburgh, R.M. Antibiotic-free plasmid stabilization by operator-repressor titration for vaccine delivery by using live Salmonella enterica serovar typhimurium. Infect. Immun. 2005, 73, 2005–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamensek, U.; Tesic, N.; Sersa, G.; Cemazar, M. Clinically Usable Interleukin 12 Plasmid without an Antibiotic Resistance Gene: Functionality and Toxicity Study in Murine Melanoma Model. Cancers 2018, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Prosen, L.; Hudoklin, S.; Cemazar, M.; Stimac, M.; Lampreht Tratar, U.; Ota, M.; Scancar, J.; Romih, R.; Sersa, G. Magnetic field contributes to the cellular uptake for effective therapy with magnetofection using plasmid DNA encoding against Mcam in B16F10 melanoma in vivo. Nanomedicine 2016, 11, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Stimac, M.; Dolinsek, T.; Lampreht, U.; Cemazar, M.; Sersa, G. Gene electrotransfer of plasmid with tissue specific promoter encoding shRNA against endoglin exerts antitumor efficacy against murine TS/A tumors by vascular targeted effects. PLoS ONE 2015, 10, e0124913. [Google Scholar] [CrossRef]
- Brezar, S.K.; Mrak, V.; Bosnjak, M.; Savarin, M.; Sersa, G.; Cemazar, M. Intratumoral gene electrotransfer of plasmid DNA encoding shRNA against melanoma cell adhesion molecule radiosensitizes tumors by antivascular effects and activation of an immune response. Vaccines 2020, 8, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Ciric, E.; Sersa, G. Radiotherapy in combination with vascular-targeted therapies. Radiol. Oncol. 2010, 44, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Mazeron, R.; Anderson, B.; Supiot, S.; Paris, F.; Deutsch, E. Current state of knowledge regarding the use of antiangiogenic agents with radiation therapy. Cancer Treat. Rev. 2011, 37, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Clémenson, C.; Chargari, C.; Deutsch, E. Combination of vascular disrupting agents and ionizing radiation. Crit. Rev. Oncol. Hematol. 2013, 86, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Znidar, K.; Bosnjak, M.; Cemazar, M.; Heller, L.C. Cytosolic DNA Sensor Upregulation Accompanies DNA Electrotransfer in B16.F10 Melanoma Cells. Mol. Ther. Nucleic Acids 2016, 5, e322. [Google Scholar] [CrossRef] [Green Version]
- Jesenko, T.; Bosnjak, M.; Markelc, B.; Sersa, G.; Znidar, K.; Heller, L.; Cemazar, M. Radiation induced upregulation of dna sensing pathways is cell-type dependent and can mediate the off-target effects. Cancers 2020, 12, 3365. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Polajzer, T.; Jarm, T.; Miklavcic, D. Analysis of damage-associated molecular pattern molecules due to electroporation of cells in vitro. Radiol. Oncol. 2020, 54, 317–328. [Google Scholar] [CrossRef]
- Liang, Y.; Peng, H. STING-cytosolic DNA sensing: The backbone for an effective tumor radiation therapy. Ann. Transl. Med. 2016, 4, 60. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Du, S.; Chen, G.; Yuan, B.; Hu, Y.; Yang, P.; Chen, Y.; Zhao, Q.; Zhou, J.; Fan, J.; Zeng, Z. DNA sensing and associated type 1 interferon signaling contributes to progression of radiation-induced liver injury. Cell. Mol. Immunol. 2020, 1–11. [Google Scholar] [CrossRef]
- Dormiani, K.; Sadeghi, H.M.M.; Sadeghi-Aliabadi, H.; Forouzanfar, M.; Baharvand, H.; Ghaedi, K.; Nasr-Esfahani, M.H. Rational development of a polycistronic plasmid with a CpG-free bacterial backbone as a potential tool for direct reprogramming. Cell J. 2016, 18, 565–581. [Google Scholar] [CrossRef]
- Qin, W.; Dion, S.L.; Kutny, P.M.; Zhang, Y.; Cheng, A.W.; Jillette, N.L.; Malhotra, A.; Geurts, A.M.; Chen, Y.G.; Wang, H. Efficient CRISPR/cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics 2015, 200, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Brzeszczynska, J.; Samuel, K.; Black, J.; Palakkan, A.; Anderson, R.A.; Gallagher, R.; Ross, J.A. Efficient episomal reprogramming of blood mononuclear cells and differentiation to hepatocytes with functional drug metabolism. Exp. Cell Res. 2015, 338, 203–213. [Google Scholar] [CrossRef]
- Liu, J.; Xia, X.; Torrero, M.; Barrett, R.; Shillitoe, E.J.; Li, S. The mechanism of exogenous B7.1-enhanced IL-12-mediated complete regression of tumors by a single electroporation delivery. Int. J. Cancer 2006, 119, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, L.L.; Mir, L.M.; André, F.M. Overcoming the specific toxicity of large plasmids electrotransfer in primary cells in vitro. Mol. Ther. Acids 2016, 5, e291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcintyre, G.J.; Arndt, A.J.; Gillespie, K.M.; Mak, W.M.; Fanning, G.C. A comparison of multiple shRNA expression methods for combinatorial RNAi. Genet. Vaccines Ther. 2011, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, G.R.; Maser, R.L.; Calvet, J.P.; Pederson, T. U6 small nuclear RNA is transcribed by RNA polymerase III. Proc. Natl. Acad. Sci. USA 1986, 83, 8575–8579. [Google Scholar] [CrossRef] [Green Version]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense oligonucleotides targeting angiogenic factors as potential cancer therapeutics. Mol. Ther. Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, C.J.; Ishii, K.J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef]
- Keating, S.E.; Baran, M.; Bowie, A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011, 32, 574–581. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: Endothelial activation and dysfunction. Cell. Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.M.; Flynn, N.J.; Raben, A.; Romak, L.; Yu, Y.; Dicker, A.P.; Mourtada, F.; Sims-Mourtada, J. The Impact of radiation on the tumor microenvironment: Effect of Dose and Fractionation Schedules. Cancer Growth Metastasis 2018, 11. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, M.S.; Jeong, Y.K.; Cho, I.; You, S.H.; Cho, S.H.; Lee, H.; Jung, W.G.; Kim, H.D.; Kim, J. Mechanisms for SU5416 as a radiosensitizer of endothelial cells. Int. J. Oncol. 2015, 47, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis analyzer for imageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savarin, M.; Kamensek, U.; Znidar, K.; Todorovic, V.; Sersa, G.; Cemazar, M. Evaluation of a Novel Plasmid for Simultaneous Gene Electrotransfer-Mediated Silencing of CD105 and CD146 in Combination with Irradiation. Int. J. Mol. Sci. 2021, 22, 3069. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063069
Savarin M, Kamensek U, Znidar K, Todorovic V, Sersa G, Cemazar M. Evaluation of a Novel Plasmid for Simultaneous Gene Electrotransfer-Mediated Silencing of CD105 and CD146 in Combination with Irradiation. International Journal of Molecular Sciences. 2021; 22(6):3069. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063069
Chicago/Turabian StyleSavarin, Monika, Urska Kamensek, Katarina Znidar, Vesna Todorovic, Gregor Sersa, and Maja Cemazar. 2021. "Evaluation of a Novel Plasmid for Simultaneous Gene Electrotransfer-Mediated Silencing of CD105 and CD146 in Combination with Irradiation" International Journal of Molecular Sciences 22, no. 6: 3069. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063069