
Neonatal Mesenchymal Stem Cell Treatment Improves Myelination Impaired by Global Perinatal Asphyxia in Rats

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Apgar Scale
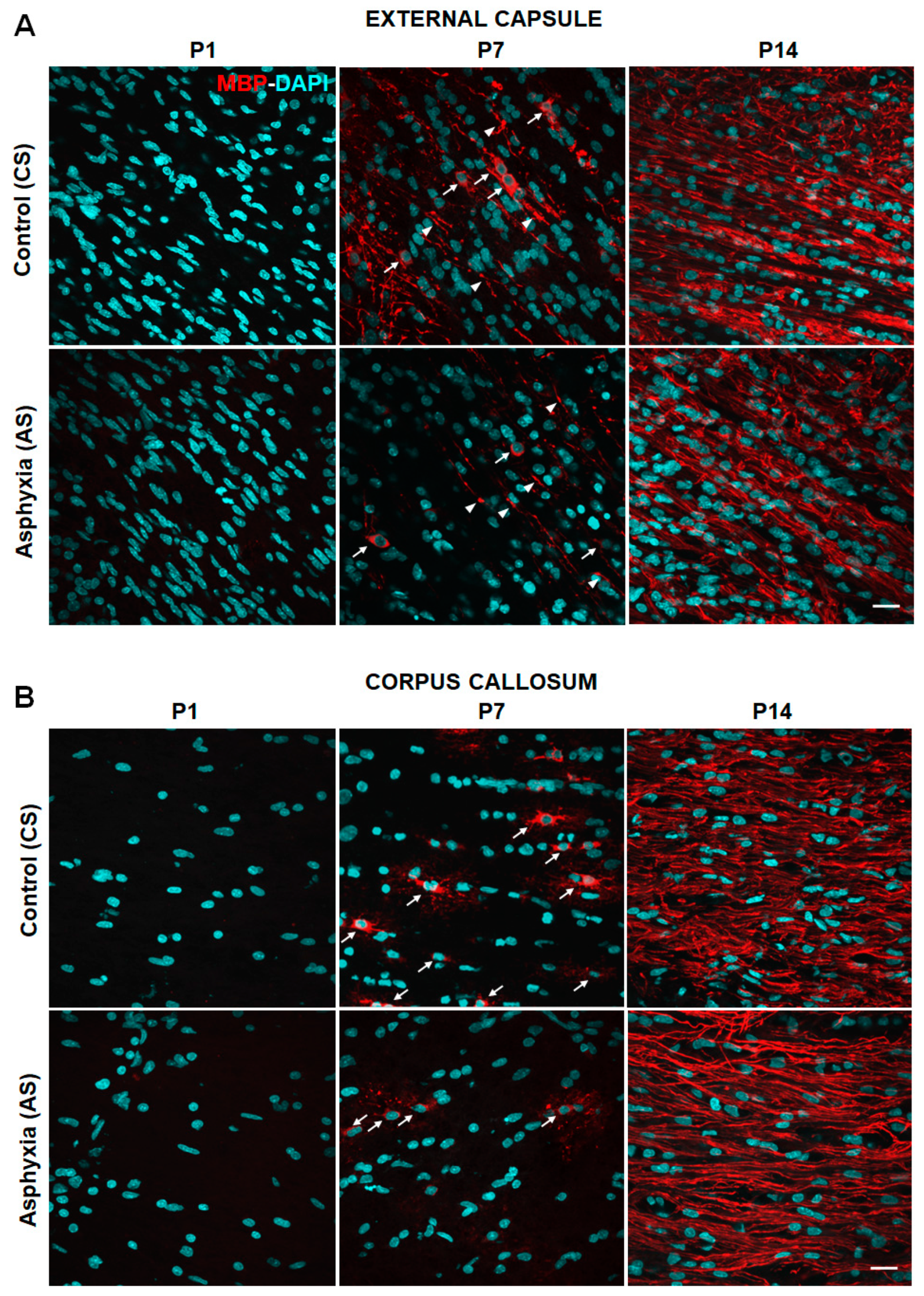
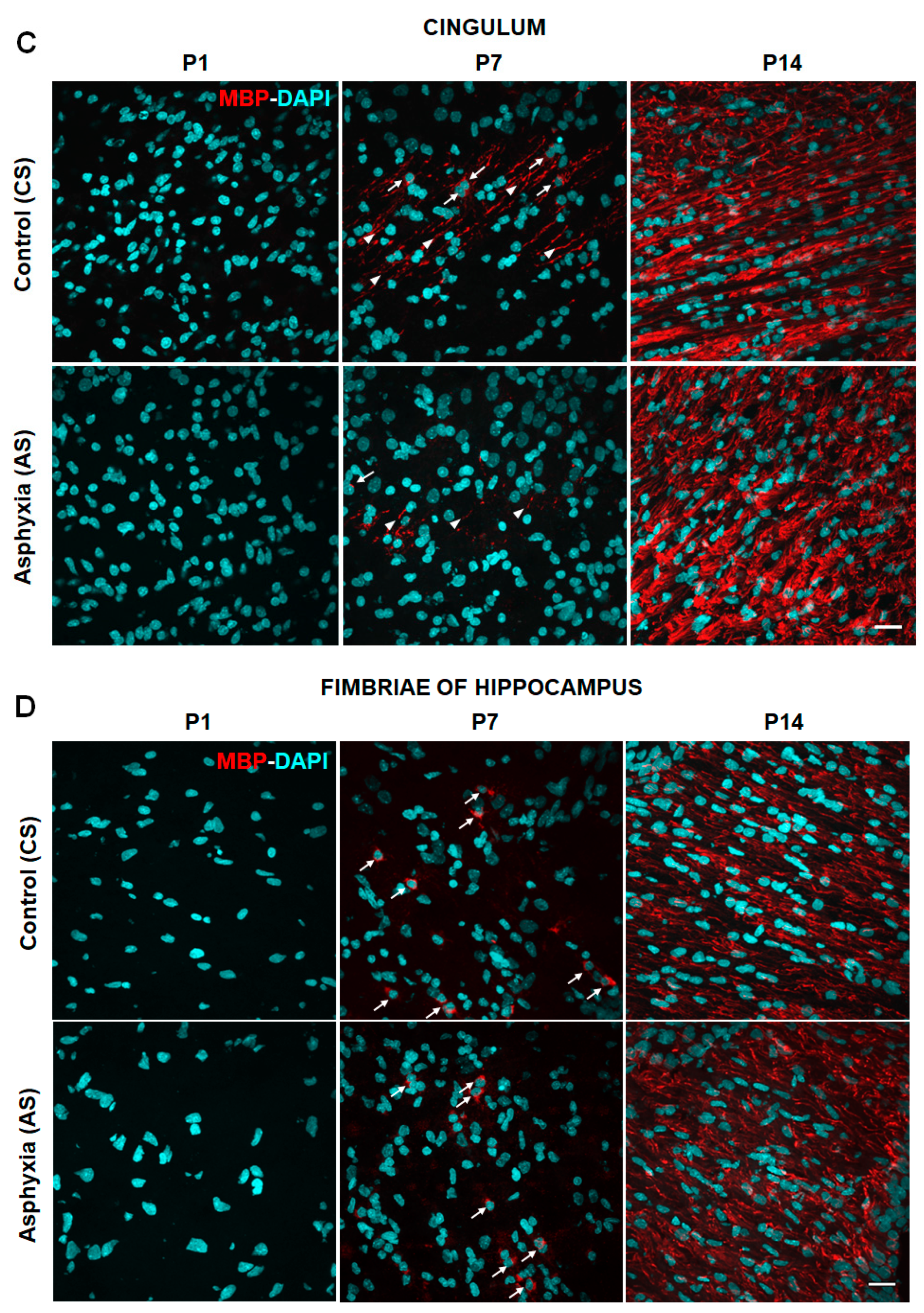
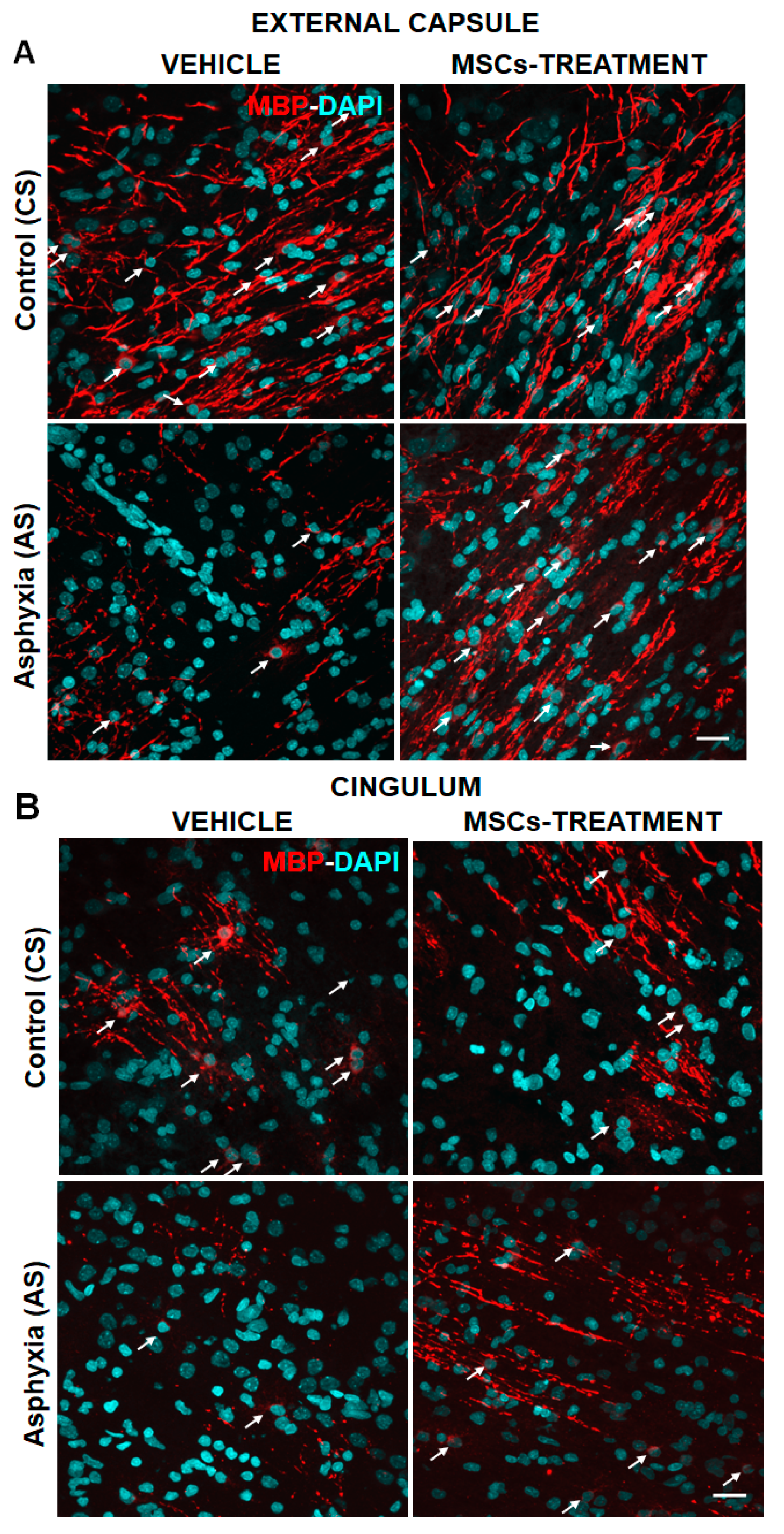
2.2. Effect of Neonatal Development and PA on Myelination in Telencephalon (External Capsule, Corpus Callosum, Cingulum) and Fimbriae of Hippocampus at P1, P7 and P14
2.3. Effect of Neonatal Development and PA on mRNA Levels (MBP, OL Transcription Factors, and Pro-Inflammatory Cytokines) Evaluated in the Telencephalon by RT-qPCR at P1, P7 and P14
2.3.1. MBP, Olig-1 and Olig-2 mRNA Levels
2.3.2. Pro-Inflammatory Cytokines (IL-1β, IL-6, TNF-α, Cox-2) mRNA Levels
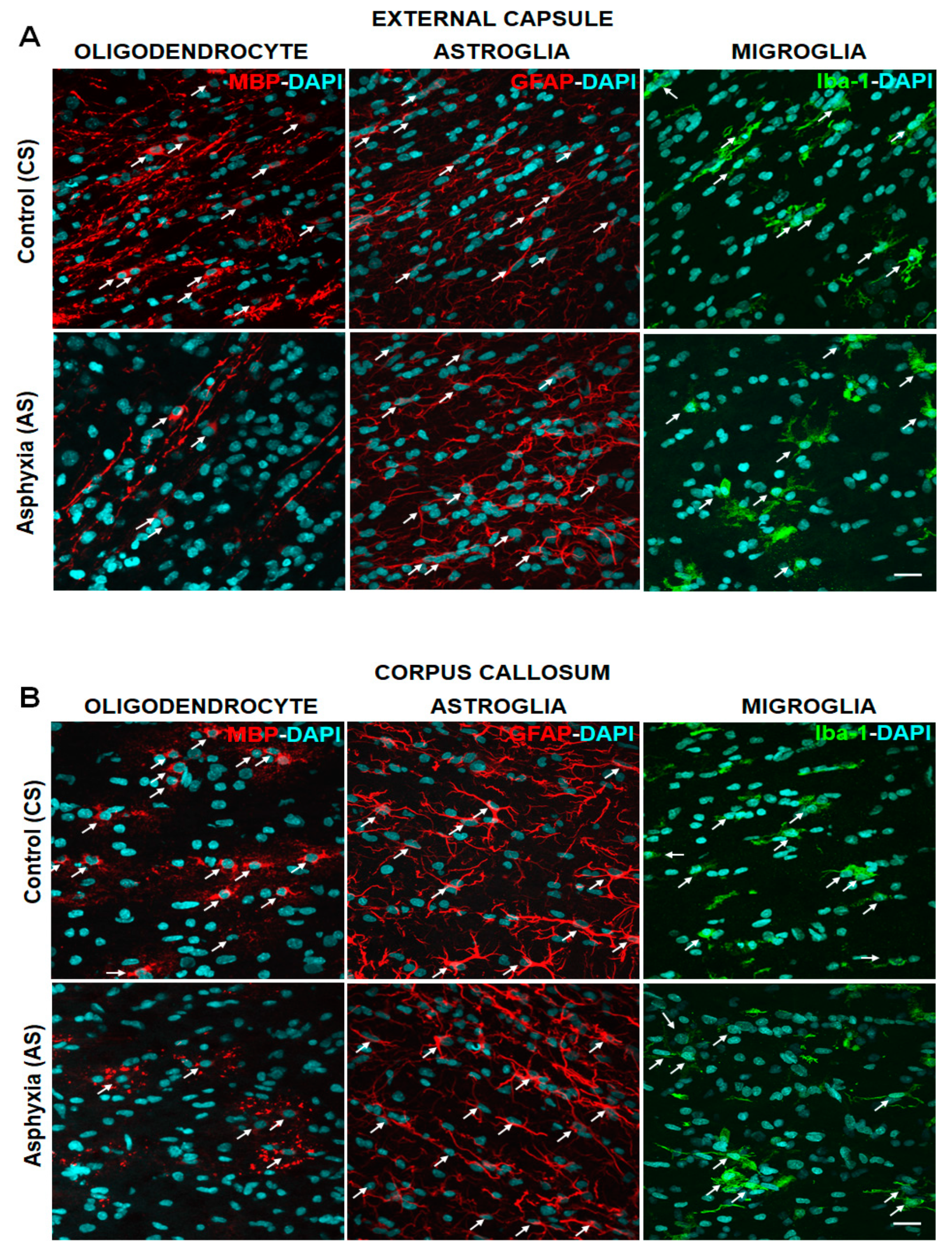
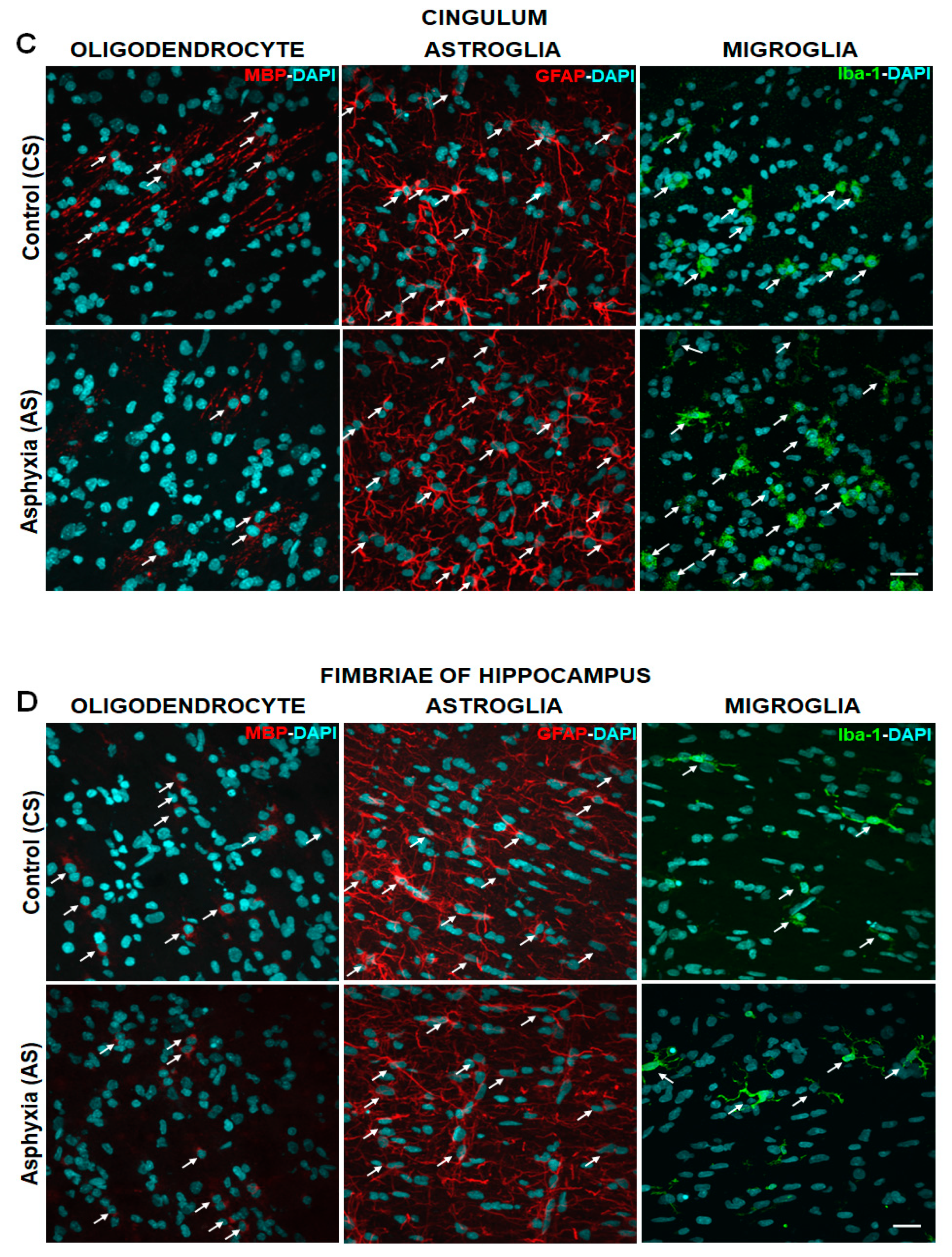
2.4. Effect of PA on Glial Cells Measured in Telencephalon and Fimbriae of Hippocampus at P7, Focusing on Mature OLs (MBP), Astrocytes (GFAP) and Microglia (Iba-1)
2.5. Effect of MSCs Treatment on Cell Death, Number of OL and Myelination at P7
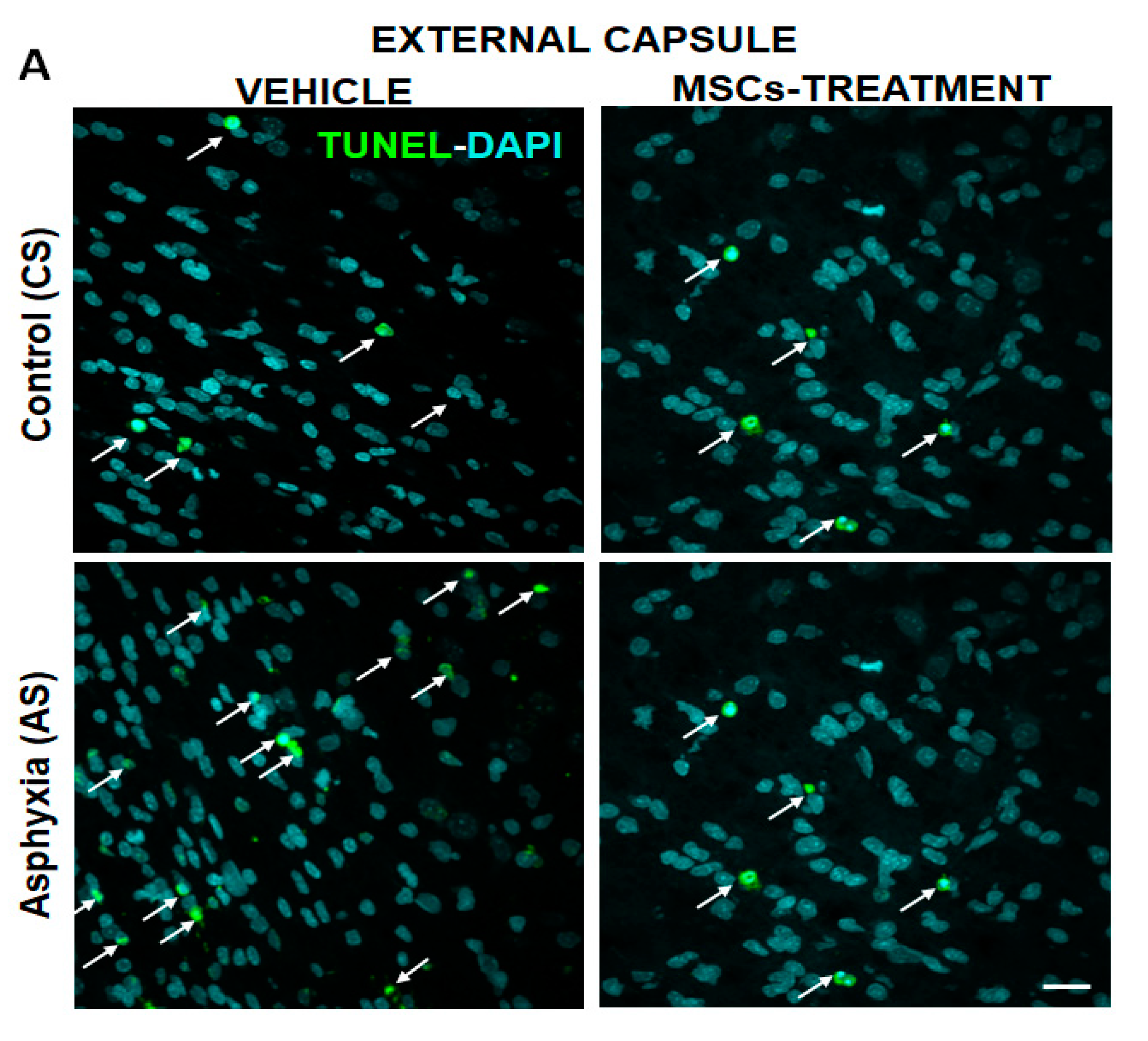
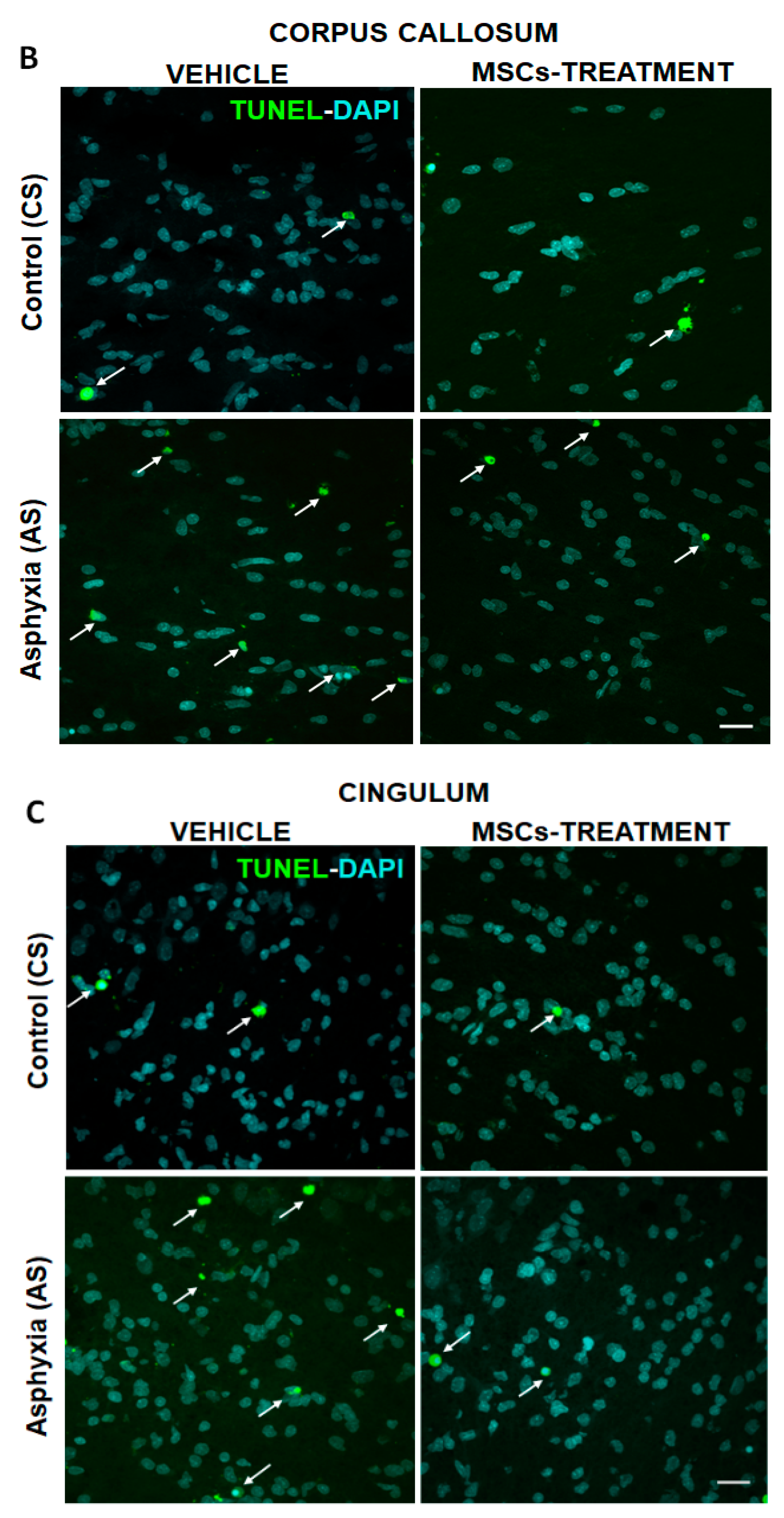
2.5.1. Effect of PA on Apoptotic-Like (TUNEL-DAPI+/mm3) and OL-Specific Cell Death (TUNEL-DAPI-MBP+/mm3) at P7
2.5.2. Effect of MSCs Treatment on Cell Death, Number of OLs and Myelination at P7
2.5.3. Effect of PA and MSCs Treatment on the Number of Mature OLs (MBP-DAPI+/mm3) and Myelination (MBP-Positive Pixels/Total Pixels)
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ethic Statement
4.3. Perinatal Asphyxia
4.4. Isolation, Expansion, and Characterization of Rat Adipose Tissue-Derived MSCs
4.5. Administration of MSCs
4.6. Tissue Sampling for Immunofluorescence
4.7. Antibodies
4.7.1. Primary Antibodies
4.7.2. Secondary Antibodies
4.8. Immunofluorescence
4.9. TUNEL Assay
4.10. Image Processing and Stereological Analysis
4.11. RT-qPCR and Enzyme-Linked Assay (ELISA)
4.11.1. Tissue Sampling
4.11.2. RT-qPCR
4.11.3. ELISA
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-MEM | Alpha-minimum essential medium |
| APC | Allophycocyanin |
| AS | Asphyxia exposed rats |
| a.u. | arbitrary units |
| BCA | Bicinchoninic acid assay |
| BSA | Bovine serum albumin |
| CNS | Central Nervous System |
| CO2 | Carbon dioxide |
| Cox-2 | Cyclooxygenase-2 |
| CS | Caesarean-delivered rat controls |
| DAPI | 4,6 diamino-2-phenylindol |
| ELISA | Enzyme-Linked ImmunoSorbent Assay |
| FBS | Foetal bovine serum |
| FITC | Fluorescein isothiocyanate |
| GFAP | Glial fibrillary acid protein |
| G22 | Gestation day 22 |
| h | Hour |
| HI | Hypoxia/ischemia |
| HIE | Hypoxic-ischemic encephalopathy |
| HRP | Horseradish peroxidase |
| H2O2 | Hydrogen peroxide |
| H2Od | Distilled water |
| Iba-1 | Calcium binding adaptor molecule 1 |
| IF | Immunofluorescence |
| IL-1β | Interleukin 1-beta |
| IL-6 | Interleukin 6 |
| i.c.v. | Intracerebroventricular |
| MAP-2 | Microtubule-associated protein 2 |
| MBP | Myelin basic protein |
| A-MEM | alpha minimum essential medium |
| min | Minute |
| MMLV-RT | Moloney Murine Leukemia Virus Reverse Transcriptase |
| MSCs | Mesenchymal stem cells |
| MSC-S | mesenchymal stem cell-derived secretome |
| NGS | Normal goat serum |
| OL | Oligodendrocytes |
| Olig-1 | Oligodendrocyte Transcription Factor 1 |
| Olig-2 | Oligodendrocyte Transcription Factor 2 |
| OPCs | Oligodendrocyte precursor cells |
| P | Postnatal day |
| PA | Perinatal asphyxia |
| PBS | Phosphate-buffered saline |
| PE | Phycoerythrin |
| preOL | Pre-oligodendrocytes |
| PVL | Periventricular leukomalacia |
| RIPA Buffer | Radio-Immune Precipitation Assay Buffer |
| rpm | revolutions per minute |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SEM | Standard error of the mean |
| SVZ | Subventricular zone |
| TdT | Terminal deoxynucleotidyl transferase |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| TNF-α | Tumor necrosis factor alpha |
| Veh | Vehicle |
References
- Miller, S.P.; Ramaswamy, V.; Michelsol, D.; Barkovich, A.J.; Holshouser, B.; Wycliffe, N.; Glidden, D.V.; Deming, D.; Partridge, J.C.; Wu, Y.W.; et al. Patterns of brain injury in term neonatal encephalopathy. J. Pediatr. 2005, 146, 453–460. [Google Scholar] [CrossRef]
- Volpe, J.J. Hypoxic-Ischemic encephalopathy: Neuropathology and pathogenesis. In Neurology of the Newborn; WB Saunders: London, UK, 1995; pp. 279–313. [Google Scholar]
- Swarte, R.; Lequin, M.; Cherian, P.; Zecic, A.; van Goudoever, J.; Govaert, P. Imaging patterns of brain injury in term-birth asphyxia. Acta Paediatr. 2009, 98, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Marschitz, M.; Morales, P.; Leyton, L.; Bustamante, D.; Klawitter, V.; Espina-Marchant, P.; Allende, C.; Lisboa, F.; Cunich, G.; Jara-Cavieres, A.; et al. Perinatal asphyxia: Current status and approaches towards neuroprotective strategies, with focus on sentinel proteins. Neurotox. Res. 2011, 19, 603–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Marschitz, M.; Neira-Peña, T.; Rojas-Mancilla, E.; Espina-Marchant, P.; Esmar, D.; Pérez, R.; Muñoz, V.; Gutierrez-Hernandez, M.; Rivera, B.; Simola, N.; et al. Perinatal asphyxia: CNS development and deficits with delayed onset. Front. Neurosci. 2014, 8, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahearne, C.E.; Boylan, G.B.; Murray, D.M. Short and long term prognosis in perinatal asphyxia: An update. World J. Clin. Pediatr. 2016, 5, 67–74. [Google Scholar] [CrossRef]
- Davidson, J.O.; Wassink, G.; van den Heuij, L.G.; Bennet, L.; Gunn, A.J. Therapeutic Hypothermia for Neonatal Hypoxic–Ischemic Encephalopathy – Where to from Here? Front. Neurol. 2015, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northington, F.J.; Ferriero, D.M.; Martin, L.J. Neurodegeneration in the thalamus following neonatal hypoxia-ischemia is programmed cell death. Dev. Neurosci. 2001, 23, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]
- Liu, F.; McCullough, L.D. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol. Sin. 2013, 34, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leaw, B.; Nair, S.; Lim, R.; Thornton, C.; Mallard, C.; Hagberg, H. Mitochondria, bioenergetics, and excitotoxicity: New therapeutic targets in perinatal brain injury. Front. Cell. Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Marschitz, M.; Perez-Lobos, R.; Lespay-Rebolledo, C.; Tapia-Bustos, A.; Casanova-Ortiz, E.; Morales, P.; Valdes, J.L.; Bustamante, D.; Cassels, B.K. Targeting sentinel proteins and extrasynaptic glutamate receptors: A therapeutic strategy for preventing the effects elicited by perinatal asphyxia? Neurotox. Res. 2018, 33, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Sävman, K. The Brain and Resuscitation. Neoreviews 2008, 9, e513–e519. [Google Scholar] [CrossRef]
- Perez-Lobos, R.; Lespay-Rebolledo, C.; Tapia-Bustos, A.; Palacios, E.; Vío, V.; Bustamante, D.; Morales, P.; Herrera-Marschitz, M. Vulnerability to a metabolic challenge following perinatal asphyxia evaluated by organotypic cultures: Neonatal nicotinamide treatment. Neurotox. Res. 2017, 32, 426–443. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, A.; Majed, H.; Layfield, R.; Compston, A.; Chandran, S. Oligodendrocytes promote neuronal survival and axonal length by distinct intracellular mechanisms: A novel role for oligodendrocyte-derived glial cell line-derived neurotrophic factor. J. Neurosci. 2003, 23, 4967–4974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Nave, K.M. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Doretto, S.; Malerba, M.; Ramos, M.; Ikrar, T.; Kinoshita, C.; De Mei, C.; Tirotta, E.; Xu, X.; Borrelli, E. Oligodendrocytes as regulators of neuronal networks during early postnatal development. PLoS ONE 2011, 6, e19849. [Google Scholar] [CrossRef]
- Guardia, M.; Pasquini, L.A.; Soto, E.F.; Pasquini, J.M. Apotransferrin-induced recovery after hypoxic/ischaemic injury on myelination. ASN Neuro 2010, 2, e00048. [Google Scholar]
- Kesssaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Baron, W.; Hoekstra, D. On the biogenesis of myelin membranes: Sorting, trafficking and cell polarity. FEBS Lett. 2010, 584, 1760–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggs, J.M. Myelin basic protein: A multifunctional protein. Cell Mol. Life Sci. 2006, 63, 1945–1961. [Google Scholar] [CrossRef]
- Ozgen, H.; Baron, W.; Hoekstra, D.; Kahya, N. Oligodendroglial membrane dynamics in relation to myelin biogenesis. Cell Mol. Life Sci. 2016, 73, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Inouye, H.; Kirschner, D.A. Folding and function of the myelin proteins from primary sequence data. J. Neurosci. Res. 1991, 28, 1–17. [Google Scholar] [CrossRef]
- Nicholas, R.S.; Wing, M.G.; Compston, A. Nonactivated Microglia Promote Oligodendrocyte Precursor Survival and Maturation Through the Transcription Factor NF-kappa B. Eur. J. Neurosci. 2001, 13, 959–967. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Freidin, M.; Fischer, E.; Scherer, S.S.; Abrams, C.K. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 2007, 27, 13949–13957. [Google Scholar] [CrossRef] [Green Version]
- Iacobas, S.; Iacobas, D.A. Astrocyte proximity modulates the myelination gene fabric of oligodendrocytes. Neuron Glia Biol. 2010, 6, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Fan, L.W.; Tien, L.T.; Dai, X.; Zheng, B.; Cai, Z.; Lin, R.C.S.; Bhatt, A. Differential roles of astrocyte and microglia in supporting oligodendrocyte development and myelination in vitro. Brain Behav. 2013, 3, 503–514. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; van Deijk, A.L.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Ling, E.A.; Kaur, C. Hypoxia and myelination deficits in the developing brain. Int. J. Dev. Neurosci. 2018, 70, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.; Fancy, S.; Franklin, R.; Rowitch, D. Olig gene function in CNS development and disease. Glia 2006, 54, 1–10. [Google Scholar] [CrossRef]
- Labombarda, F.; González, A.; Lima, P.; Roig, R.; Guennoun, M.; Schumacher, A.F.; De Nicola, A. Effects of progesterone on oligodendrocyte progenitors, oligodendrocyte transcription factors, and myelin proteins following spinal cord injury. Glia 2009, 57, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The developing oligodendrocyte: Key cellular target in brain injury in the premature infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef]
- Bain, J.R.; Ziegler, A.; Yang, Z.; Levison, S.W.; Sen, E. TGFβ1 stimulates the over-production of white matter astrocytes from precursors of the “brain marrow” in a rodent model of neonatal encephalopathy. PLoS ONE 2010, 5, e9567. [Google Scholar] [CrossRef] [PubMed]
- Mc Tigue, D.M.; Tripathi, R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Oligodendrocytes: Susceptibility to injury and involvement in neurologic disease. Neurology 2009, 72, 1779–1785. [Google Scholar] [CrossRef]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Dimou, L.; Simon, C.; Kirchhoff, F.; Takebayashi, H.; Götz, M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 2008, 28, 10434–10442. [Google Scholar] [CrossRef]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nait-Oumesmar, B.; Picard-Riéra, N.; Kerninon, C.; Baron-Van, E. The role of SVZ-derived neural precursors in demyelinating diseases: From animal models to multiple sclerosis. J. Neurol. Sci. 2008, 265, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Volpe, J.J. Cellular and molecular pathogenesis of periventricular white matter injury. Ment. Retard. Dev. Disabil. Res. 1997, 3, 96–107. [Google Scholar] [CrossRef]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, J.K.; Romanko, M.J.; Rothestein, R.P.; Wood, T.L.; Levison, S.W. Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev. Neurosci. 2001, 23, 203–208. [Google Scholar] [CrossRef]
- Bonestroo, H.J.; Heijnen, C.J.; Groenendaal, F.; van Bel, F.; Nijboer, C.H. Development of cerebral gray and White matter injury and cerebral inflammation over time after inflammatory perinatal asphyxia. Dev. Neurosci. 2015, 37, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C. Arrested oligodendrocyte lineage progression during human cerebral white matter development: Dissociation between the timing of progenitor differentiation and myelinogenesis. J. Neuropathol. Exp. Neurol. 2002, 61, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Ziemka-Nalecz, M.; Janowska, J.; Strojek, L.; Jaworska, J.; Zalewska, T.; Frontczak-Baniewicz, M.; Sypecka, J. Impact of neonatal hypoxia-ischaemia on oligodendrocyte survival, maturation and myelinating potential. J. Cell. Mol. Med. 2018, 22, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Skoff, R.P.; Bessert, D.A.; Barks, J.D.; Song, D.; Cerghet, M.; Silverstein, F. Hypoxic-ischemic injury results in acute disruption of myelin gene expression and death of oligodendroglial precursors in neonatal mice. Int. J. Dev. Neurosci. 2001, 19, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Kohlhauser, C.; Mosgöller, W.; Höger, H.; Lubec, B. Myelination deficits in brain of rats following perinatal asphyxia. Life Sci. 2000, 67, 2355–2368. [Google Scholar] [CrossRef]
- Drury, P.P.; Gunn, E.R.; Bennet, L.; Gunn, A.J. Mechanism of Hypothermic Neuroprotection. Clin. Perinatol. 2014, 41, 161–175. [Google Scholar] [CrossRef]
- Ezquer, M.; Urzua, C.A.; Montecino, S.; Leal, K.; Conget, P.; Ezquer, F. Intravitreal administration of multipotent mesenchymal stromal cells triggers a cytoprotective microenvironment in the retina of diabetic mice. Stem Cell Res. Ther. 2016, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Janowska, J.; Sypecka, J. Therapeutic strategies for leukodysthrophic disorders resulting from perinatal asphyxia: Focus on myelinating oligodendrocytes. Mol. Neurobiol. 2018, 55, 4388–4402. [Google Scholar]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Mesenchymal stem cell treatment after neonatal hypoxic-ischemic brain injury improves behavioral outcome and induces neuronal and oligodendrocyte regeneration. Brain Behav. Immun. 2010, 24, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Repeated mesenchymal stem cell treatment after neonatal hypoxia-ischemia has distinct effects on formation and maturation of new neurons and oligodendrocytes leading to restoration of damage, corticospinal motor tract activity, and sensorimotor function. J. Neurosci. 2010, 30, 9603–9611. [Google Scholar] [CrossRef] [Green Version]
- Drago, D.; Cossetti, C.; Iraci, N.; Gaude, E.; Musco, G.; Bachi, A.; Pluchino, S. The stem cell secretome and its role in brain repair. Biochimie 2013, 95, 2271–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wei, Z.Z.; Gu, X.; Zhang, J.Y.; Zhang, Y.; Li, J.; Wei, L. Intranasal delivery of hypoxia-preconditioned bone marrow-derived mesenchymal stem cells enhanced regenerative effects after intracerebral hemorrhagic stroke in mice. Exp. Neurol. 2015, 272, 78–87. [Google Scholar] [CrossRef]
- Wei, Z.Z.; Gu, X.; Ferdinand, A.; Lee, J.H.; Ji, X.; Ji, X.M.; Yu, S.P.; Wei, L. Intranasal delivery of bone marrow mesenchymal stem cells improved neurovascular regeneration and rescued neuropsychiatric deficits after neonatal stroke in rats. Cell Transplant. 2015, 24, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Nagai, A.; Wakabayashi, K.; Narantuya, D.; Kobayashi, S.; Yamaguchi, S.; Kim, S.U. Mesenchymal stem cell transplantation modulates neuroinflammation in focal cerebral ischemia: Contribution of fractalkine and IL-5. Neurobiol. Dis. 2011, 41, 717–724. [Google Scholar] [CrossRef]
- Donega, V.; Nijboer, C.H.; Braccioli, L.; Slaper-Cortenbach, I.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Intranasal administration of human MSC for ischemic brain injury in the mouse: In vitro and in vivo neurodegenerative functions. PLoS ONE 2014, 9, e112339. [Google Scholar]
- Voorn, P.; Kalsbeek, A.; Jorritsma-Byham, B.; Groenewegen, H.J. The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 1988, 25, 857–887. [Google Scholar] [CrossRef]
- Vitalis, T.; Cases, O.; Parnavelas, J.G. Development of the dopaminergic neurons in the rodent brainstem. Exp. Neurol. 2005, 191, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anna, E.; Chen, Y.; Engidawork, E.; Andersson, K.; Lubec, G.; Luthman, J.; Herrera-Marschitz, M. Delayed neuronal death following perinatal asphyxia rat. Exp. Brain Res. 1997, 115, 105–115. [Google Scholar] [CrossRef]
- Downes, N.; Mullins, P. The development of myelin in the brain of the juvenile rat. Toxicol. Pathol. 2014, 42, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Vannucci, R.G.; Vannucci, S.J. Perinatal hypoxic-ischemic brain damage evolution of an animal model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.; Collin, L.; Borrelli, E. Oligodendrocyte ablation impairs cerebellum development. Development 2003, 130, 4709–4718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, L.; Doretto, S.; Malerba, M.; Ruat, M.; Borrelli, E. Oligodendrocyte ablation affects the coordinated interaction between granule and Purkinje neurons during cerebellum development. Exp. Cell Res. 2007, 313, 2946–2957. [Google Scholar] [CrossRef]
- Van Tilborg, E.; de Theije, C.; van Hal, M.; Wagenaar, N.; de Vries, L.; Benders, M.; Rowitch, D.; Nijboer, C. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Craig, A.; Ling Luo, N.; Beardsley, D.J.; Wingate-Pearse, N.; Walker, D.W.; Hohimer, A.R.; Back, S.A. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 2003, 181, 231–240. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Connor, J.R.; Mauger, D.T.; Palmer, C.; Smith, M.B.; Towfighi, J.; Vannucci, S.J. Rat model of perinatal hypoxic ischemic brain damage. J. Neurosci. Res. 1999, 55, 158–163. [Google Scholar] [CrossRef]
- Dean, J.M.; Moravec, M.D.; Grafe, M.; Abend, N.; Ren, J.; Gong, X.; Volpe, J.J.; Jensen, F.E.; Hohimer, A.R.; Back, S.A. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev. Neurosci. 2011, 33, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Ferreira, E.; Hristova, M. Plasticity in the Neonatal Brain following Hypoxic-Ischaemic Injury. Neural. Plast. 2016, 2016, 4901014. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Fiedler, J.L.; Andrés, S.; Berrios, C.; Huaiquin, P.; Bustamante, D.; Cardenas, S.; Parra, E.; Herrera-Marschitz, M. Plasticity of hippocampus following perinatal asphyxia: Effects on postnatal apoptosis and neurogenesis. J. Neurosci. Res. 2008, 86, 2650–2662. [Google Scholar] [CrossRef]
- Morales, P.; Reyes, P.; Klawitter, V.; Huaiquin, P.; Bustamante, D.; Fiedler, J.L.; Herrera-Marschitz, M. Effects of perinatal asphyxia on cell proliferation and neuronal phenotype evaluated with organotypic hippocampal cultures. Neuroscience 2005, 135, 421–431. [Google Scholar] [CrossRef]
- Morales, P.; Huaiquin, P.; Bustamante, D.; Fiedler, J.L.; Herrera-Marschitz, M. Perinatal asphyxia induces neurogenesis in hippocampus: An organotypic culture study. Neurotox. Res. 2007, 12, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Bustos, A.; Perez-Lobos, R.; Vío, V.; Lespay-Rebolledo, C.; Palacios, E.; Chiti-Morales, A.; Bustamante, D.; Herrera-Marschitz, M.; Morales, P. Modulation of Postnatal Neurogenesis by Perinatal Asphyxia: Effect of D1 and D2 Dopamine Receptor Agonists. Neurotox. Res. 2017, 31, 109–121. [Google Scholar] [CrossRef]
- Meijer, D.H.; Kane, M.F.; Mehta, S.; Liu, H.; Harrington, E.; Taylor, C.M.; Stiles, C.D.; Rowitch, D.H. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat. Rev. Neurosci. 2012, 13, 819–831. [Google Scholar] [CrossRef]
- Arnett, H.A.; Fancy, S.P.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.; Stiles, C.D. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 2004, 306, 2111–2115. [Google Scholar] [CrossRef]
- Mei, F.; Wang, H.; Liu, S.; Niu, J.; Wang, L.; He, Y.; Etxeberria, A.; Chan, J.R.; Xiao, L. Stage-specific deletion of Olig2 conveys opposing functions on differentiation and maturation of oligodendrocytes. J. Neurosci. 2013, 33, 8454–8462. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; Wassink, G.; Galinsky, R.; Xu, B.; Mathai, S.; Dhillon, S.; van den Heuik, L.; Davidson, J.; Weaver-Mikaere, L.; Bennet, L.; et al. Protective effects of delayed intraventricular TLR7 agonist administration on cerebral white and gray matter following asphyxia in the preterm fetal sheep. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.R.; Sun, T.; Zhu, Z.; Ma, N.; Garcia, M.; Stiles, C.D.; Rowitch, D.H. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 2002, 109, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.E.; Greenberg, M.E.; Stiles, C.D. Basic helix-loop-helix factors in cortical development. Neuron 2003, 39, 13–25. [Google Scholar] [CrossRef] [Green Version]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative Stress Disrupts Oligodendrocyte Maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Xue, X.; Fu, J. Effect of OLIG1 on the development of oligodendrocytes and myelination in a neonatal rat PVL model induced by hypoxia-ischemia. Mol. Med. Rep. 2015, 11, 2379–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerwald, K.D.; Popko, B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-gamma. J. Neurosci. Res. 1998, 52, 230–239. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arevalo-Martina, A.; Guaza, C. LPS/IFNγ cytotoxicity in oligodendroglial cells: Role of nitric oxide and protection by the anti-inflammatory cytokine IL-10. Eur. J. Neurosci. 2001, 13, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, K.; Genc, S.; Baskin, H.; Semin, I. Erythropoietin decreases cytotoxicity and nitric oxide formation induced by inflammatory stimuli in rat oligodendrocytes. Physiol. Res. 2006, 55, 33–38. [Google Scholar] [PubMed]
- Neira-Peña, T.; Rojas-Mancilla, E.; Muñoz-Vio, V.; Perez, R.; Gutierrez-Hernandez, M.; Bustamante, D.; Morales, P.; Hermoso, M.A.; Gebicke-Haerter, P.; Herrera-Marschitz, M. Perinatal Asphyxia Leads to PARP-1 Overactivity, p65 Translocation, IL-1β and TNF-α Overexpression, and Apoptotic-Like Cell Death in Mesencephalon of Neonatal Rats: Prevention by Systemic Neonatal Nicotinamide Administration. Neurotox. Res. 2015, 27, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neira-Peña, T.; Espina-Marchant, P.; Rojas-Mancilla, E.; Esmar, D.; Kraus, C.; Muñoz, V.; Pérez, R.; Rivera, B.; Bustamante, D.; Valdés, J.L.; et al. Molecular, Cellular, and Behavioral effects produced by perinatal asphyxia: Protection by Poly(ADP-ribose) Polymerase 1 (PARP1) Inhibition. In Handbook of Neurotoxicity; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Tjalkens, R.B.; Popichak, K.A.; Kirkley, K.A. Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv. Neurobiol. 2017, 18, 159–181. [Google Scholar] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [PubMed]
- O’Callaghan, J.P.; Sriram, K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin. Drug Saf. 2005, 4, 433–442. [Google Scholar] [CrossRef]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.; Schousboe, A.; Haydon, P.G.; Stout, R.F.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial cells in (patho)physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Robertson, N.J.; Lingam, I.; Meehan, C.; Martinello, K.A.; Avdic-Belltheus, A.; Stein, L.; Tachrount, M.; Price, D.; Sokolska, M.; Bainbridge, A.; et al. High-dose melatonin and ethanol excipient combined with therapeutic hypothermia in a newborn piglet asphyxia model. Sci. Rep. 2020, 10, 3898. [Google Scholar] [CrossRef] [Green Version]
- Labat-Moleur, F.; Guillermet, C.; Lorimier, P.; Robert, C.; Lantuejoul, S.; Bramilla, E.; Negoescu, A. TUNEL apoptotic cell detection in tissue sections: Critical evaluation and improvement. J. Histochem. Cytochem. 1998, 46, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Casaccia-Bonnefil, P.; Tikoo, R.; Kiyokawa, H.; Friedrich, V.; Chao, M.V.; Koff, A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27kip1. Genes Dev. 1997, 11, 2335–2346. [Google Scholar] [CrossRef] [Green Version]
- Durand, B.; Gao, F.B.; Raff, M. Accumulation of the cyclin-dependent kinase inhibitor p27/kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997, 16, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Tikoo, R.; Osterhout, D.J.; Casaccia-Bonnefil, P.; Seth, P.; Koff, A.; Chao, M.V. Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell-cycle growth arrest. J. Neurobiol. 1998, 36, 431–440. [Google Scholar] [CrossRef]
- Ohtaki, H.; Ylostalo, J.H.; Foraker, J.E.; Robinson, A.P.; Reger, R.L.; Shioda, S.; Prockop, D.J. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc. Natl. Acad. Sci. USA 2008, 105, 14638–14643. [Google Scholar] [CrossRef] [Green Version]
- Van Velthoven, C.T.; Kavelaars, A.; van Bel, F.; Heijnen, C.J. Regeneration of the ischemic brain by engineered stem cells: Fuelling endogenous repair processes. Brain Res. Rev. 2009, 61, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Farfan, N.; Carril, J.; Redel, M.; Zamorano, M.; Araya, M.; Monzon, E.; Alvarado, R.; Contreras, N.; Tapia-Bustos, A.; Quintanilla, M.E.; et al. Intranasal Administration of Mesenchymal Stem Cell Secretome Reduces Hippocampal Oxidative Stress, Neuroinflammation and Cell Death, Improving the Behavioral Outcome Following Perinatal Asphyxia. Int. J. Mol. Sci. 2020, 21, 680. [Google Scholar] [CrossRef] [PubMed]
- Vizoso, F.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal Stem Cell Secretome: Toward Cell-Free Therapeutic Strategies in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Halabian, R.; Fooladi, A.A.I. A revealing review of mesenchymal stem cells therapy, clinical perspectives and Modification strategies. Stem Cell Investig. 2019, 6, 34–52. [Google Scholar] [CrossRef]
- Ezquer, F.; Giraud-Billoud, M.; Carpio, D.; Cabezas, F.; Conget, P.; Ezquer, M. Pro-regenerative microenvironment triggered by donor mesenchymal stem cells preserves renal function and structure in mice with severe diabetes mellitus. BioMed Res. Int. 2015, 2015, 164703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, Y.; Ezquer, F.; Quintanilla, M.E.; Morales, P.; Ezquer, M.; Herrera-Marschitz, M. Intracerebral Stem Cell Administration Inhibits Relapse-like Alcohol Drinking in Rats. Alcohol Alcohol. 2017, 52, 1–4. [Google Scholar] [CrossRef]
- Monin, P.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2015, 20, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. CS (n = 75; m = 20) | 40 min | P1 | P7 | P14 |
|---|---|---|---|---|
| Survival (%) | 100 | 100 | 100 | 100 |
| Body weight (g) | 6.07 ± 0.08 | 6.60 ± 0.09 | 14.86 ± 0.93 | 23.05 ± 3.36 |
| Gasping (%) | 1.053 ± 1.05 | 0 | 0 | 0 |
| Respiratory frequency (events/min) | 74.29 ± 1.36 | 81.33 ± 3.77 | 94.67 ± 4.92 | 100 ± 5.59 |
| Skin color (P%) | 98 ± 1 | 100 | 100 | 100 |
| Spontaneous Movements (0–4) | 3.74 ± 0.07 | 4 | 4 | 4 |
| Vocalizations (%) | 98 ± 1 | 100 | 100 | 100 |
| B. AS (n = 72; m = 20) | ||||
| Survival (%) | 63 ± 6 (by ~40%)b **** | 100 | 100 | 100 |
| Body weight (g) | 7.38 ± 0.64 | 7.73 ± 0.91 | 13.90 ± 0.63 | 16.54 ± 2.49 |
| Gasping (%) | 36.76 ± 10.51 (> 35×)b ** | 0 | 0 | 0 |
| Respiratory frequency (events/min) | 32.53 ± 2.00 (by ~40%)b **** | 59.00 ± 1.92 (by ~20%)b **** | 75.20 ± 6.47 (by ~20%)b **** | 86.00 ± 2.00 (by ~14%)b **** |
| Skin color (P%) | 0 | 100 | 100 | 100 |
| Spontaneous Movements (0–4) | 0.50 ± 0.10 (by ~90%)b **** | 4 | 4 | 4 |
| Vocalizations (%) | 44.72 ± 10.55 (by ~55%)b **** | 100 | 100 | 100 |
| Experimental Groups | P1 | P7 | P14 | |||
|---|---|---|---|---|---|---|
| A. Caesarean-Delivered (CS) | DAPI (cells/ mm3) | MBP (Pixels/Total Pixels) | DAPI (cells/ mm3) | MBP (Pixels/Total Pixels) | DAPI (cells/mm3) | MBP (Pixels/Total Pixels) |
| External capsule | 374,444 ± 45,042 | nd | 355,067 ± 17,917 | 20.54 ± 1.14 | 367,240 ± 21,229 | 49.76 ± 0.12 (>2×)a **** |
| Corpus callosum | 262,787 ± 68,397 | nd | 235,916 ± 24,303 | 4.27 ± 1.03 | 325,644 ± 41,589 | 49.97 ± 0.15 (>10×)a **** |
| Cingulum | 378,496 ± 105,892 | nd | 300,722 ± 20,739 | 7.92 ± 0.82 | 388,988 ± 27,324 | 49.39 ± 0.33 (>6×)a **** |
| Fimbriae of hippocampus | 240,763 ± 67,331 | nd | 264,911 ± 19,963 | 1.94 ± 0.59 | 449,593 ± 51,446 | 49.97 ± 0.16 (>25×)a **** |
| B. Asphyxia-Exposed (AS); 21 Min Asphyxia | ||||||
| External capsule | 521,962 ± 114,815 | nd | 392,315 ± 29,773 | 9.88 ± 2.73 (by 50%)b * | 398,727 ± 40,493 | 49.67 ± 0.07 (>5×)a **** |
| Corpus callosum | 199,665 ± 55,743 | nd | 285,083 ± 19,983 | 1.51 ± 0.47 (by 60%)b * | 310,779 ± 37,973 | 49.43 ± 0.14 (>30×)a **** |
| Cingulum | 438,835 ± 95,565 | nd | 346,825 ± 22,842 | 2.29 ± 0.62 (by~70%)b *** | 411,625 ± 27,658 | 49.61 ± 0.09 (>20×)a **** |
| Fimbriae of hippocampus | 269,789 ± 45,231 | nd | 286,527 ± 24,323 | 1.49 ± 0.62 | 344,219 ± 31,841 | 48.65 ± 1.19 (>30×)a **** |
| Gene Transcripts (mRNA) (Fold/Change) | P1 | P7 | P14 |
|---|---|---|---|
| A. Caesarean Delivered (CS) | CS | CS | CS |
| MBP | 1.000 ± 0.191 | 23.23 ± 4.01 (>20×)a **** | 1165 ± 258.90 (>1000×)a **** |
| Olig-1 | 1.000 ± 0.098 | 1.37 ± 0.05 | 2.315 ± 0.13 (>2×)a **** |
| Olig-2 | 1.000 ± 0.076 | 1.24 ± 0.03 | 1.826 ± 0.181 (> 1.8×)a **** |
| IL-1β | 1.000 ± 0.101 | 1.00 ± 0.13 | 2.36 ± 0.18 (>2×)a ** |
| IL-6 | 1.000 ± 0.201 | 0.70 ± 0.03 | 1.86 ± 0.17 (>1.8×)a *** |
| TNF-α | 1.000 ± 0.208 | 2.14 ± 0.38 | 7.02 ± 3.63 |
| Cox-2 | 1.000 ± 0.255 | 5.84 ± 1.50 (>5×)a ** | 33.56 ± 8.76 (>30×)a ** |
| B. Asphyxia-Exposed (AS) | AS | AS | AS |
| MBP | 0.91 ± 0.04 | 35.63 ± 6.76 (>35×)a **** | 768.90 ± 201.30 (>800×)a *** |
| Olig-1 | 1.23 ± 0.05 | 2.06 ± 0.36 (>1.6×)a * (>1.4×)b * | 2.40 ± 0.25 (>1.9×)a **** |
| Olig-2 | 1.33 ± 0.15 | 1.61 ± 0.29 | 1.81 ± 0.21 |
| IL-1β | 1.50 ± 0.27 | 1.10 ± 0.08 | 2.46 ± 0.47 |
| IL-6 | 0.78 ± 0.12 | 1.16 ± 0.13 (>1.6×)b ** | 1.46 ± 0.20 (>1.8×)a * |
| TNF-α | 2.22 ± 0.67 | 8.32 ± 3.63 | 40.42 ± 25.11 (>18×)a * |
| Cox-2 | 1.16 ± 0.34 | 9.80 ± 1.59 (>8×)a **** | 23.03 ± 6.00 (>20×)a ** |
| Experimental Groups | P7 | |||
|---|---|---|---|---|
| A. Caesarean Delivered (CS) | DAPI cells/mm3 | MBP-DAPI cells/mm3 | GFAP-DAPI cells/mm3 | Iba-1-DAPI cells/mm3 |
| External capsule | 346,895 ± 12,632 | 18,932 ± 1960 | 18,890 ± 1648 | 14,020 ± 1808 |
| Corpus callosum | 214,600 ± 14,080 | 10,810 ± 1668 | 19,090 ± 3229 | 8519 ± 561.6 |
| Cingulum | 294,902 ± 14,140 | 12,439 ± 1616 | 19,510 ± 3277 | 10,360 ± 955.6 |
| Fimbriae of hippocampus | 275,700 ± 15,350 | 8811 ± 2405 | 13,210 ± 2487 | 5957 ± 1027 |
| B. Asphyxia-Exposed (AS) | ||||
| External capsule | 370,346 ± 24,821 | 8603 ± 982.7 (by~50%)b ** | 21,150 ± 3637 | 15,840 ± 1429 |
| Corpus callosum | 237,200 ± 20,170 | 3413 ± 637.7 (by~65%)b ** | 21,280 ± 579.6 | 7380 ± 848.8 |
| Cingulum | 334,385 ± 18,199 | 6643 ± 1121 (by~45%)b * | 30,150 ± 5358 | 17,800 ± 3047 (>1.5×)b * |
| Fimbriae of hippocampus | 270,800 ± 19,810 | 4101 ± 2144 | 10,640 ± 591.70 | 6014 ± 909.1 |
| Experimental Groups | P7 | ||
|---|---|---|---|
| A. CS-Vehicle | DAPI+/mm3 | TUNEL-DAPI+/mm3 | TUNEL-MBP-DAPI+/mm3 |
| External capsule | 382,789 ± 29,017 | 8145 ± 3543 | 152 ± 92.74 |
| Corpus callosum | 196,023 ± 12,124 | 5239 ± 1007 | nd |
| Cingulum | 346,834 ± 7511 | 7310 ± 2231 | 1138 ± 915.80 |
| B. CS-MSCs | P7 | ||
| External capsule | 378,613 ± 16,522 | 9198 ± 2064 | 277 ± 191.20 |
| Corpus callosum | 203,899 ± 7019 | 3975 ± 476 | nd |
| Cingulum | 379,610 ± 4365 | 4402 ± 1541 | nd |
| C. AS-Vehicle | P7 | ||
| External capsule | 341,872 ± 12,628 | 22,837 ± 3732 (> 2.5×)a *** | 1034 ±132.20 (>6.5×)a **** |
| Corpus callosum | 241,023 ± 21,694 | 9276 ± 1942 (>1.7×)a * | 261 ± 124.30 |
| Cingulum | 332,495 ± 28,634 | 14,333 ± 2035 (>1.9×)a * | 786 ± 423.90 |
| D. AS-MSCs | P7 | ||
| External capsule | 404,578 ± 21,398 | 8145 ± 3543 (by ~60%)b ** | 143 ± 89.23 (by 85%)b **** |
| Corpus callosum | 220,892 ± 8903 | 4523 ± 642 (by ~50%)b * | nd |
| Cingulum | 324,002 ± 44,070 | 7147 ± 1790 (by ~50%)b ** | nd |
| Experimental Groups | P7 | |
|---|---|---|
| A. CS-Vehicle | MBP-DAPI+/mm3 | MBP (Pixels/Total Pixels) |
| External capsule | 13,833 ± 1801 | 19.60 ± 1.32 |
| Corpus callosum | 6126 ± 1345 | 2.66 ± 0.44 |
| Cingulum | 9172 ± 2078 | 9.27 ± 1.61 |
| B. CS-MSCs | ||
| External capsule | 13,632 ± 1783 | 16.36 ± 3.05 |
| Corpus callosum | 5972 ± 2775 | 2.24 ± 0.46 |
| Cingulum | 6572 ± 1585 | 8.91 ± 1.49 |
| C. AS-Vehicle | ||
| External capsule | 6690 ± 2173 (by ~50%)a * | 13.08 ± 0.81 (by ~30%)a ** |
| Corpus callosum | 3942 ± 1255 | 1.54 ± 0.42 |
| Cingulum | 3840 ± 509.30 (by ~55%)a * | 4.52 ± 0.10 (by 40%)a * |
| D. AS-MSCs | ||
| External capsule | 13,628 ± 2237 (>2×)b * | 17.53 ± 1.70 (>1.3×)b * |
| Corpus callosum | 7835 ± 2339 | 2.73 ± 0.62 |
| Cingulum | 6194 ± 2145 (>1.6×)b * | 7.07 ± 1.47 (>1.5×)b * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapia-Bustos, A.; Lespay-Rebolledo, C.; Vío, V.; Pérez-Lobos, R.; Casanova-Ortiz, E.; Ezquer, F.; Herrera-Marschitz, M.; Morales, P. Neonatal Mesenchymal Stem Cell Treatment Improves Myelination Impaired by Global Perinatal Asphyxia in Rats. Int. J. Mol. Sci. 2021, 22, 3275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063275
Tapia-Bustos A, Lespay-Rebolledo C, Vío V, Pérez-Lobos R, Casanova-Ortiz E, Ezquer F, Herrera-Marschitz M, Morales P. Neonatal Mesenchymal Stem Cell Treatment Improves Myelination Impaired by Global Perinatal Asphyxia in Rats. International Journal of Molecular Sciences. 2021; 22(6):3275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063275
Chicago/Turabian StyleTapia-Bustos, Andrea, Carolyne Lespay-Rebolledo, Valentina Vío, Ronald Pérez-Lobos, Emmanuel Casanova-Ortiz, Fernando Ezquer, Mario Herrera-Marschitz, and Paola Morales. 2021. "Neonatal Mesenchymal Stem Cell Treatment Improves Myelination Impaired by Global Perinatal Asphyxia in Rats" International Journal of Molecular Sciences 22, no. 6: 3275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063275