
The GNAQ T96S Mutation Affects Cell Signaling and Enhances the Oncogenic Properties of Hepatocellular Carcinoma

 ,
,
Abstract
:1. Introduction
2. Results
2.1. The Cancer Genome Atlas (TCGA) Genotype Data in Liver Cancer Patients with HCC
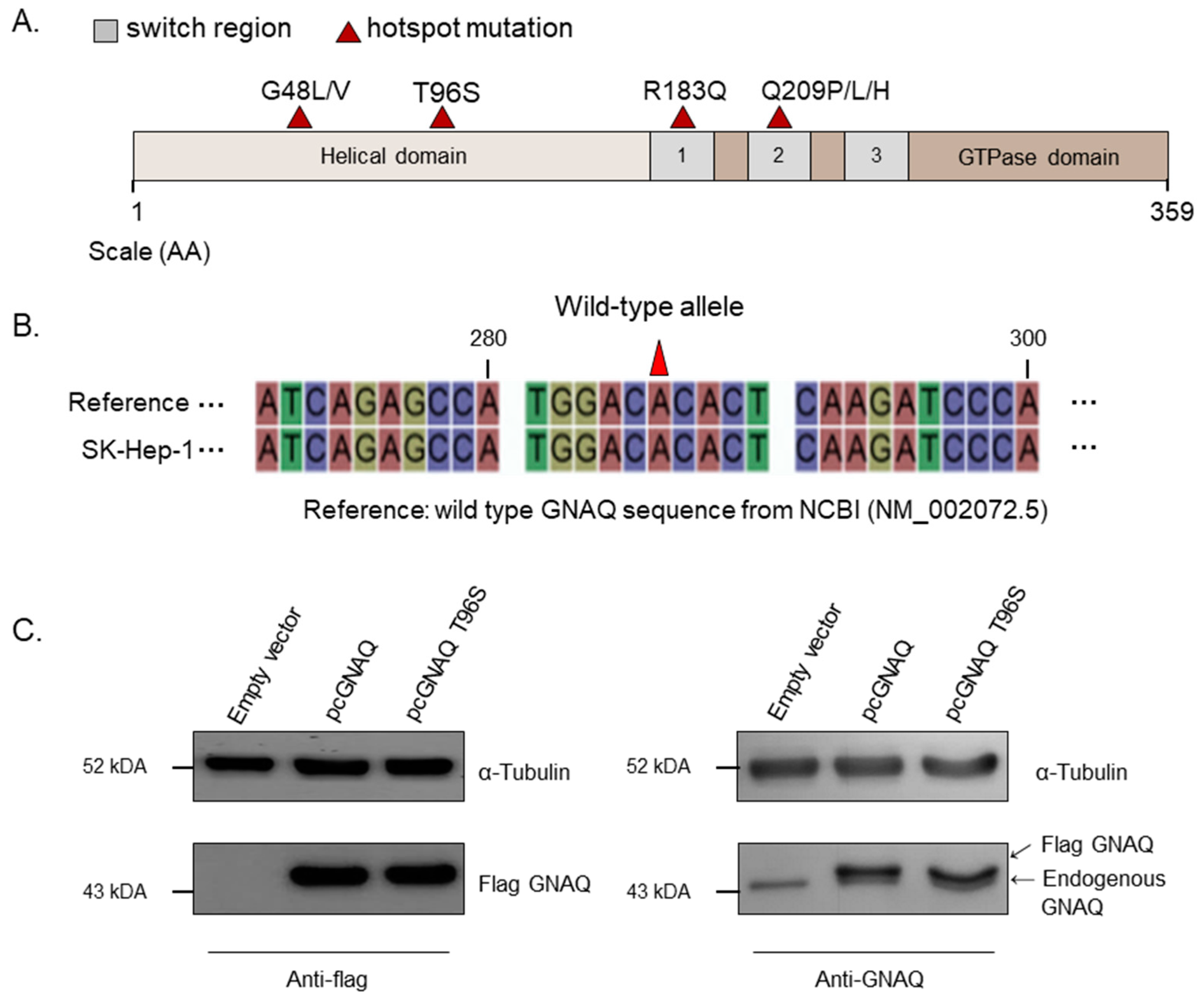
2.2. Sequencing and Overexpression of the GNAQ T96S Hotspot Mutation in SK-Hep-1 Cells
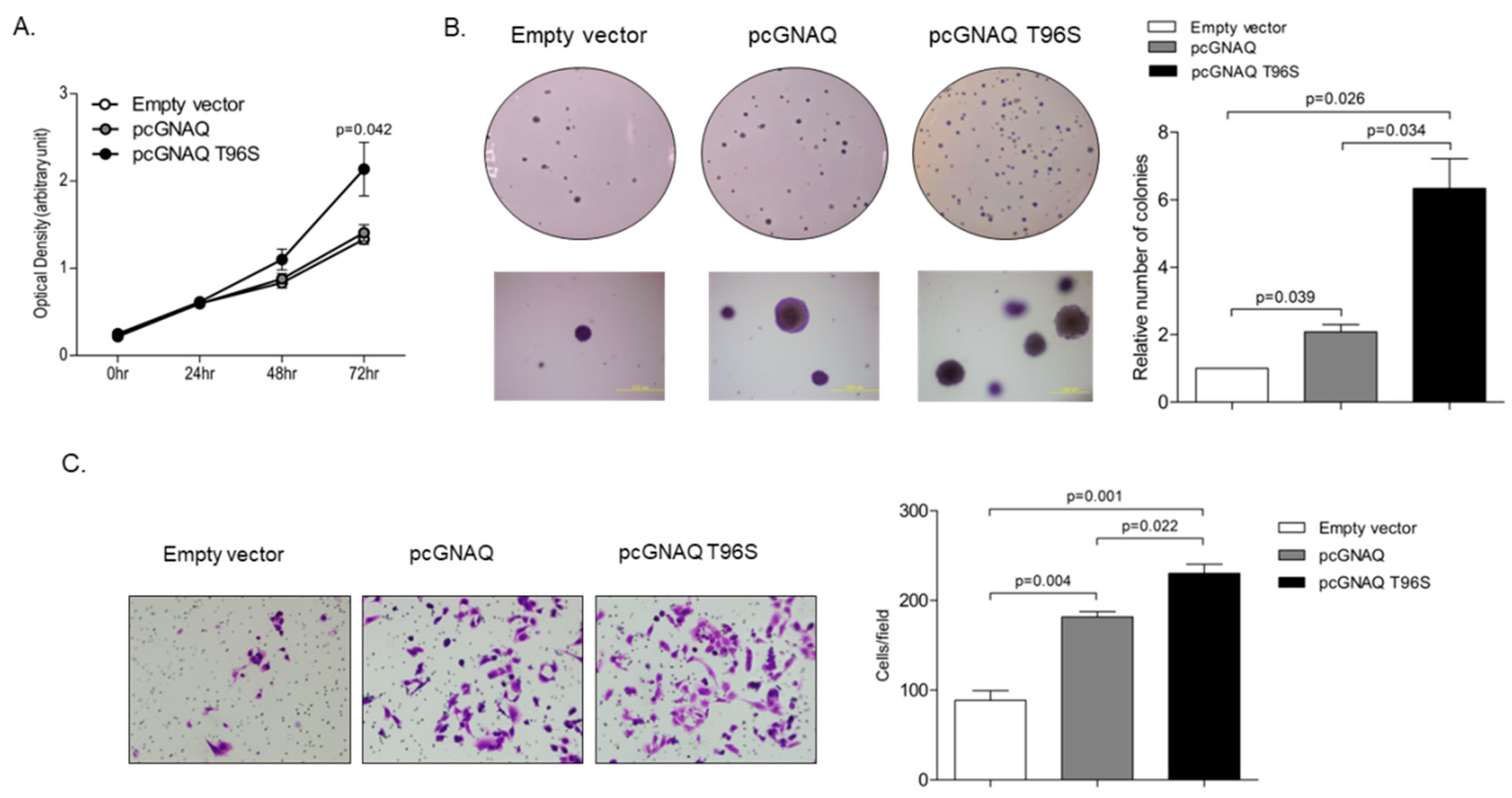
2.3. Effect of GNAQ and GNAQ T96S on Cell Proliferation, Anchorage-Independent Growth, and Migration in SK-Hep-1 Cells
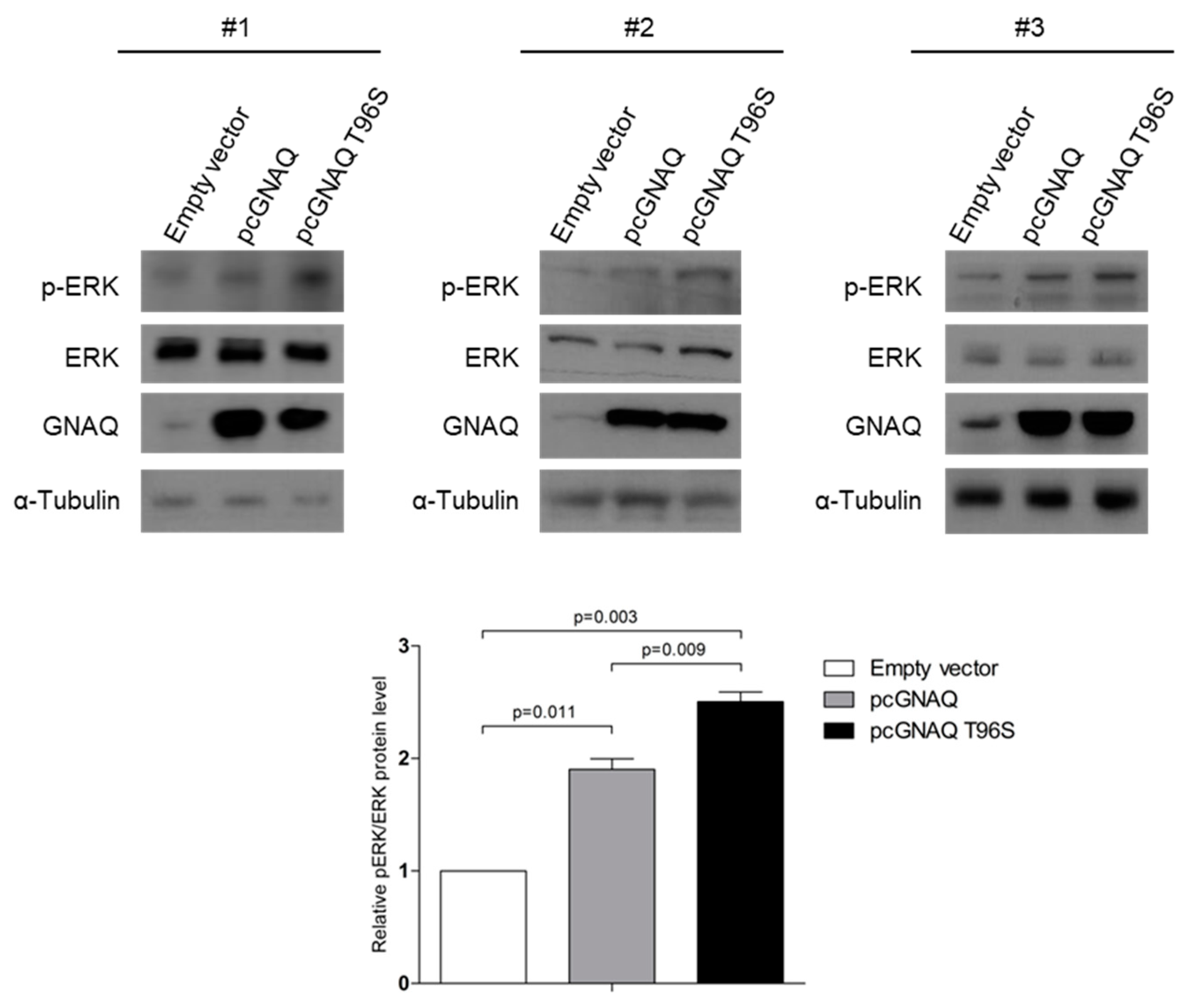
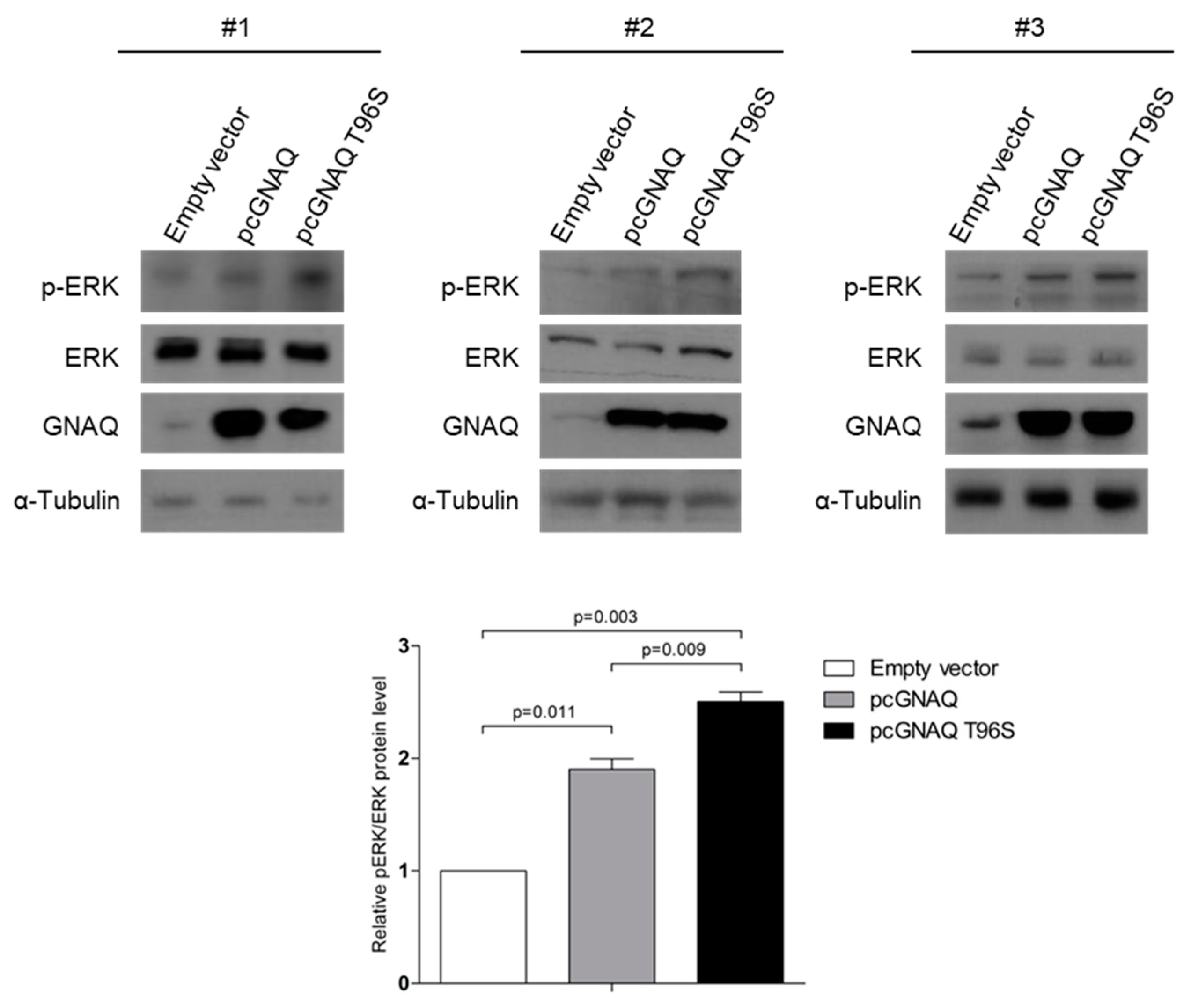
2.4. GNAQ and GNAQ T96S Enhance ERK Signaling in SK-Hep-1 Cells
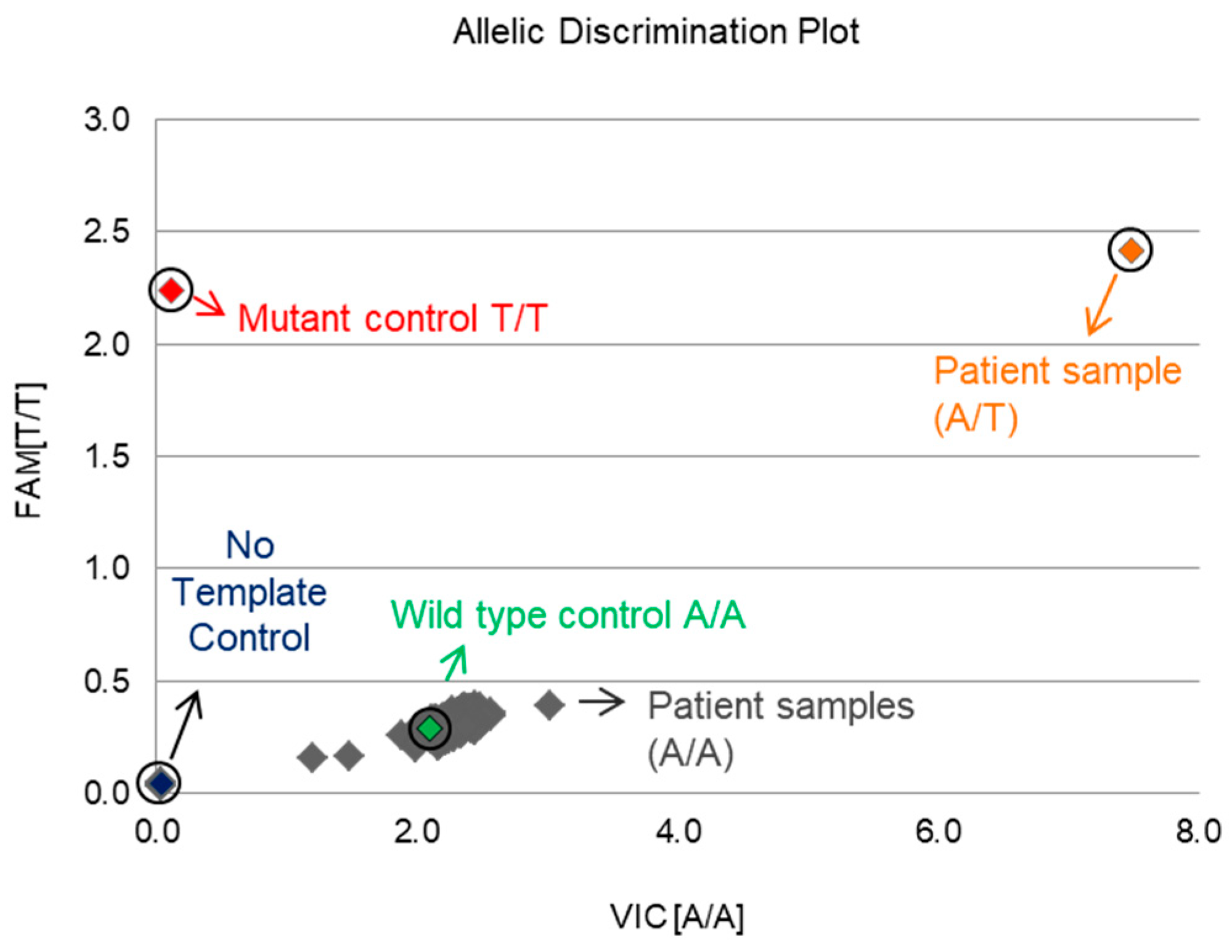
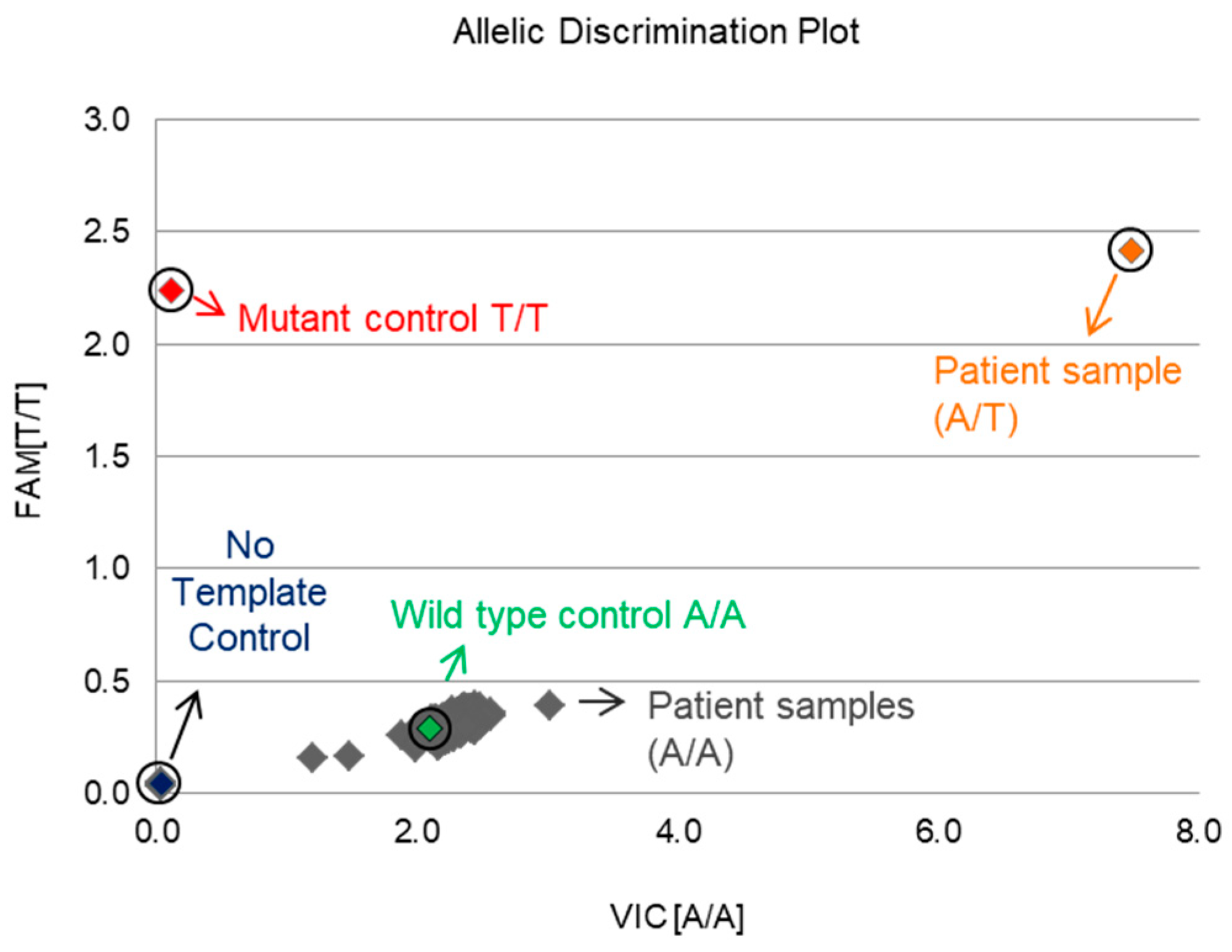
2.5. Screening for the GNAQ T96S (rs753716491) Mutation in the Korean HCC Population
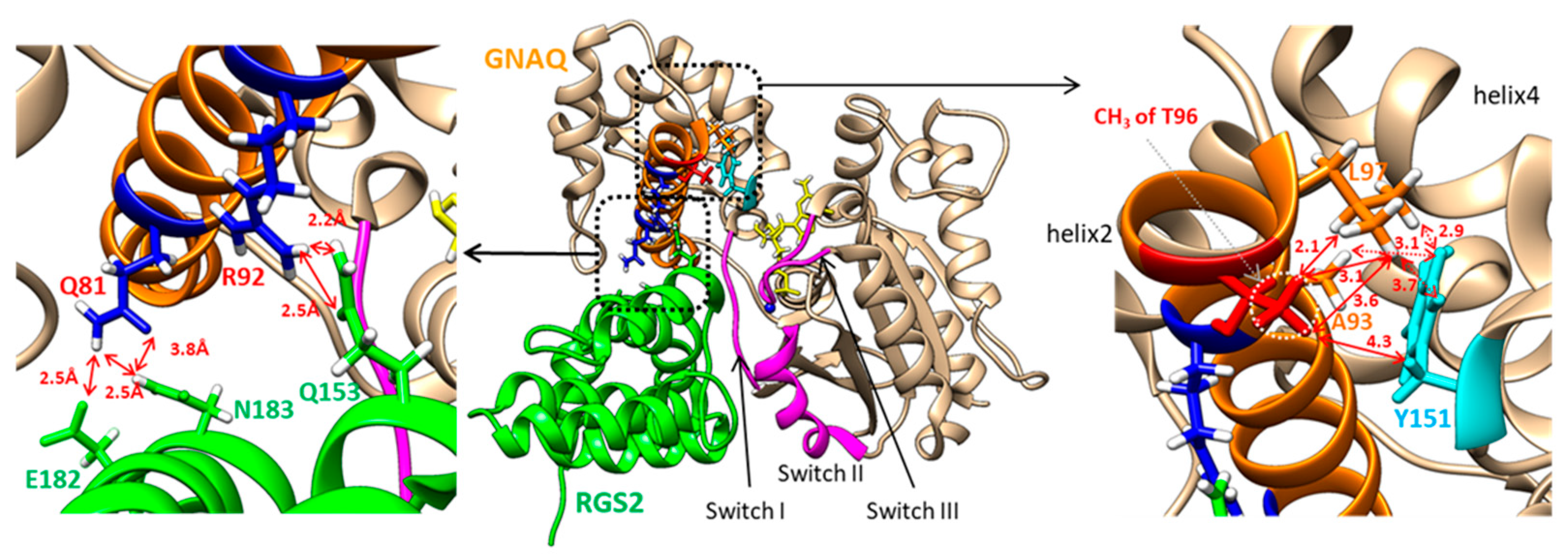
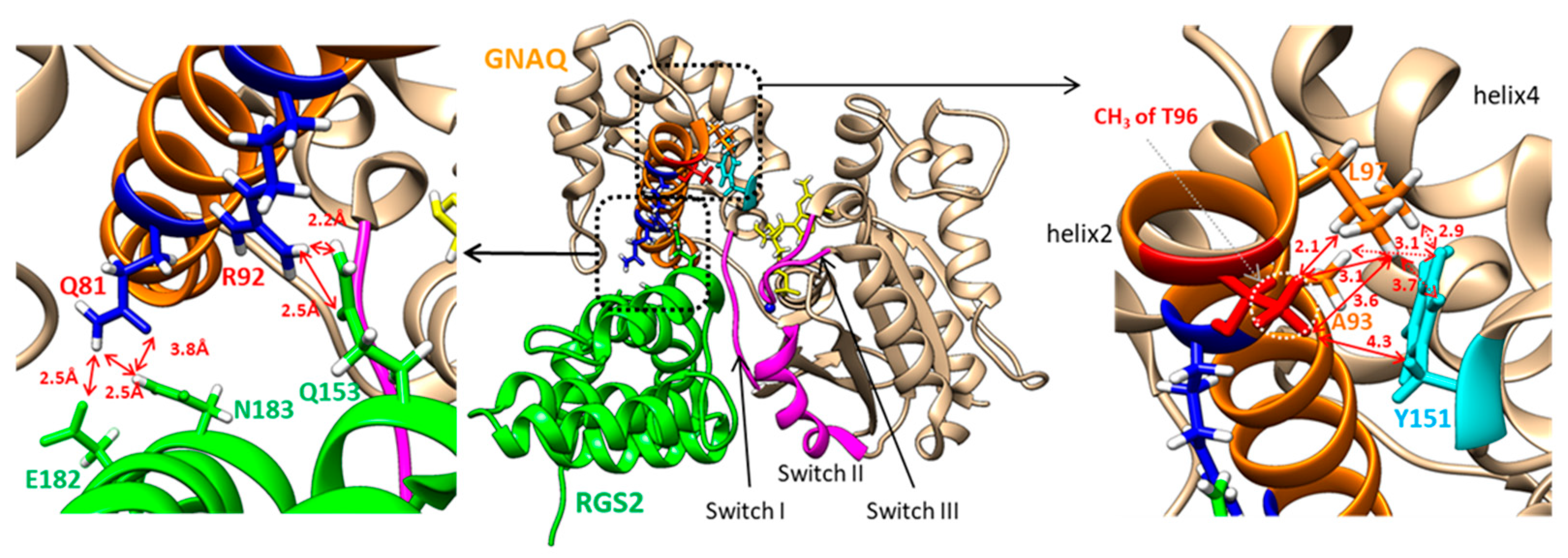
2.6. In Silico Structural Analysis of the GNAQ T96 and RGS2 Protein Complex
3. Discussion
4. Materials and Methods
4.1. TCGA Analysis
4.2. Cell Lines and Culture Conditions
4.3. Clinical Samples
4.4. GNAQ Expression Vectors
4.5. Sequencing of GNAQ Exons in SK-Hep-1 Cells
4.6. Transfection of the Expression Vectors
4.7. MTT Assay (Cell Viability Assay)
4.8. Soft Agar Colony Formation Assay
4.9. Transwell Migration Assay
4.10. Western Blot
4.11. Genomic DNA Extraction and SNP Genotyping
4.12. In Silico Structure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, I.C.; Chong, C.C.; Cheung, T.T.; Yeung, P.C.; Ng, K.K.; Lai, P.B.; Chan, S.L.; Chan, A.W.; Tang, P.M.; Cheung, S.T. Genetic variation in ABCB5 associates with risk of hepatocellular carcinoma. J. Cell. Mol. Med. 2020, 24, 10705–10713. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Feng, X.; Shi, H.; Wang, Y.; Wang, L.; Shen, H.; Zha, Q.; Chen, Y.; Jiang, C. MIR17HG polymorphism (rs7318578) is associated with liver cancer risk in the Chinese Han population. Biosci. Rep. 2020, 40, BSR20193312. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kobayashi, T.; Mashima, H.; Miki, D.; Kuroda, S.; Hamaoka, M.; Aikata, H.; Chayama, K.; Ohdan, H. PD1 gene polymorphism is associated with a poor prognosis in hepatocellular carcinoma following liver resection, cohort study. Int. J. Surg. 2020, 80, 84–90. [Google Scholar] [CrossRef]
- Yang, M.-H.; Yen, C.-H.; Chen, Y.-F.; Fang, C.-C.; Li, C.-H.; Lee, K.-J.; Lin, Y.-H.; Weng, C.-H.; Liu, T.-T.; Huang, S.-F.; et al. Somatic mutations of PREX2 gene in patients with hepatocellular carcinoma. Sci. Rep. 2019, 9, 2552. [Google Scholar] [CrossRef] [PubMed]
- Landerer, S.; Kalthoff, S.; Paulusch, S.; Strassburg, C.P. UDP-glucuronosyltransferase polymorphisms affect diethylnitrosamine-induced carcinogenesis in humanized transgenic mice. Cancer Sci. 2020, 111, 4266–4275. [Google Scholar] [CrossRef]
- Taki, K.; Ohmuraya, M.; Tanji, E.; Komatsu, H.; Hashimoto, D.; Semba, K.; Araki, K.; Kawaguchi, Y.; Baba, H.; Furukawa, T. GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene 2016, 35, 2407–2412. [Google Scholar] [CrossRef] [PubMed]
- Maziarz, M.; Federico, A.; Zhao, J.; Dujmusic, L.; Zhao, Z.; Monti, S.; Varelas, X.; Garcia-Marcos, M. Naturally occurring hotspot cancer mutations in Gα(13) promote oncogenic signaling. J. Biol. Chem. 2020, 295, 16897–16904. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Brunt, E.M.; Marginean, C.; Nalbantoglu, I.; Snover, D.C.; Thung, S.N.; Yeh, M.M.; Umetsu, S.E.; Ferrell, L.D.; Gill, R.M. Frequent GNAQ and GNA14 Mutations in Hepatic Small Vessel Neoplasm. Am. J. Surg. Pathol. 2018, 42, 1201–1207. [Google Scholar] [CrossRef]
- Jang, S.; Chun, S.M.; Hong, S.M.; Sung, C.O.; Park, H.; Kang, H.J.; Kim, K.P.; Lee, Y.J.; Yu, E. High throughput molecular profiling reveals differential mutation patterns in intrahepatic cholangiocarcinomas arising in chronic advanced liver diseases. Mod. Pathol. 2014, 27, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Sprang, S.R. G protein mechanisms: Insights from structural analysis. Annu. Rev. Biochem. 1997, 66, 639–678. [Google Scholar] [CrossRef] [Green Version]
- Dohlman, H.G.; Jones, J.C. Signal activation and inactivation by the Gα helical domain: A long-neglected partner in G protein signaling. Sci. Signal. 2012, 5, re2. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Lee, Y.S.; Shim, D.M.; Seo, S.W. Effect of GNAQ alteration on RANKL-induced osteoclastogenesis in human non-small-cell lung cancer. Bone Jt. Res. 2020, 9, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, X.; Xue, W.; Zhang, Y.; Li, C.; Song, Y.; Mei, M.; Lu, L.; Wang, Y.; Zhou, Z.; et al. Recurrent GNAQ mutation encoding T96S in natural killer/T cell lymphoma. Nat. Commun. 2019, 10, 4209. [Google Scholar] [CrossRef] [Green Version]
- Della Rocca, G.J.; van Biesen, T.; Daaka, Y.; Luttrell, D.K.; Luttrell, L.M.; Lefkowitz, R.J. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J. Biol. Chem. 1997, 272, 19125–19132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhao, W.; Wu, J.; Lu, J.; Ding, Y.; Wu, S.; Wang, H.; Ding, D.; Mo, F.; Zhou, Z.; et al. Neoantigens Derived from Recurrently Mutated Genes as Potential Immunotherapy Targets for Gastric Cancer. BioMed Res. Int. 2019, 2019, 8103142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.M.; Jung, S.H.; Baek, I.P.; Lee, S.H.; Choi, Y.J.; Lee, J.Y.; Chung, Y.J.; Lee, S.H. Regional biases in mutation screening due to intratumoural heterogeneity of prostate cancer. J. Pathol. 2014, 233, 425–435. [Google Scholar] [CrossRef]
- Gaeta, R.; Lessi, F.; Mazzanti, C.; Modena, M.; Garaventa, A.; Boero, S.; Michelis, M.B.; Capanna, R.; Aretini, P.; Franchi, A. Diffuse bone and soft tissue angiomatosis with GNAQ mutation. Pathol. Int. 2020, 70, 452–457. [Google Scholar] [CrossRef]
- Li, Y.; Tang, Z.Y.; Hou, J.X. Hepatocellular carcinoma: Insight from animal models. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 32–43. [Google Scholar] [CrossRef]
- Nance, M.R.; Kreutz, B.; Tesmer, V.M.; Sterne-Marr, R.; Kozasa, T.; Tesmer, J.J. Structural and functional analysis of the regulator of G protein signaling 2-gαq complex. Structure 2013, 21, 438–448. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, H.; Wu, H.; Yao, C.; He, H.; Wang, C.; Li, W. G protein subunit α q regulates gastric cancer growth via the p53/p21 and MEK/ERK pathways. Oncol. Rep. 2017, 37, 1998–2006. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell 2017, 31, 685–696.e686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Min, K.; Lee, S.K. Epstein-Barr Virus miR-BART17-5p Promotes Migration and Anchorage-Independent Growth by Targeting Kruppel-Like Factor 2 in Gastric Cancer. Microorganisms 2020, 8, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, A.S.; Murugan, A.K.; Qasem, E.; Alswailem, M.M.; AlGhamdi, B.; Moria, Y.; Al-Hindi, H. Correction to: Absence of EIF1AX, PPM1D, and CHEK2 mutations reported in Thyroid Cancer Genome Atlas (TCGA) in a large series of thyroid cancer. Endocrine 2019, 65, 226. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Johnson, C.; Hu, Q.; Yan, L.; Liu, B.; Ambrosone, C.B.; Wang, J.; Liu, S. Differences in somatic mutation landscape of hepatocellular carcinoma in Asian American and European American populations. Oncotarget 2016, 7, 40491–40499. [Google Scholar] [CrossRef] [Green Version]
- Day, P.W.; Tesmer, J.J.; Sterne-Marr, R.; Freeman, L.C.; Benovic, J.L.; Wedegaertner, P.B. Characterization of the GRK2 binding site of Galphaq. J. Biol. Chem. 2004, 279, 53643–53652. [Google Scholar] [CrossRef] [Green Version]
- Lyon, A.M.; Dutta, S.; Boguth, C.A.; Skiniotis, G.; Tesmer, J.J.G. Full-length Gαq–phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 2013, 20, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Tesmer, V.M.; Kawano, T.; Shankaranarayanan, A.; Kozasa, T.; Tesmer, J.J.G. Snapshot of Activated G Proteins at the Membrane: The Gαq-GRK2-Gßγ Complex. Science 2005, 310, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Shankaranarayanan, A.; Coco, C.; Ridilla, M.; Nance, M.R.; Vettel, C.; Baltus, D.; Evelyn, C.R.; Neubig, R.R.; Wieland, T.; et al. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 2007, 318, 1923–1927. [Google Scholar] [CrossRef]
- Anger, T.; Klintworth, N.; Stumpf, C.; Daniel, W.G.; Mende, U.; Garlichs, C.D. RGS protein specificity towards Gq- and Gi/o-mediated ERK 1/2 and Akt activation, in vitro. J. Biochem. Mol. Biol. 2007, 40, 899–910. [Google Scholar] [CrossRef]
- Oberg, J.A.; Glade Bender, J.L.; Sulis, M.L.; Pendrick, D.; Sireci, A.N.; Hsiao, S.J.; Turk, A.T.; Dela Cruz, F.S.; Hibshoosh, H.; Remotti, H.; et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: Moving beyond actionable alterations. Genome Med. 2016, 8, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.B.; Xu, J.; Skanderup, A.J.; Dong, Y.; Brannon, A.R.; Wang, L.; Won, H.H.; Wang, P.I.; Nanjangud, G.J.; Jungbluth, A.A.; et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat. Commun. 2016, 7, 13131. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Alvarez, J.J.S.; Lu, B.; Lim, J.Q.; Takano, A.; et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Li, X.; Ye, J.; Wu, X.; Tan, Z.; Liu, C.; Shen, B.; Wang, X.A.; Wu, W.; et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014, 46, 872–876. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, M.; Miyajima, M.; Sugano, H.; Iimura, Y.; Kato, M.; Tsurusaki, Y.; Miyake, N.; Saitsu, H.; Arai, H.; Matsumoto, N. The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge–Weber syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Sample | Matched Normal | Race | Age | Sex |
|---|---|---|---|---|
| CC-A8HS-01A-11D-A35Z-10 | CC-A8HS-10A-01D-A35Z-10 | Asian | 18 | M |
| DD-A4NF-01A-11D-A27I-10 | DD-A4NF-10A-01D-A27I-10 | White | 72 | M |
| DD-AACW-01A-11D-A40R-10 | DD-AACW-10A-01D-A40U-10 | Asian | 43 | M |
| DD-AADG-01A-11D-A40R-10 | DD-AADG-10A-01D-A40U-10 | Asian | 70 | M |
| DD-AADV-01A-11D-A38X-10 | DD-AADV-10A-01D-A38X-10 | Asian | 50 | M |
| DD-AADW-01A-11D-A38X-10 | DD-AADW-10A-01D-A38X-10 | Asian | 48 | M |
| DD-AAE8-01A-11D-A40R-10 | DD-AAE8-10A-01D-A40U-10 | Asian | 45 | M |
| DD-AAEK-01A-11D-A40R-10 | DD-AAEK-10A-01D-A40U-10 | Asian | 51 | M |
| ED-A97K-01A-21D-A382-10 | ED-A97K-10A-01D-A385-10 | Asian | 54 | M |
| G3-A3CG-01A-11D-A20W-10 | G3-A3CG-10A-01D-A20W-10 | White | 80 | M |
| K7-A5RF-01A-11D-A28X-10 | K7-A5RF-10B-01D-A28X-10 | White | 64 | M |
| MI-A75G-01A-11D-A32G-10 | MI-A75G-10A-01D-A32G-10 | White | 63 | M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, E.; Park, S.J.; Lee, G.; Yoon, S.K.; Lee, M.; Lee, S.K. The GNAQ T96S Mutation Affects Cell Signaling and Enhances the Oncogenic Properties of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063284
Choi E, Park SJ, Lee G, Yoon SK, Lee M, Lee SK. The GNAQ T96S Mutation Affects Cell Signaling and Enhances the Oncogenic Properties of Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(6):3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063284
Chicago/Turabian StyleChoi, Eugene, Sung Jean Park, Gunhee Lee, Seung Kew Yoon, Minho Lee, and Suk Kyeong Lee. 2021. "The GNAQ T96S Mutation Affects Cell Signaling and Enhances the Oncogenic Properties of Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 6: 3284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22063284