
BRAF Gene and Melanoma: Back to the Future

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. BRAF Gene Biology
3. BRAF Mutations in Melanoma: Epidemiology and Clinic-Pathological Correlations
3.1. BRAF V600 Mutations
3.2. BRAF Non-V600 Mutations
3.3. BRAF Fusions
4. BRAF Mutational Assessment: State of Art of Companion Diagnostic and Laboratory-Developed Tests
4.1. Immunohistochemistry
4.2. Sanger Sequencing
4.3. Pyrosequencing
4.4. Real-Time PCR
4.5. Next-Generation Sequencing
5. BRAF Mutations as Pharmacological Targets in Melanoma
5.1. Dabrafenib Plus Trametinib (COMBI-d and COMBI-v)
5.2. Vemurafenib Plus Cobimetinib (coBRIM)
5.3. Encorafenib Plus Binimetinib (COLUMBUS)
5.4. Pembrolizumab and Nivolumab in Monotherapy
5.5. Ipilimumab Plus Nivolumab
5.6. Adjuvant Therapies
6. From the Past to the Future of BRAF-Mutant Melanoma Treatment
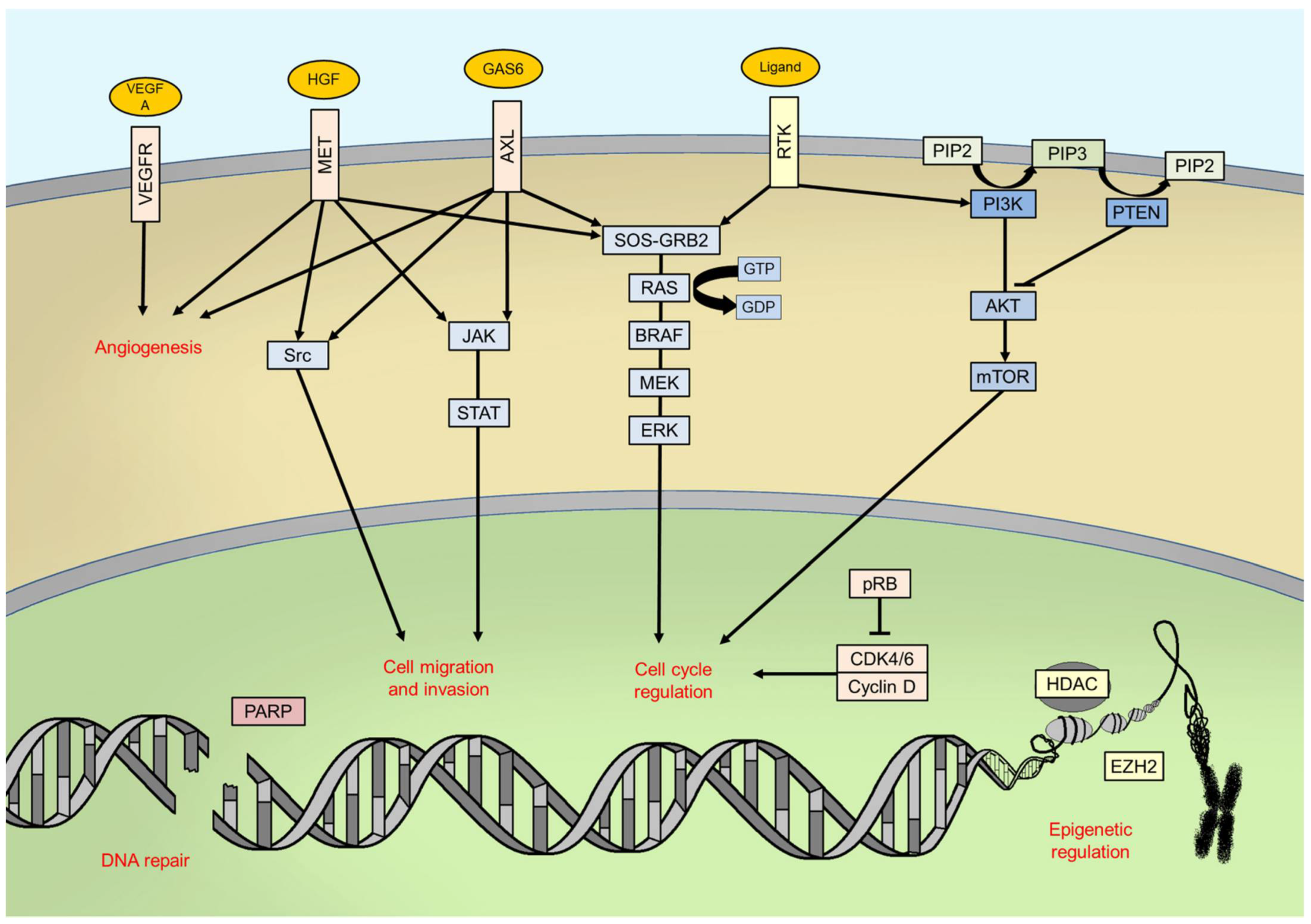
6.1. Combining Immunotherapy with BRAF and MEK Inhibitors
6.2. MAPK Pathway
6.3. PI3K/AKT/mTOR Pathway and PTEN
6.4. Cell Cycle Regulation
6.5. Genomic Instability
6.6. Epigenetics
6.7. Angiogenesis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palumbo, G.; Di Lorenzo, G.; Ottaviano, M.; Damiano, V. The future of melanoma therapy: Developing new drugs and improving the use of old ones. Future Oncol. 2016, 12, 2531–2534. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Funck-Brentano, E.; Malissen, N.; Roger, A.; Lebbé, C.; Deilhes, F.; Frénard, C.; Dréno, B.; Meyer, N.; Grob, J.J.; Tétu, P.; et al. Which adjuvant treatment for patients with BRAFV600-mutant cutaneous melanoma? In Annales de Dermatologie et de Vénéréologie; Cribier, B., Ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Giunta, E.F.; De Falco, V.; Napolitano, S.; Argenziano, G.; Brancaccio, G.; Moscarella, E.; Ciardiello, D.; Ciardiello, F.; Troiani, T. Optimal treatment strategy for metastatic melanoma patients harboring BRAF-V600 mutations. Ther. Adv. Med. Oncol. 2020, 12, 1758835920925219. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Arch. 2019, 474, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Morrison, D.K. Map kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Cuevas, B.D.; Abell, A.N.; Johnson, G.L. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene 2007, 26, 3159–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramini, J.M.; Vorobiev, S.M.; Tuberty, L.M.; Janjua, H.; Campbell, E.T.; Seetharaman, J.; Su, M.; Huang, Y.J.; Acton, T.B.; Xiao, R.; et al. The RAS-binding domain of human BRAF protein serine/threonine kinase exhibits allosteric conformational changes upon binding HRAS. Structure 2015, 23, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2010, 575, 545–550. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Dahlman, K.B. Class matters: Sensitivity of BRAF-mutant melanoma to MAPK inhibition. Clin. Cancer Res. 2018, 24, 6107–6109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Buttner, P.; Murali, R.; Okamoto, I.; Kolaitis, N.A.; Landi, M.T.; Scolyer, R.A.; Bastian, B.C. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res. 2011, 24, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.; Sarihan, E.I.; Chie, E.K.; Kuzmishin, G.; Doken, S.; Pennell, N.A.; Raymond, D.P.; Murthy, S.C.; Ahmad, U.; Raja, S.; et al. Clonal selection confers distinct evolutionary trajectories in BRAF-driven cancers. Nat. Commun. 2019, 10, 5143. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, J.C.; Sznol, M.; Pavlick, A.C.; Ariyan, S.; Cheng, E.; Bacchiocchi, A.; Kluger, H.M.; Narayan, D.; Halaban, R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J. Transl. Med. 2010, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Heinzerling, L.; Kühnapfel, S.; Meckbach, D.; Baiter, M.; Kaempgen, E.; Keikavoussi, P.; Schuler, G.; Agaimy, A.; Bauer, J.; Hartmann, A.; et al. Rare BRAF mutations in melanoma patients: Implications for molecular testing in clinical practice. Br. J. Cancer 2013, 108, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Pires da Silva, I.; Wang, K.Y.X.; Wilmott, J.S.; Holst, J.; Carlino, M.S.; Park, J.J.; Quek, C.; Wongchenko, M.; Yan, Y.; Mann, G.; et al. Distinct molecular profiles and immunotherapy treatment outcomes of V600E and V600K BRAF-mutant melanoma. Clin. Cancer Res. 2019, 25, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Umbach, D.M.; Li, L. Putative genomic characteristics of BRAF V600K versus V600E cutaneous melanoma. Melanoma Res. 2017, 27, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ny, L.; Hernberg, M.; Nyakas, M.; Koivunen, J.; Oddershede, L.; Yoon, M.; Wang, X.; Guyot, P.; Geisler, J. BRAF mutational status as a prognostic marker for survival in malignant melanoma: A systematic review and meta-analysis. Acta Oncol. 2020, 59, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, N.R.; Wolfe, R.; Kelly, J.W.; Haydon, A.; McArthur, G.A.; McLean, C.A.; Mar, V.J. Tumour mutation status and sites of metastasis in patients with cutaneous melanoma. Br. J. Cancer 2017, 117, 1026–1035. [Google Scholar] [CrossRef]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.M.; Johnson, D.B.; Meiss, F.; et al. Targeted therapy in advanced melanoma with rare BRAF mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; Hicks, D.J.; Bozon, V.; et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Tseng, L.; Chen, G.; Haley, L.; Illei, P.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer 2015, 15, 779. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Wu, X.; Yan, J.; Dai, J.; Ma, M.; Tang, H.; Yu, J.; Xu, T.; Yu, H.; Si, L.; Chi, Z.; et al. Mutations in BRAF codons 594 and 596 predict good prognosis in melanoma. Oncol. Lett. 2017, 14, 3601–3605. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Villafane, N.; Dogruluk, T.; Grzeskowiak, C.L.; Kong, K.; Tsang, Y.H.; Zagorodna, O.; Pantazi, A.; Yang, L.; Neill, N.J.; et al. Engineering and functional characterization of fusion genes identifies novel oncogenic drivers of cancer. Cancer Res. 2017, 77, 3502–3512. [Google Scholar] [CrossRef] [Green Version]
- Botton, T.; Talevich, E.; Mishra, V.K.; Zhang, T.; Shain, A.H.; Berquet, C.; Gagnon, A.; Judson, R.L.; Ballotti, R.; Ribas, A.; et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep. 2019, 29, 573–588. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, T.; He, J.; Yelensky, R. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malapelle, U.; Rossi, G.; Pisapia, P.; Barberis, M.; Buttitta, F.; Castiglione, F.; Cecere, F.L.; Grimaldi, A.M.; Iaccarino, A.; Marchetti, A.; et al. BRAF as a positive predictive biomarker: Focus on lung cancer and melanoma patients. Crit. Rev. Oncol. Hematol. 2020, 156, 103118. [Google Scholar] [CrossRef]
- Hyams, D.M.; Cook, R.W.; Buzaid, A.C. Identification of risk in cutaneous melanoma patients: Prognostic and predictive markers. J. Surg. Oncol. 2019, 119, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buder-Bakhaya, K.; Machiraju, D.; Hassel, J.C. Liquid biopsy: Value for melanoma therapy. Oncol. Res. Treat. 2017, 40, 430–434. [Google Scholar] [CrossRef]
- Gaiser, M.R.; von Bubnoff, N.; Gebhardt, C.; Utikal, J.S. Liquid biopsy to monitor melanoma patients. J. Dermatol. Ges. 2018, 16, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [Green Version]
- Schirosi, L.; Strippoli, S.; Gaudio, F.; Graziano, G.; Popescu, O.; Guida, M.; Simone, G.; Mangia, A. Is immunohistochemistry of BRAF V600E useful as a screening tool and during progression disease of melanoma patients? BMC Cancer 2016, 16, 905. [Google Scholar] [CrossRef] [Green Version]
- Colomba, E.; Hélias-Rodzewicz, Z.; Von Deimling, A.; Marin, C.; Terrones, N.; Pechaud, D.; Surel, S.; Côté, J.F.; Peschaud, F.; Capper, D.; et al. Detection of BRAF p.V600E mutations in melanomas: Comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J. Mol. Diagn. 2013, 15, 94–100. [Google Scholar] [CrossRef]
- Eriksson, H.; Zebary, A.; Vassilaki, I.; Omholt, K.; Ghaderi, M.; Hansson, J. BRAFV600E protein expression in primary cutaneous malignant melanomas and paired metastases. JAMA Dermatol. 2015, 151, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Wilmott, J.S.; Capper, D.; Preusser, M.; Zhang, Y.E.; Thompson, J.F.; Kefford, R.F.; von Deimling, A.; Scolyer, R.A. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am. J. Surg. Pathol. 2013, 37, 61–65. [Google Scholar] [CrossRef]
- Pearlstein, M.V.; Zedek, D.C.; Ollila, D.W.; Treece, A.; Gulley, M.L.; Groben, P.A.; Thomas, N.E. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J. Cutan. Pathol. 2014, 41, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, L.; Meyer, N.; Tournier, E.; Grand, D.; Uro-Coste, E.; Rochaix, P.; Brousset, P.; Lamant, L. Highly concordant results between immunohistochemistry and molecular testing of mutated V600E BRAF in primary and metastatic melanoma. Acta Derm. Venereol. 2016, 96, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Thiel, A.; Moza, M.; Kytölä, S.; Orpana, A.; Jahkola, T.; Hernberg, M.; Virolainen, S.; Ristimäki, A. Prospective immunohistochemical analysis of BRAF V600E mutation in melanoma. Hum. Pathol. 2015, 46, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kaku-Ito, Y.; Murata, M.; Ichiki, T.; Kuma, Y.; Tanaka, Y.; Ide, T.; Ohno, F.; Wada-Ohno, M.; Yamada, Y.; et al. Intra- and inter-tumor BRAF heterogeneity in acral melanoma: An immunohistochemical analysis. Int. J. Mol. Sci. 2019, 20, 6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, E. Is the DNA sequence the gold standard in genetic testing? Quality of molecular genetic tests assessed. Clin. Chem. 2006, 52, 557–558. [Google Scholar] [CrossRef] [Green Version]
- Jurkowska, M.; Gos, A.; Ptaszy’nski, K.; Michej, W.; Tysarowski, A.; Zub, R.; Siedlecki, J.A.; Rutkowski, P. Comparison between two widely used laboratory methods in BRAF V600 mutation detection in a large cohort of clinical samples of cutaneous melanoma metastases to the lymph nodes. Int. J. Clin. Exp. Pathol. 2015, 8, 8487–8493. [Google Scholar]
- Lopez-Rios, F.; Angulo, B.; Gomez, B.; Mair, D.; Martinez, R.; Conde, E.; Shieh, F.; Vaks, J.; Langland, R.; Lawrence, H.J.; et al. Comparison of testing methods for the detection of BRAF V600E mutations in malignant melanoma: Pre-approval validation study of the companion diagnostic test for vemurafenib. PLoS ONE 2013, 8, e53733. [Google Scholar]
- Qu, K.; Pan, Q.; Zhang, X.; Rodriguez, L.; Zhang, K.; Li, H.; Ho, A.; Sanders, H.; Sferruzza, A.; Cheng, S.M.; et al. Detection of BRAF V600 mutations in metastatic melanoma: Comparison of the Cobas 4800 and Sanger sequencing assays. J. Mol. Diagn. 2013, 15, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Marchant, J.; Mange, A.; Larrieux, M.; Costes, V.; Solassol, J. Comparative evaluation of the new FDA approved THxID™-BRAF test with high resolution melting and sanger sequencing. BMC Cancer 2014, 14, 519. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bloom, K.J.; Vallera, D.U.; Rueschoff, J.; Meldrum, C.; Schilling, R.; Kovach, B.; Lee, J.R.; Ochoa, P.; Langland, R.; et al. Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch. Pathol. Lab. Med. 2012, 136, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Mourah, S.; Denis, M.G.; Narducci, F.E.; Solassol, J.; Merlin, J.L.; Sabourin, J.C.; Scoazec, J.Y.; Ouafik, L.; Emile, J.F.; Heller, R.; et al. Detection of BRAF V600 mutations in melanoma: Evaluation of concordance between the Cobas® 4800 BRAF V600 mutation test and the methods used in French National Cancer Institute (INCa) platforms in a real-life setting. PLoS ONE 2015, 10, e0120232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.L.; Zhou, L.; Sadri, N. Comparison of targeted next generation sequencing (NGS) versus isolated BRAF V600E analysis in patients with metastatic melanoma. Virchows Arch. 2018, 473, 371–377. [Google Scholar] [CrossRef]
- de Unamuno Bustos, B.; Murria Estal, R.; Pérez Simó, G.; de Juan Jimenez, I.; Escutia Muñoz, B.; Rodríguez Serna, M.; Alegre de Miquel, V.; Llavador Ros, M.; Ballester Sánchez, R.; Nagore Enguídanos, E.; et al. Towards personalized medicine in melanoma: Implementation of a clinical next generation sequencing panel. Sci. Rep. 2017, 7, 495. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Michelini, S.; Di Fraia, M.; Mambrin, A.; Petrozza, V.; Porta, N.; Pacini, L.; Calogero, A.; Skroza, N.; Potenza, C. A rare BRAF V600E mutation detected by next-generation sequencing in a superficial spreading melanoma: Case report and potential diagnostic implications. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e393–e395. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovkiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Hauschild, A.; Larkin, J.; Ribas, A.; Dreno, B.; Flaherty, K.T.; Ascierto, P.A.; Lewis, K.D.; McKenna, E.; Zhu, Q.; Mun, Y.; et al. Modeled prognostic subgroups for survival and treatment outcomes in BRAF V600-mutated metastatic melanoma: Pooled analysis of 4 randomized clinical trials. JAMA Oncol. 2018, 4, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Flaherty, K.T.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Update on tolerability and overall survival in COLUMBUS: Landmark analysis of a randomised phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAF V600-mutant melanoma. Eur. J. Cancer 2020, 126, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma Long-Term Therapeutic Outcomes in BRAF-Mutated Melanoma 503 (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Minor, D.; D’Angelo, S.; Neyns, B.; Smylie, M.; Miller, W.H.M., Jr.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D.; et al. Overall survival in patients with advanced melanoma who received nivolumab versus investigator’s choice chemotherapy in CheckMate 037: A randomized, controlled, open-label phase III trial. J. Clin. Oncol. 2018, 36, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A. Melanoma staging: American Joint Committee onCancer (AJCC) 8th edition and beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maio, M.; Lewis, K.; Demidov, L.; Mandalà, M.; Bondarenko, I.; Ascierto, P.A.; Herbert, C.; Mackiewicz, A.; Rutkowski, P.; Guminski, A.; et al. Adjuvant vemurafenib in resected, BRAF V600 mutation-positive melanoma(BRIM8): A randomised, double-blind, placebo-controlled, multicentre, phase3 trial. Lancet Oncol. 2018, 19, 510–520. [Google Scholar] [CrossRef]
- Hauschild, A.; Dummer, R.; Schadendorf, D.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Longer follow-up confirms relapse-free survival benefit with adjuvant dabrafenib plus trametinib in patients with resected BRAF V600—Mutant stage III melanoma. J. Clin. Oncol. 2018, 36, 3441–3449. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garrett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitner, D.; Santinami, M.; Atkinson, V.; Mandalà, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAFV600-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Rossi, C.R.; Nitti, D. Interferon alpha adjuvant therapy inpatients with high-risk melanoma: A systematic review and meta-analysis. J. Natl. Cancer Inst. 2010, 102, 493–501. [Google Scholar]
- Wheatley, K.; Ives, N.; Hancock, B.; Gore, M.; Eggermont, A.; Suciu, S. Does adjuvant interferon for high-risk melanoma provide a worthwhile bene-fit? A meta-analysis of the randomised trials. Cancer Treat. Rev. 2003, 29, 241–252. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Prolonged survival in stage III melanoma with ipilimumab adjuvanttherapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB–C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.G.; Dalle, S.; Haydon, A.M.; Meshcheryakov, A.; Khattak, A.; Carlino, M.S.; et al. Follow-up confirms recurrence-free survival benefit of adjuvant pembrolizumab in high-risk stage III melanoma: Updated results from the EORTC 1325-MG/KEYNOTE-054 Trial. J. Clin. Oncol. 2020, 38, 3925–3936. [Google Scholar] [CrossRef]
- Zimmer, L.; Livingstone, E.; Hassel, J.C.; Fluck, M.; Eigentler, T.; Loquai, C.; Haferkamp, S.; Gutzmer, R.; Meier, F.; Mohr, P.; et al. Adjuvant nivolumab plus ipilimumab or nivolumab monotherapy versus placebo inpatients with resected stage IV melanoma with no evidence of disease (IMMU-NED): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 395, 1558–1568. [Google Scholar] [CrossRef]
- Ziogas, D.C.; Konstantinou, F.; Bouros, S.; Gogas, H. Identifying the optimum first-line therapy in BRAF-mutant metastatic melanoma. Expert Rev. Anticancer Ther. 2020, 20, 53–62. [Google Scholar] [CrossRef]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.L.; et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Nathan, P.; Dummer, R.; Long, G.V.; Ascierto, P.A.; Tawbi, H.A.; Robert, C.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; Mandala, M.; et al. LBA43—Spartalizumab plus dabrafenib and trametinib (Sparta-DabTram) in patients (pts) with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: Results from the randomized part 3 of the phase III COMBI-i trial. Ann. Oncol. 2020, 31, S1142–S1215. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Mandala, M.; Ferrucci, P.F.; Rutkowski, P.; Guidoboni, M.; Arance, A.M.; Ferraresi, V.; Maiello, E.; Guida, M.; Del Vecchio, M.; et al. LBA45 First report of efficacy and safety from the phase II study SECOMBIT (SEquential COMBo immuno and targeted therapy study). Ann. Oncol. 2020, 31, S1173–S1174. [Google Scholar] [CrossRef]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.V.; Sullivan, R.J. Developments in the space of new MAPK pathway inhibitors for BRAF-mutant melanoma. Clin. Cancer Res. 2019, 25, 5735–5742. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Czyz, M. Dissecting mechanisms of melanoma resistance to BRAF and MEK inhibitors revealed genetic and non-genetic patient- and drug-specific alterations and remarkable phenotypic plasticity. Cells 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK signaling pathway in melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Kirouac, D.C.; Schaefer, G.; Chan, J.; Merchant, M.; Orr, C.; Huang, S.A.; Moffat, J.; Liu, L.; Gadkar, K.; Ramanujan, S. Clinical responses to ERK inhibition in BRAFV600E-mutant colorectal cancer predicted using a computational model. NPJ Syst. Biol. Appl. 2017, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2017, 12, e0185862. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, S.V.; Shen, W.; Zhao, B.; Cai, S.; Kindler, L.; McMillen, W.T.; Yuen, E.; McCann, D.; Manro, J.; Dropsey, A.J.; et al. Abstract 5225: Temporal inhibition of ERK is sufficient for tumor growth inhibition in KRAS-mutant or BRAF-mutant tumors. Cancer Res. 2020, 80, 5225. [Google Scholar]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Kröckel, D.; Ringborg, U.; Hansson, J. Mutations of PIK3CA are rare in cutaneous melanoma. Melanoma Res. 2006, 16, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Tsao, H.; Goel, V.; Wu, H.; Yang, G.; Haluska, F.G. Genetic interaction between NRAS and BRAF mutations and PTEN//MMAC1 inactivation in melanoma. J. Investig. Dermatol. 2004, 122, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Mitra, S.; Sanna, A.; Ekedahl, H.; Lövgren, K.; Olsson, H.; Ingvar, C.; Isaksson, K.; Lauss, M.; Carneiro, A.; et al. The role of PTEN loss in immune escape, melanoma prognosis and therapy response. Cancers 2020, 12, 742. [Google Scholar] [CrossRef] [Green Version]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef]
- Silva, J.M.; Bulman, C.; McMahon, M. BRAFV600E cooperates with PI3K signaling, independent of AKT, to regulate melanoma cell proliferation. Mol. Cancer Res. 2014, 12, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.H.; Peng, W.; Phillips, S.; Milton, D.R.; Amaria, R.N.; Diab, A.; Glitza, I.C.; Patel, S.P.; Wong, M.K.K.; Yee, C.; et al. Safety results from phase I/II study of the PI3Kβ inhibitor GSK2636771 (G) in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. J. Clin. Oncol. 2020, 38, e22000. [Google Scholar] [CrossRef]
- Dummer, R.; Sandhu, S.K.; Miller, W.H.; Butler, M.O.; Blank, C.U.; Muñoz-Couselo, E.; Burris, H.A., III; Postow, M.A.; Chmielowski, B.; Middleton, M.R.; et al. A phase II, multicenter study of encorafenib/binimetinib followed by a rational triple-combination after progression in patients with advanced BRAF V600-mutated melanoma (LOGIC2). J. Clin. Oncol. 2020, 38, 10022. [Google Scholar] [CrossRef]
- Algazi, A.P.; Rotow, J.; Posch, C.; Ortiz-Urda, S.; Pelayo, A.; Munster, P.N.; Daud, A. A dual pathway inhibition strategy using BKM120 combined with vemurafenib is poorly tolerated in BRAF V600E/K mutant advanced melanoma. Pigment Cell Melanoma Res. 2019, 32, 603–606. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; McArthur, G. Cell Cycle Regulation and Melanoma. Curr. Oncol. Rep. 2016, 18, 34. [Google Scholar] [CrossRef]
- Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK-crosstalk as opportunity for cancer treatment. Pharmaceuticals 2020, 13, 418. [Google Scholar] [CrossRef]
- Wongchenko, M.J.; McArthur, G.A.; Dréno, B.; Larkin, J.; Ascierto, P.A.; Sosman, J.; Andries, L.; Kockx, M.; Hurst, S.D.; Caro, I.; et al. Gene expression profiling in BRAF-mutated melanoma reveals patient subgroups with poor outcomes to vemurafenib that may be overcome by cobimetinib plus vemurafenib. Clin. Cancer Res. 2017, 23, 5238–5245. [Google Scholar] [CrossRef] [Green Version]
- Julve, M.; Clark, J.J.; Lythgoe, M.P. Advances in cyclin-dependent kinase inhibitors for the treatment of melanoma. Expert Opin. Pharmacother. 2021, 22, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Burke, T.F.; Huber, L.; Van Horn, R.D.; Zhang, Y.; Buchanan, S.G.; Chan, E.M.; Starling, J.J.; Beckmann, R.P.; Peng, S.B. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol. Cancer Ther. 2014, 13, 2253–2263. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Martin, A.; Simpson, L.; Algazi, A.P.; Chawla, P.; Kim, D.W.; Santra, S.; Patel, V.; Jadhav, N.; Abhyankar, D.; et al. Phase I trial of the CDK 4/6 inhibitor, P1446A-05 (voruciclib) in combination with the BRAF inhibitor (BRAFi), vemurafenib in advanced, BRAF-mutant melanoma. J. Clin. Oncol. 2015, 33, 9076. [Google Scholar] [CrossRef]
- Taylor, M.; Sosman, J.; Gonzalez, R.; Carlino, M.S.; Kittaneh, M.; Lolkema, M.P.; Miller, W.; Marino, A.; Zhang, V.; Bhansali, S.G.; et al. 1086O—Phase Ib/II Study of Lee011 (Cdk4/6 Inhibitor) and Lgx818 (Braf Inhibitor) in Braf-Mutant Melanoma. Ann. Oncol. 2014, 25, iv374. [Google Scholar] [CrossRef]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Tang, S.; Poorman, K.; Olszanski, A.J.; Eisenberg, B.L.; Mehmi, I.; Farma, J.M.; In, G.K.; Amin, A.; Rapisuwon, S.; et al. Associations of age, PD-L1 status, BRAF mutation and tumor mutational burden (TMB) in advanced melanoma. J. Clin. Oncol. 2018, 36, e21609. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA damaging revolution: PARP inhibitors and beyond. ASCO Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Kim, K.B.; Millis, S.Z.; Ross, J.; Gay, L.M.; Vosoughi, E.; Moretto, J.; Leong, S.P.; Singer, M.I.; Parrett, B.M.; Minor, D.R.; et al. Abstract 320: Frequency and patient characteristics of homologous recombination deficiency in metastatic cutaneous melanoma. Cancer Res. 2018, 78, 320. [Google Scholar]
- Plummer, R.; Lorigan, P.; Steven, N.; Scott, L.; Middleton, M.R.; Wilson, R.H.; Mulligan, E.; Curtin, N.; Wang, D.; Dewji, R.; et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother. Pharmacol. 2013, 71, 1191–1199. [Google Scholar] [CrossRef]
- Middleton, M.R.; Friedlander, P.; Hamid, O.; Daud, A.; Plummer, R.; Falotico, N.; Chyla, B.; Jiang, F.; McKeegan, E.; Mostafa, N.M.; et al. Randomized phase II study evaluating veliparib (ABT-888) with temozolomide in patients with metastatic melanoma. Ann. Oncol. 2015, 26, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef] [Green Version]
- Fratangelo, F.; Camerlingo, R.; Carriero, M.V.; Pirozzi, G.; Palmieri, G.; Gentilcore, G.; Ragone, C.; Minopoli, M.; Ascierto, P.A.; Motti, M.L. Effect of ABT-888 on the apoptosis, motility and invasiveness of BRAFi-resistant melanoma cells. Int. J. Oncol. 2018, 53, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef]
- Straume, O.; Smeds, J.; Kumar, R.; Hemminki, K.; Akslen, L.A. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am. J. Pathol. 2002, 161, 229–237. [Google Scholar] [CrossRef]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epi Genet. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Tanemura, A.; Terando, A.M.; Sim, M.S.; van Hoesel, A.Q.; de Maat, M.F.; Morton, D.L.; Hoon, D.S. CpG island methylator phenotype predicts progression of malignant melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Hutchinson, L.; Deng, A.; Green, M.R. Common BRAF(V600E)-directed pathway mediates widespread epigenetic silencing in colorectal cancer and melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1250–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, A.M.; Covre, A.; Finotello, F.; Rieder, D.; Danielli, R.; Sigalotti, L.; Giannarelli, D.; Petiprez, F.; Lacroix, L.; Valente, M.; et al. Guadecitabine plus ipilimumab in unresectable melanoma: The NIBIT-M4 clinical trial. Clin. Cancer Res. 2019, 14, 4681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Mehta, G.; Aras, S.; Trivedi, A.; de la Serna, I.L. SWI/SNF chromatin remodeling enzymes in melanocyte differentiation and melanoma. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef] [Green Version]
- Emmons, M.F.; Faião-Flores, F.; Sharma, R.; Thapa, R.; Messina, J.L.; Becker, J.C.; Schadendorf, D.; Seto, E.; Sondak, V.K.; Koomen, J.M.; et al. HDAC8 regulates a stress response pathway in melanoma to mediate escape from BRAF inhibitor therapy. Cancer Res. 2019, 79, 2947–2961. [Google Scholar] [CrossRef] [Green Version]
- Madorsky Rowdo, F.P.; Barón, A.; Gallagher, S.J.; Hersey, P.; Emran, A.A.; Von Euw, E.M.; Barrio, M.M.; Mordoh, J. Epigenetic inhibitors eliminate senescent melanoma BRAFV600E cells that survive long‑term BRAF inhibition. Int. J. Oncol. 2020, 56, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.; Niebel, D.; Aymans, P.; Ferring-Schmitt, S.; Dietrich, D.; Landsberg, J. H3K27me3 and EZH2 expression in melanoma: Relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epi Genet. 2020, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef]
- Watnick, R.S. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef] [PubMed]
- Dewing, D.; Emmett, M.; Pritchard Jones, R. The roles of angiogenesis in malignant melanoma: Trends in basic science research over the last 100 years. ISRN Oncol. 2012, 2012, 546927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Xiao, M.; Ge, Y.; Krepler, C.; Belser, E.; Lopez-Coral, A.; Xu, X.; Zhang, G.; Azuma, R.; Liu, Q.; et al. BRAF inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin. Cancer Res. 2015, 21, 1652–1664. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol Cancer. 2019, 18, 153. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Corrie, P.G.; Marshall, A.; Nathan, P.D.; Lorigan, P.; Gore, M.; Tahir, S.; Faust, G.; Kelly, C.G.; Marples, M.; Danson, S.J.; et al. Adjuvant bevacizumab for melanoma patients at high risk of recurrence: Survival analysis of the AVAST-M trial. Ann. Oncol. 2018, 29, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Minchella, I.; Mosconi, M.; Gandini, S.; Verrecchia, F.; Cocorocchio, E.; Passoni, C.; Pari, C.; Testori, A.; Coco, P.; et al. Dacarbazine in combination with bevacizumab for the treatment of unresectable/metastatic melanoma: A phase II study. Melanoma Res. 2015, 25, 239–245. [Google Scholar] [CrossRef]
- de Aguiar, R.B.; de Moraes, J.Z. Exploring the immunological mechanisms underlying the anti-vascular endothelial growth factor activity in tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozeman, E.A.; Blank, C.U. Combining checkpoint inhibition and targeted therapy in melanoma. Nat. Med. 2019, 25, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Mutation | Frequency (among BRAF V600 Mutations) | DFS | PFS (with TT) | TMB | Response to ICIs |
|---|---|---|---|---|---|
| V600E | 70–88% | Longer | Longer | Lower | Good |
| V600K | 10–20% | Shorter | Shorter | Higher | Better |
| Phase 3 Clinical Trial | % of BRAF V600-Mutant Patients | Experimental Arm | Standard Arm | Primary Endpoint in BRAF V600-Mutant Patients | Reference |
|---|---|---|---|---|---|
| COMBI-d | 100% | D + T | D | PFS: 9.3 months vs. 8.8 months (p = 0.03) | [59] |
| COMBI-v | 100% | D + T | V | 1-year OS: 72% vs. 65% (p = 0.005) | [60] |
| coBRIM | 100% | V + C | V | PFS: 9.9 months vs. 6.2 months (p < 0.001) | [63,64] |
| COLUMBUS | 100% | E + B | V | PFS: 14.9 months vs. 7.3 months (p < 0.0001) | [66,67] |
| KEYNOTE-006 | 36.2% | P | I | HR for PFS: 0.44–0.87 | [4,69] |
| CheckMate 067 | 31% | N + I | I | 5-year OS: 60% vs. 30% | [70,71] |
| Phase 3 Clinical Trial | % of BRAF V600-Mutant Patients | Stage (AJCC VII Edition) | Experimental Arm | Standard Arm | Primary Endpoint in BRAF V600-Mutant Patients | Reference |
|---|---|---|---|---|---|---|
| BRIM-8 | 100% | IIC-IIIB | V | placebo | RFS: 23.1 months vs. 15.4 months (p = 0.026) | [74] |
| COMBI-AD | 100% | III | D+T | placebo | 4-year RFS: 54% vs. 38% | [73,75] |
| CheckMate-238 | 42.1% | IIIB-IV | N | I | HR for 2-year RFS: 0.79 | [81] |
| KEYNOTE-054 | 43.3% | III | P | placebo | HR for 1-year RFS: 0.59 | [80] |
| Pathway/Cancer Hallmark | Molecular Target | Drugs under Investigation | Phase of Development | Reference (or NCT Number) |
|---|---|---|---|---|
| MAPK | ERK1/2 | MK-8353 | Phase 1 trial | NCT01358331 |
| Ulixertinib | Phase 1trial | NCT01781429 | ||
| PI3K-AKT-mTOR | PI3K | GSK2636771 | Phase 1/2 trial | [115] |
| Buparlisib | Phase 2 trial | [116] | ||
| AKT | Uprosertib | Phase 1 trial | [118] | |
| MK2206 | Phase 2 trial | NCT01519427 | ||
| Cell cycle regulation | CDK4/6 | LY2835219 | Preclinical data | [126] |
| Voruciclib | Phase 1 trial | [127] | ||
| Ribociclib | Phase 1b/2 trial | [128] | ||
| Phase 2 trial | [116] | |||
| Palbociclib | Preclinical data | [129] | ||
| DNA repair | PARP | Rucaparib | Phase 2 trial | [142] |
| Veliparib | Preclinical data | [145] | ||
| Phase 2 trial | [143] | |||
| Niraparib | Phase 2 trial | NCT03925350 | ||
| Olaparib | Phase 2 trial | NCT04633902 | ||
| Epigenetic regulation | HDAC | Vorinostat | Phase 1/2 trial | NCT02836548 |
| EZH2 | Tazemetostat | Phase 1/2 trial | NCT04557956 | |
| Angiogenesis | VEGF-A | Bevacizumab | Phase 2 trial | NCT04356729 |
| Phase 2 trial | NCT02681549 | |||
| VEGFR, PDGFR | Lenvatinib | Phase 2 trial | NCT01136967 | |
| VEGFR, AXL, MET | Cabozantinib | Phase 2 trial | NCT04091750 | |
| Phase 1b/2 trial | NCT03957551 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073474
Ottaviano M, Giunta EF, Tortora M, Curvietto M, Attademo L, Bosso D, Cardalesi C, Rosanova M, De Placido P, Pietroluongo E, et al. BRAF Gene and Melanoma: Back to the Future. International Journal of Molecular Sciences. 2021; 22(7):3474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073474
Chicago/Turabian StyleOttaviano, Margaret, Emilio Francesco Giunta, Marianna Tortora, Marcello Curvietto, Laura Attademo, Davide Bosso, Cinzia Cardalesi, Mario Rosanova, Pietro De Placido, Erica Pietroluongo, and et al. 2021. "BRAF Gene and Melanoma: Back to the Future" International Journal of Molecular Sciences 22, no. 7: 3474. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073474