
Growth and Viability of Cutaneous Squamous Cell Carcinoma Cell Lines Display Different Sensitivities to Isoform-Specific Phosphoinositide 3-Kinase Inhibitors


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
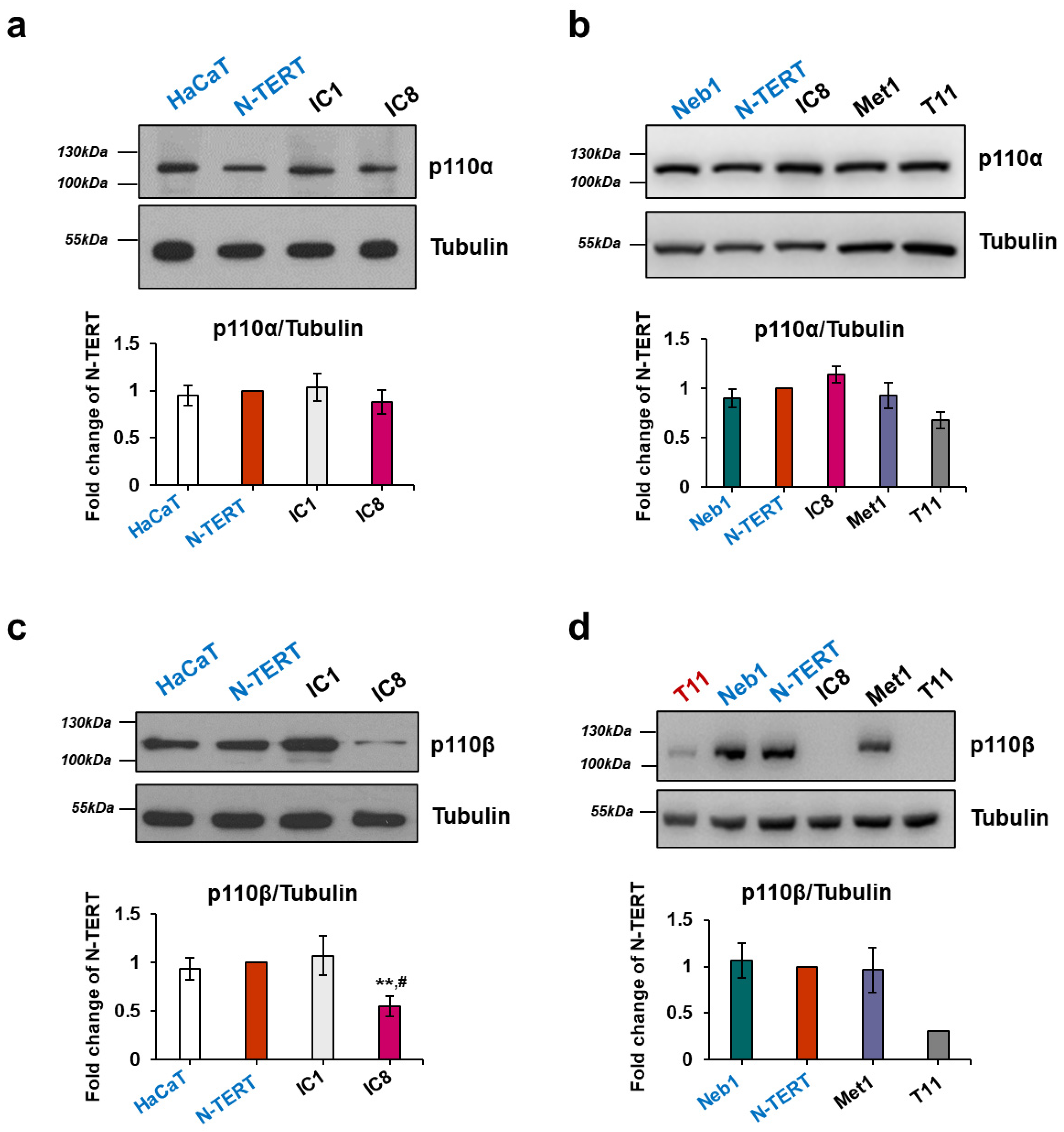
2.1. Expression of Class I PI3K Isoforms in Keratinocytes and cSCC Cell Lines
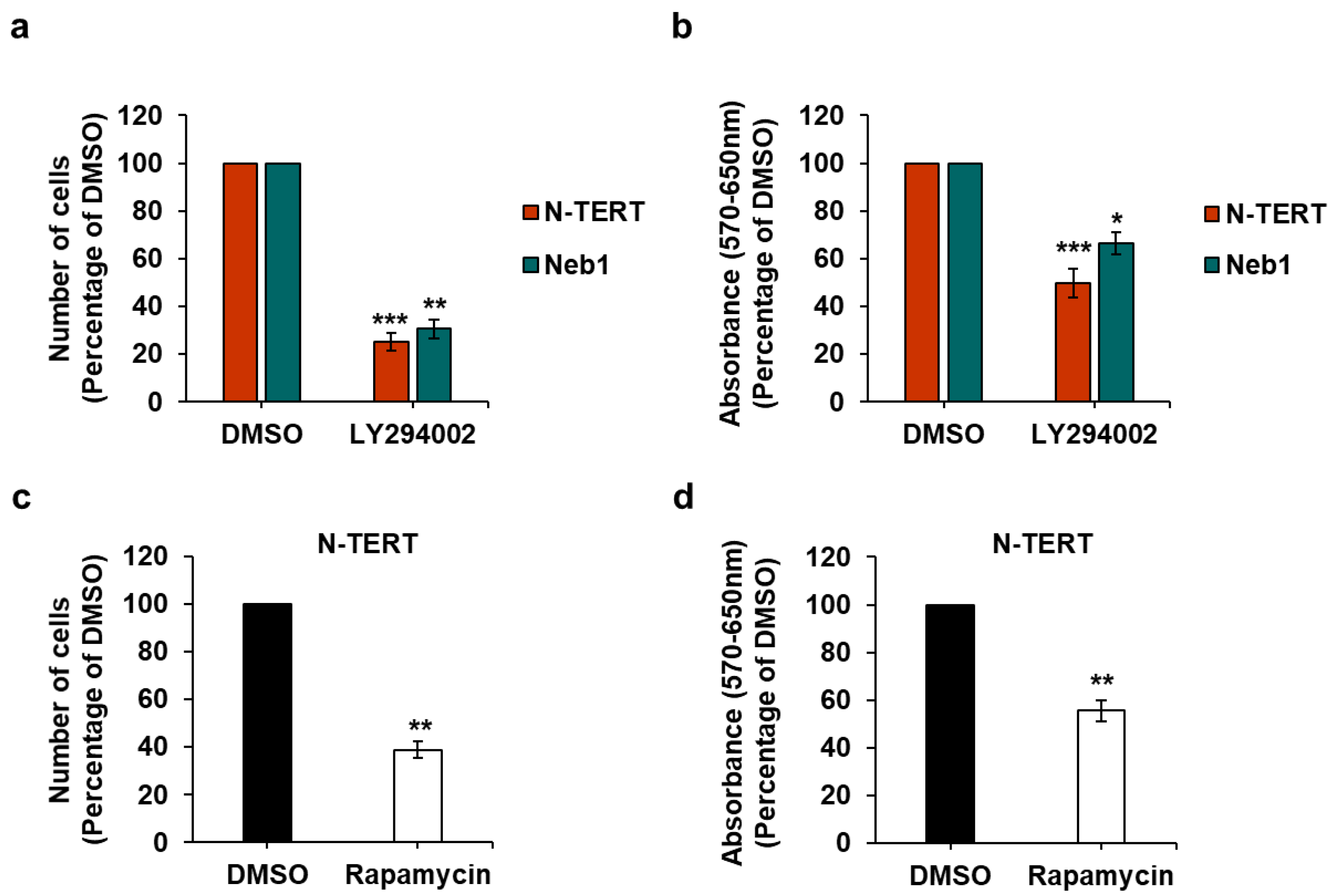
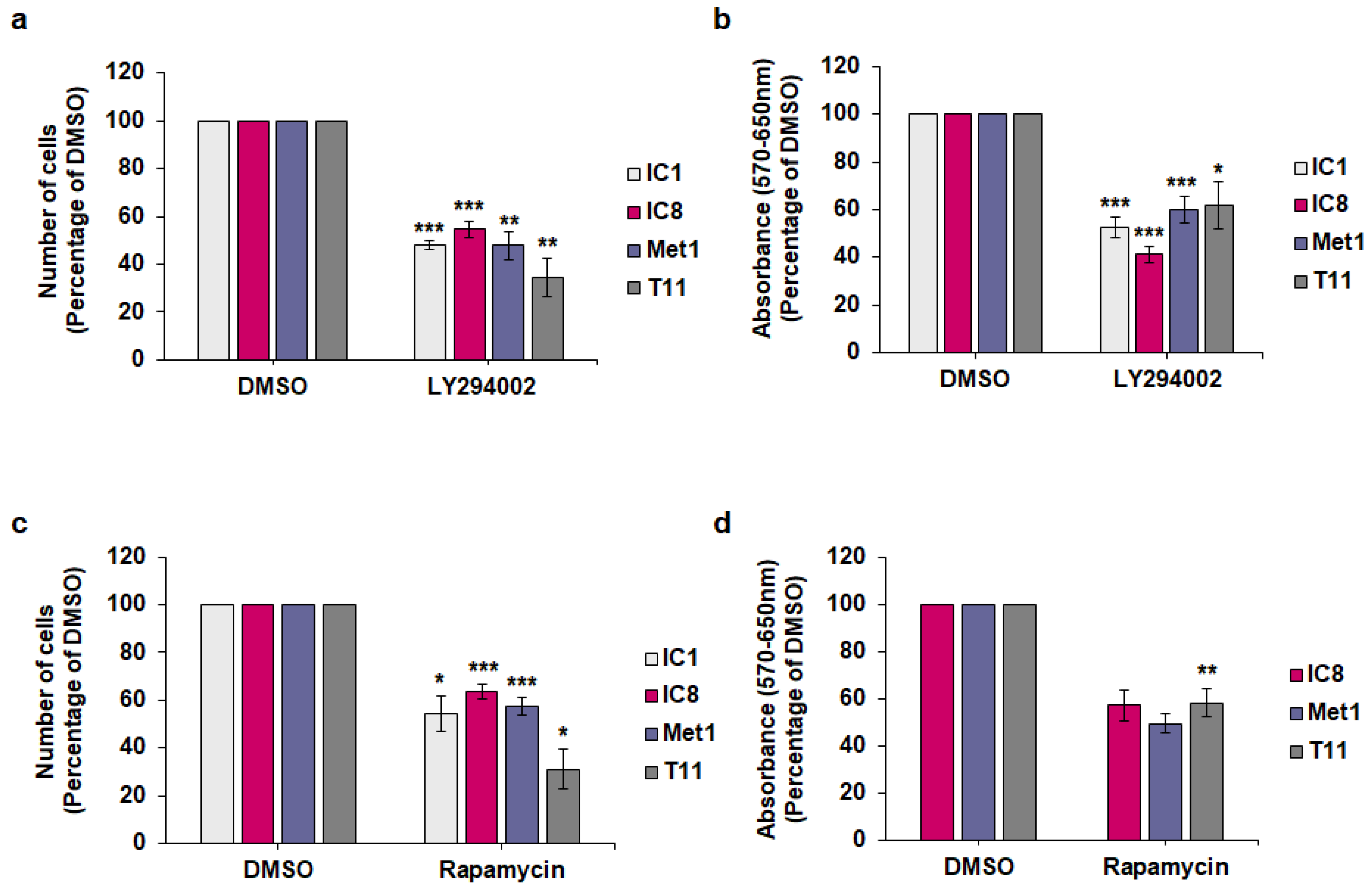
2.2. Effect of PI3K/Akt/mTOR Pathway Inhibition on Keratinocytes and cSCC Cell Lines
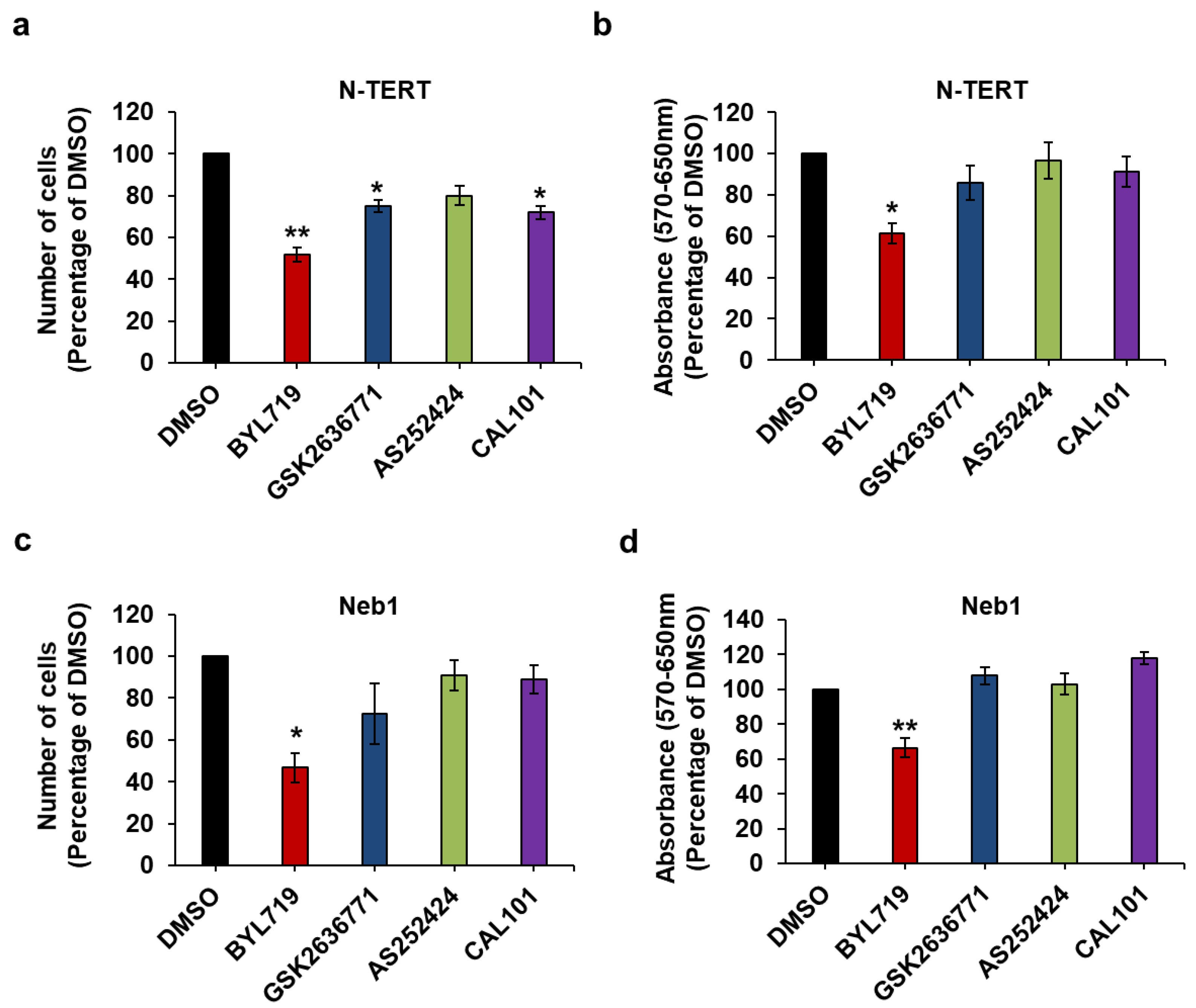
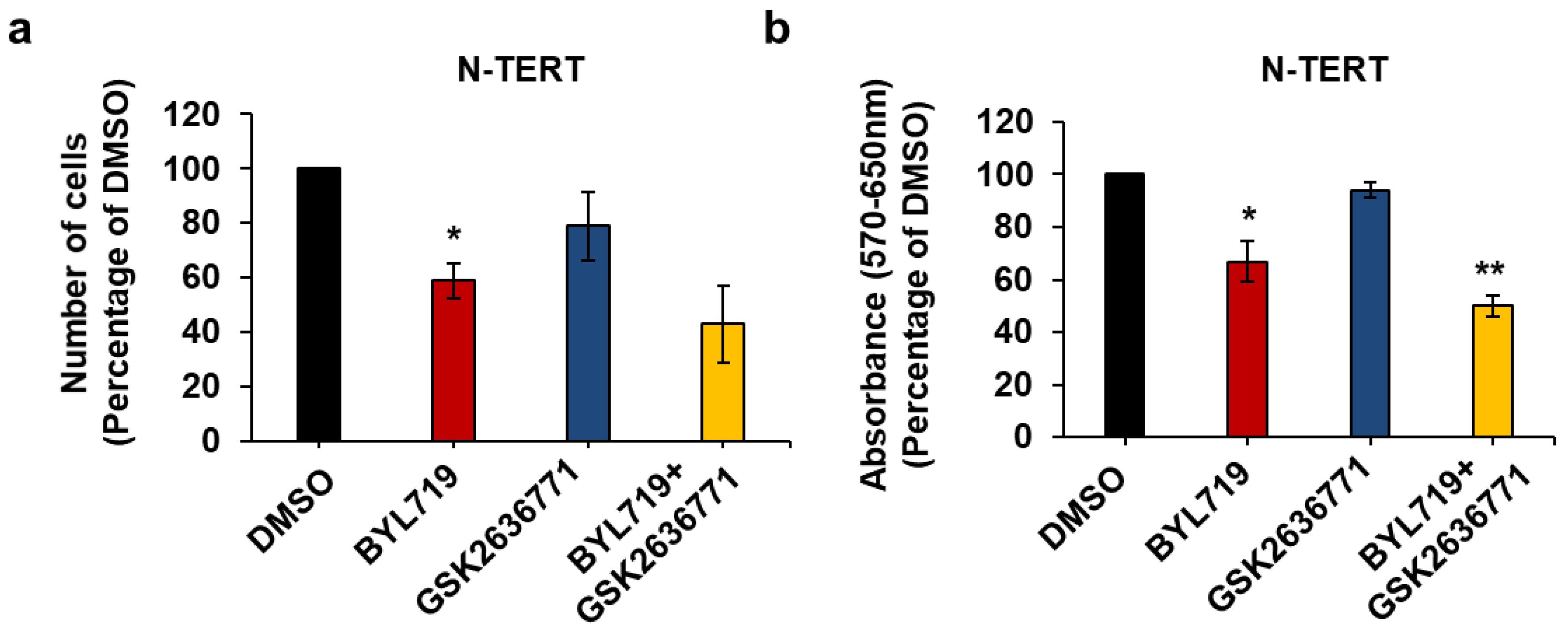
2.3. Effect of Isoform-Specific PI3K Inhibitors on Keratinocytes Growth and Viability
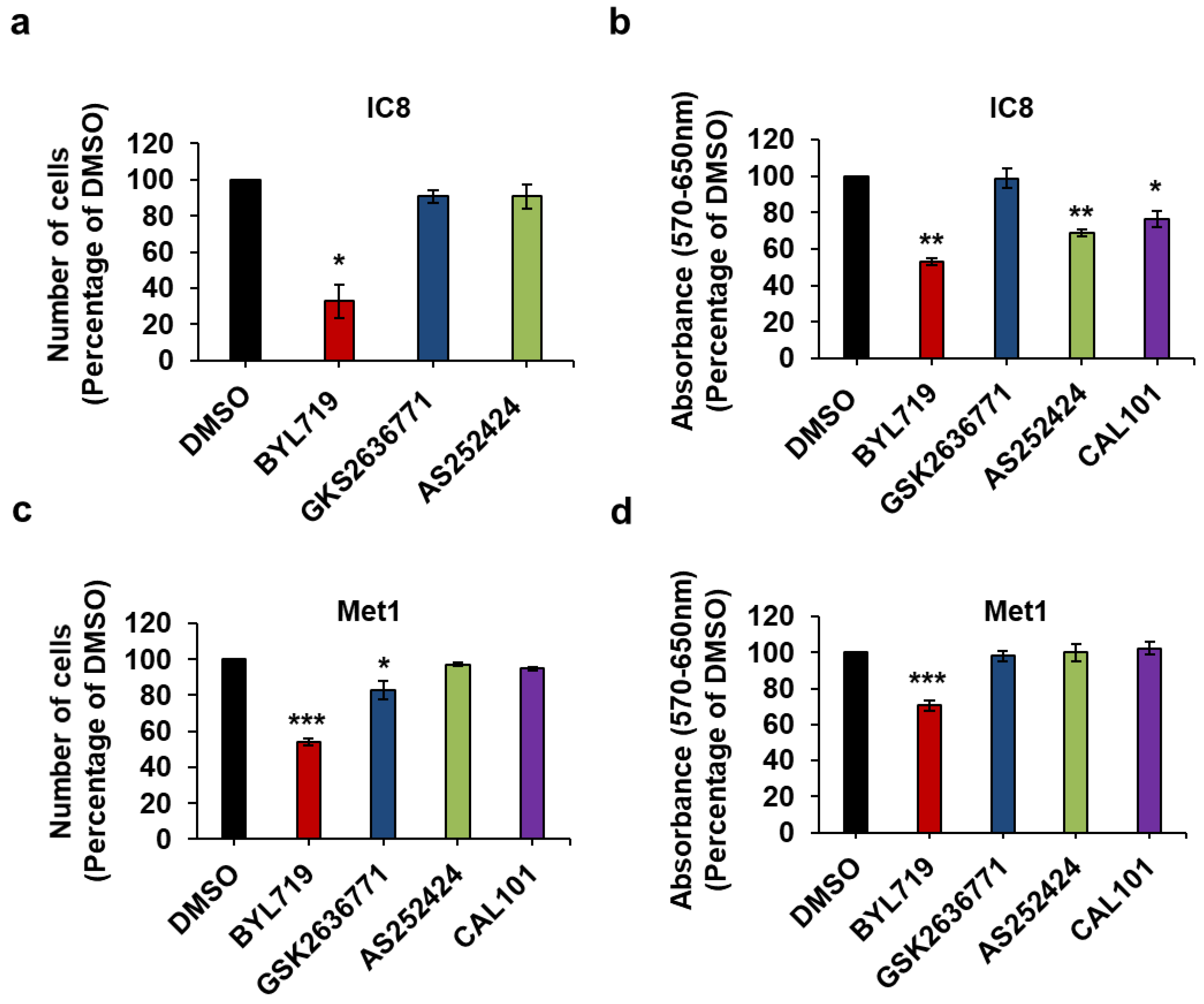
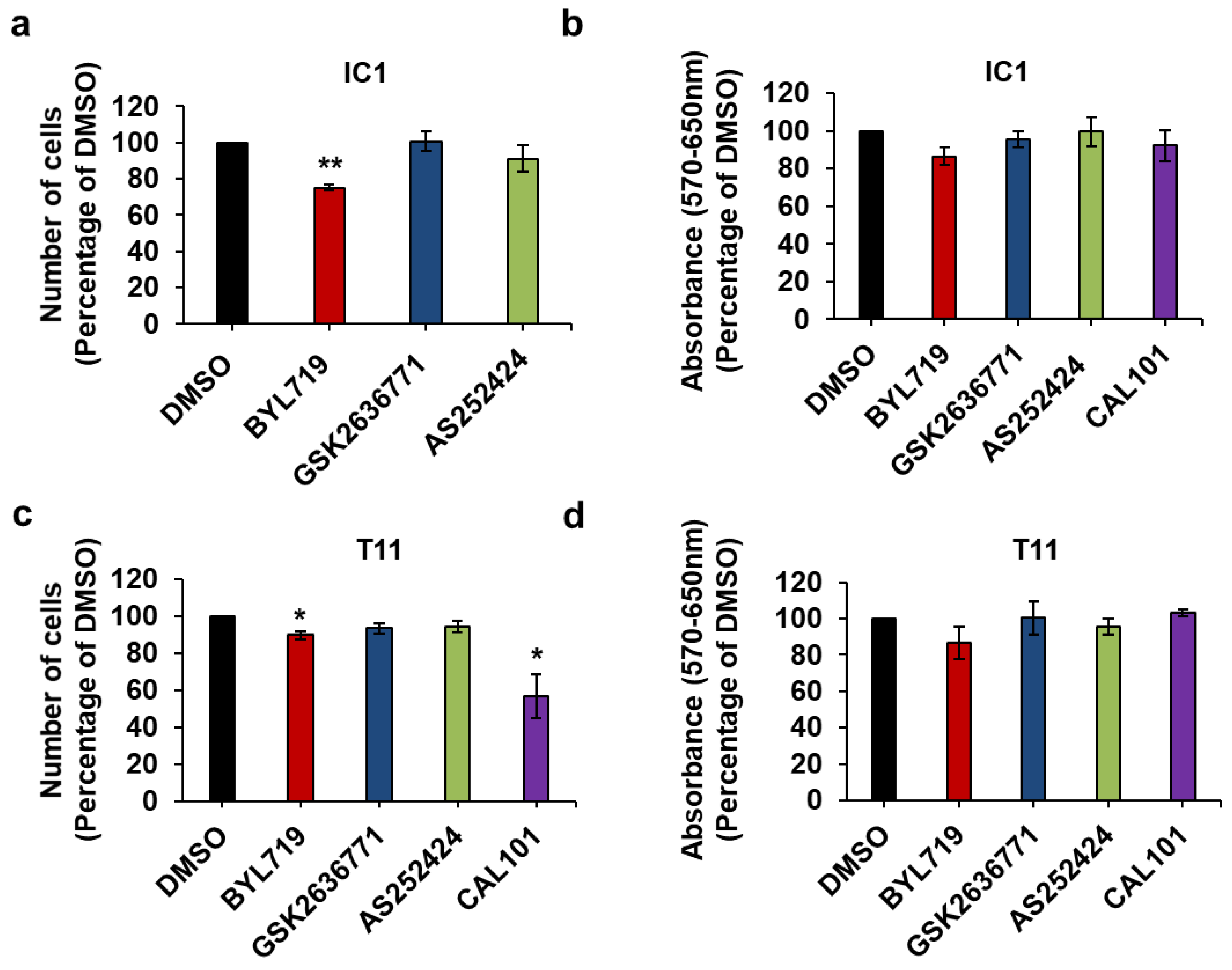
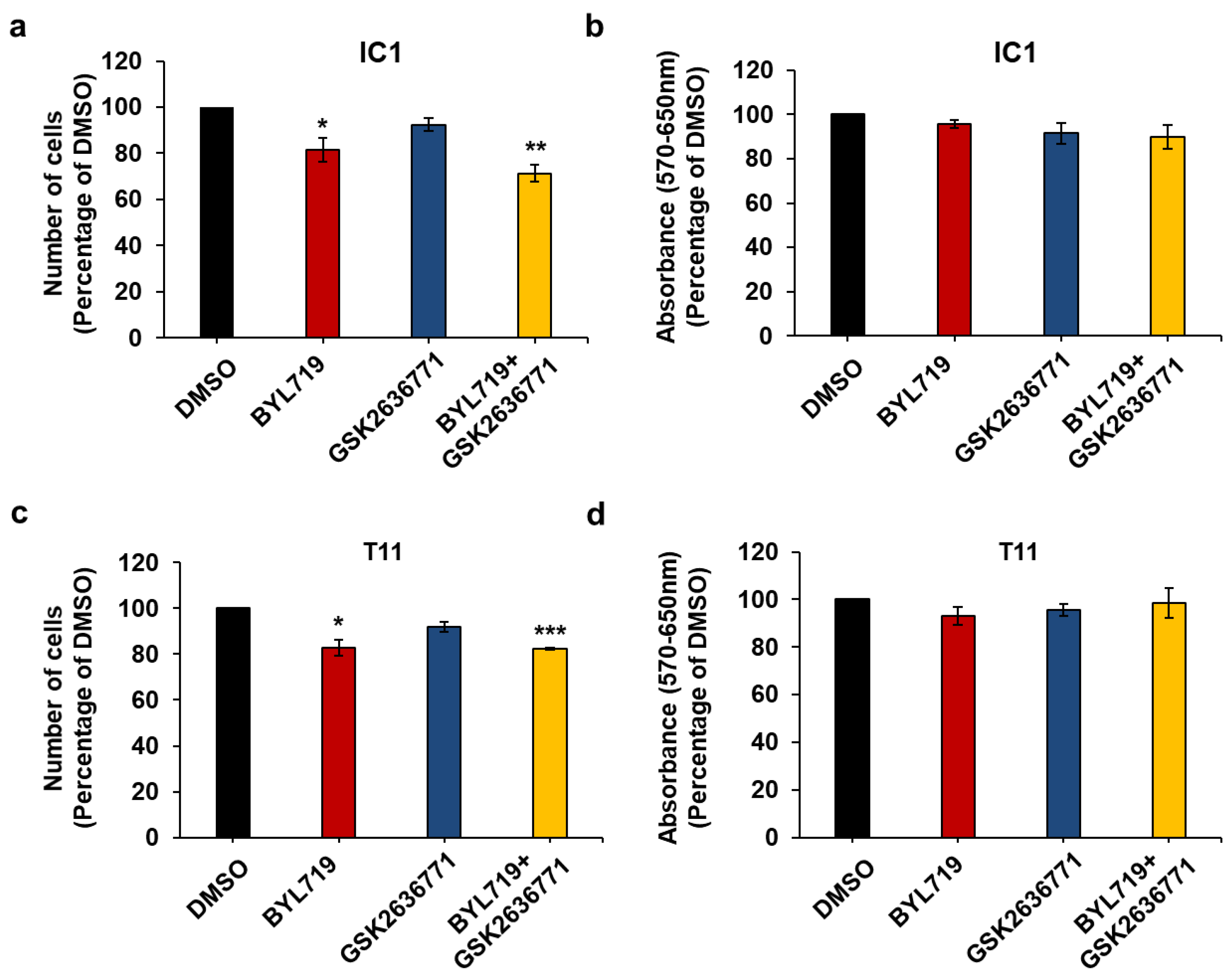
2.4. Effect of Isoform-Specific PI3K Inhibitors on cSCC Cells Growth and Viability
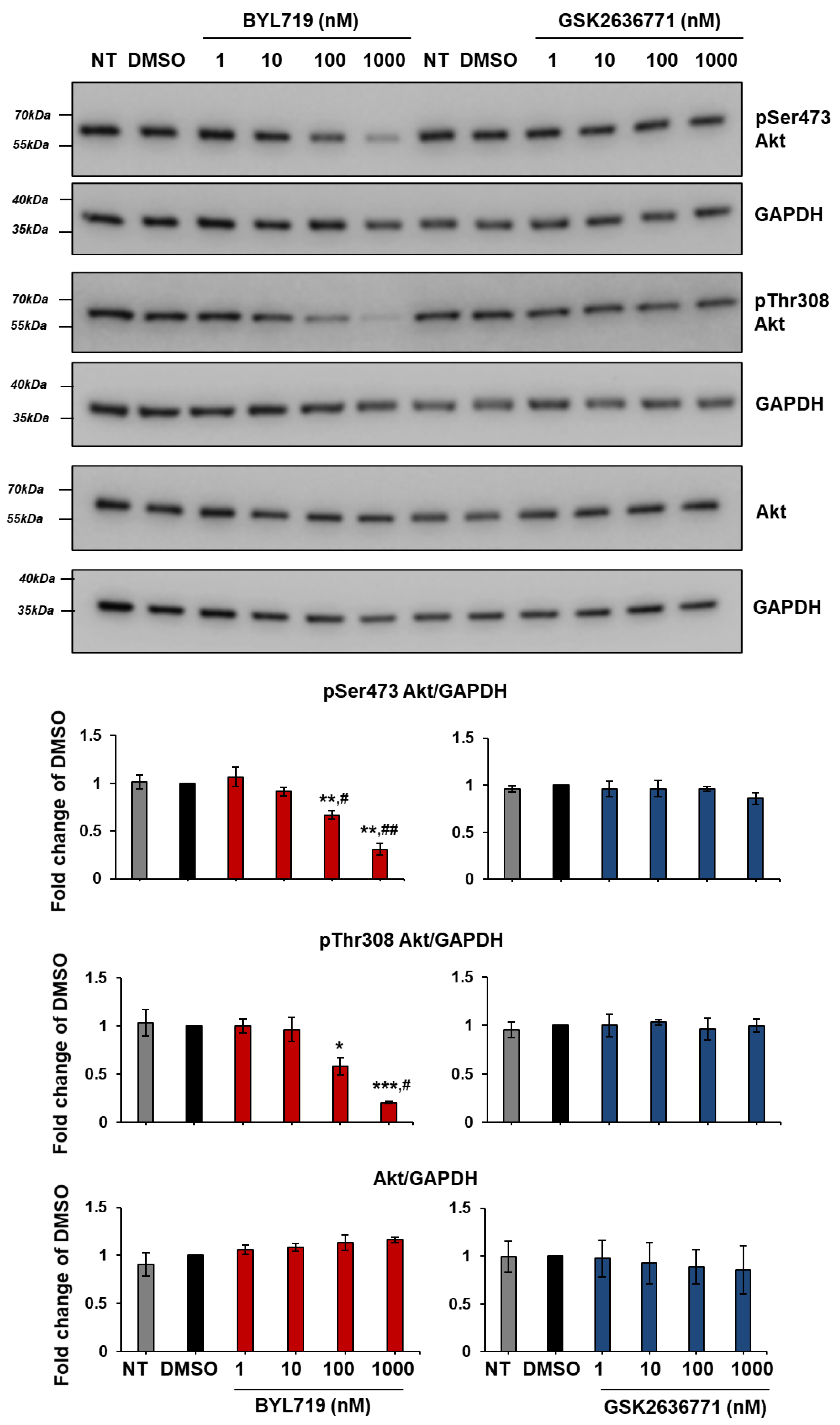
2.5. Effect of Isoform-Specific PI3K Inhibitors on Akt Activation in cSCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Cell Counting and MTT Assays
4.3. Western Blotting Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNI | Calcineurin inhibitors |
| cSCC | Cutaneous squamous cell carcinoma |
| DMEM | Dulbecco’s Modified Eagle Medium |
| ESCC | Esophageal squamous cell carcinoma |
| FBS | Fetal Bovine Serum |
| HNSCC | Head and neck squamous cell carcinoma |
| ICI | Immune checkpoints inhibitors |
| KCs | Keratinocyte carcinomas |
| mTOR | Mechanistic (or mammalian) target of rapamycin |
| MTT | Thiazolyl Blue Tetrazolium Blue (MTT) |
| PBS-T | Phosphate-buffered saline supplemented with 0.05% Tween 20 |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphate and tensin homolog |
| PtdIns(3,4,5)P3 | Phosphatidylinositol 3,4,5-trisphosphate |
References
- Venables, Z.; Nijsten, T.; Wong, K.; Autier, P.; Broggio, J.; Deas, A.; Harwood, C.; Hollestein, L.; Langan, S.; Morgan, E.; et al. Epidemiology of basal and cutaneous squamous cell carcinoma in the U.K. 2013–15: A cohort study. Br. J. Dermatol. 2019, 181, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Asgari, M.M.; Green, A.C.; Guhan, S.M.; Arron, S.T.; Proby, C.M.; Rollison, D.E.; Harwood, C.A.; Toland, A.E. Keratinocyte Carcinomas: Current Concepts and Future Research Priorities. Clin. Cancer Res. 2019, 25, 2379–2391. [Google Scholar] [CrossRef] [Green Version]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am. J. Clin. Dermatol. 2016, 17, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Harwood, A.C.; Proby, C.M.; Inman, G.J.; Leigh, I.M. The Promise of Genomics and the Development of Targeted Therapies for Cutaneous Squamous Cell Carcinoma. Acta Derm. Venereol. 2016, 96, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Lebbe, C.; Bataille, V.; Bastholt, L.; Dreno, B.; Concetta Fargnoli, M.; Forsea, A.M.; Frenard, C.; et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part Treatment. Eur. J. Cancer. 2020, 128, 60–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keohane, S.; Botting, J.; Budny, P.; Dolan, O.; Fife, K.; Harwood, C.; Mallipeddi, R.; Marsden, J.; Motley, R.; Newlands, C.; et al. British Association of Dermatologists guidelines for the management of people with cutaneous squamous cell carcinoma 2020. Br. J. Dermatol. 2021, 184, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Willenbrink, T.J.; Jambusaria-Pahlajani, A.; Arron, S.; Seckin, D.; Harwood, C.A.; Proby, C.M. Treatment approaches in im-munosuppressed patients with advanced cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 57–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P.T. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef]
- Ghigo, A.; Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-Kinases in Health and Disease. Subcellular Biochem. 2012, 58, 183–213. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabo-lism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worby, C.A.; Dixon, J.E. PTEN. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Sayama, K.; Yamasaki, K.; Hanakawa, Y.; Shirakata, Y.; Tokumaru, S.; Ijuin, T.; Takenawa, T.; Hashimoto, K. Phosphatidyl-inositol 3-kinase is a key regulator of early phase differentiation in keratinocytes. J. Biol. Chem. 2002, 277, 40390–40396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankow, S.; Bamberger, C.; Klippel, A.; Werner, S. Regulation of epidermal homeostasis and repair by phosphoinositide 3-kinase. J. Cell Sci. 2006, 119, 4033–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-Kinase Signaling to Akt Promotes Keratinocyte Differentiation Versus Death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobert, J.A.; Eng, C. PTEN hamartoma tumor syndrome: An overview. Genet. Med. 2009, 11, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Itami, S.; Ohishi, M.; Hamada, K.; Inoue, T.; Komazawa, N.; Senoo, H.; Sasaki, T.; Takeda, J.; Manabe, M.; et al. Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumour formation. Cancer Res. 2003, 63, 674–681. [Google Scholar] [PubMed]
- Peng, X.-D.; Xu, P.-Z.; Chen, M.-L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Bloch, W.; Iden, S.; Rüegg, M.A.; Hall, M.N.; Leptin, M.; Partridge, L.; Eming, S.A. mTORC1 and mTORC2 regulate skin morphogenesis and epidermal barrier formation. Nat. Commun. 2016, 7, 13226. [Google Scholar] [CrossRef] [Green Version]
- Janus, J.M.; O’Shaughnessy, R.F.L.; Harwood, C.A.; Maffucci, T. Phosphoinositide 3-Kinase-Dependent Signalling Pathways in Cutaneous Squamous Cell Carcinomas. Cancers 2017, 9, 86. [Google Scholar] [CrossRef] [Green Version]
- Einspahr, J.G.; Calvert, V.; Alberts, D.S.; Curiel-Lewandrowski, C.; Warneke, J.; Krouse, R.; Stratton, S.P.; Liotta, L.; Longo, C.; Pellicani, G.; et al. Functional Protein Pathway Activation Mapping of the Progression of Normal Skin to Squamous Cell Carcinoma. Cancer Prev. Res. 2012, 5, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-J.; Nakahara, T.; Takahara, M.; Kido, M.; Dugu, L.; Uchi, H.; Takeuchi, S.; Tu, Y.-T.; Moroi, Y.; Furue, M. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br. J. Dermatol. 2009, 160, 442–445. [Google Scholar] [CrossRef]
- O’Shaughnessy, R.F.; Akgül, B.; Storey, A.; Pfister, H.; Harwood, C.A.; Byrne, C. Cutaneous Human Papillomaviruses Down-regulate AKT1, whereas AKT2 Up-regulation and Activation Associates with Tumours. Cancer Res. 2007, 67, 8207–8215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segrelles, C.; Ruiz, S.; Pérez, P.; Murga, C.; Santos, M.; Budunova, I.V.; Martínez, J.; Larcher, F.; Slaga, T.J.; Gutkind, J.S.; et al. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene 2002, 21, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilker, E.; Lu, J.; Rho, O.; Carbajal, S.; Beltrán, L.; DiGiovanni, J. Role of PI3K/Akt signaling in insulin-like growth factor-1 (IGF-1) skin tumor promotion. Mol. Carcinog. 2005, 44, 137–145. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanni, J.; Bol, D.K.; Wilker, E.; Beltrán, L.; Carbajal, S.; Moats, S.; Ramirez, A.; Jorcano, J.; Kiguchi, K. Constitutive ex-pression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000, 60, 1561–1570. [Google Scholar] [PubMed]
- Segrelles, C.; Lu, J.; Hammann, B.; Santos, M.; Moral, M.; Cascallana, J.L.; Lara, M.F.; Rho, O.; Carbajal, S.; Traag, J.; et al. Deregulated Activity of Akt in Epithelial Basal Cells Induces Spontaneous Tumors and Heightened Sensitivity to Skin Carcinogenesis. Cancer Res. 2007, 67, 10879–10888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, S.; Purdie, K.J.; Wang, J.; Harwood, C.A.; Proby, C.M.; Pourreyron, C.; Mladkova, N.; Nagano, A.; Dhayade, S.; Athineos, D.; et al. A Unique Panel of Patient-Derived Cutaneous Squamous Cell Carcinoma Cell Lines Provides a Preclinical Pathway for Therapeutic Testing. Int. J. Mol. Sci. 2019, 20, 3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Ebi, H.; Martini, M.; Beausoleil, S.A.; Faber, A.C.; Jakubik, C.T.; Huang, A.; Wang, Y.; Nishtala, M.; Hall, B.; et al. Measurement of PIP3 Levels Reveals an Unexpected Role for p110β in Early Adaptive Responses to p110α-Specific Inhibitors in Luminal Breast Cancer. Cancer Cell 2015, 27, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nat. Cell Biol. 2015, 518, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balagula, Y.; Kang, S.; Patel, M.J. Synergism between mTOR pathway and ultraviolet radiation in the pathogenesis of squamous cell carcinoma and its implication for solid-organ transplant recipients. Photodermatol. Photoimmunol. Photomed. 2014, 31, 15–25. [Google Scholar] [CrossRef] [Green Version]
- De Gruijl, F.; Koehl, G.; Voskamp, P.; Strik, A.; Rebel, H.; Gaumann, A.; De Fijter, J.; Tensen, C.; Bavinck, J.B.; Geissler, E. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int. J. Cancer 2010, 127, 796–804. [Google Scholar] [CrossRef]
- Wulff, B.C.; Kusewitt, D.F.; VanBuskirk, A.M.; Thomas-Ahner, J.M.; Jason, D.F.; Oberyszyn, T.M.; Duncan, F.J. Sirolimus Reduces the Incidence and Progression of UVB-Induced Skin Cancer in SKH Mice even with Co-administration of Cyclosporine A. J. Investig. Dermatol. 2008, 128, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Rho, O.; Kiguchi, K.; Jiang, G.; DiGiovanni, J. Impact of mTORC1 inhibition on keratinocyte proliferation during skin tumor promotion in wild-type and BK5.AktWT mice. Mol. Carcinog. 2013, 53, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Amornphimoltham, P.; Leelahavanichkul, K.; Molinolo, A.; Patel, V.; Gutkind, J.S. Inhibition of Mammalian target of ra-pamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin. Cancer Res. 2008, 14, 8094–8101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, H.M.; Cherikh, W.S.; Cheng, Y.; Hanto, D.W.; Kahan, B.D. Maintenance immunosuppression with tar-get-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation 2005, 80, 883–889. [Google Scholar] [CrossRef]
- Euvrard, S.; Morelon, E.; Rostaing, L.; Goffin, E.; Brocard, A.; Tromme, I.; Broeders, E.N.; Del Marmol, V.; Chatelet, V.; Dompmartin, A.; et al. Sirolimus and Secondary Skin-Cancer Prevention in Kidney Transplantation. N. Engl. J. Med. 2012, 367, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.B.; Walker, R.; Tai, S.S.; Jian, Q.; Russ, G.R. Randomized controlled trial of sirolimus for renal transplant recipients at high risk of melanoma skin cancer. Am. J. Transpl. 2012, 12, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Akker, J.M.H.-V.D.; Harden, P.N.; Hoitsma, A.J.; Proby, C.M.; Wolterbeek, R.; Bavinck, J.N.B.; De Fijter, J.W. Two-Year Randomized Controlled Prospective Trial Converting Treatment of Stable Renal Transplant Recipients with Cutaneous Invasive Squamous Cell Carcinomas to Sirolimus. J. Clin. Oncol. 2013, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Alberú, J.; Pascoe, M.D.; Campistol, J.M.; Schena, F.P.; Polinsky, M.; Neylan, J.F.; Korth-Bradley, J.; Goldberg-Alberts, R.; Maller, E.S.; Maria Del Carmen Sirolimus Convert Trial Study Group. Lower Malignancy Rates in Renal Allograft Recipients Converted to Sirolimus-Based, Calcineurin Inhibitor-Free Immunotherapy: 24-Month Results from the CONVERT Trial. Transplantation 2011, 92, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cisse, O.; Quraishi, M.; Gulluni, F.; Guffanti, F.; Mavrommati, I.; Suthanthirakumaran, M.; Oh, L.C.R.; Schlatter, J.N.; Sarvananthan, A.; Broggini, M.; et al. Downregulation of class II phosphoinositide 3-kinase PI3K-C2β delays cell division and potentiates the effect of docetaxel on cancer cell growth. J. Exp. Clin. Cancer Res. 2019, 38, 472. [Google Scholar] [CrossRef]
- Edling, C.E.; Selvaggi, F.; Buus, R.; Maffucci, T.; Di Sebastiano, P.; Friess, H.; Innocenti, P.; Kocher, H.M.; Falasca, M. Key Role of Phosphoinositide 3-Kinase Class IB in Pancreatic Cancer. Clin. Cancer Res. 2010, 16, 4928–4937. [Google Scholar] [CrossRef] [Green Version]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorganic Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Shanker, K.-S.; Swales, K.; Decordova, S.; Deyoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomel, V.; Klicic, J.; Covini, D.; Church, D.D.; Shaw, J.P.; Roulin, K.; Burgat-Charvillon, F.; Valognes, D.; Camps, M.; Chabert, C.; et al. Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase gamma. J. Med. Chem. 2006, 49, 3857–3871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ng, P.K.-S.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; De Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, C.; Landthaler, M.; Vogt, T. Activation of the PI3K/AKT signalling pathway in non-melanoma skin cancer is not mediated by oncogenic PIK3CA and AKT1 hotspot mutations. Exp. Dermatol. 2010, 19, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational Landscape of Aggressive Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Bs, A.J.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.-A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2015, 122, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic Analysis of Metastatic Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.; Ashford, B.; Thind, A.S.; Gauthier, M.E.; Minaei, E.; Major, G.; Iyer, N.G.; Gupta, R.; Clark, J.; Ranson, M. Compre-hensive Mutational and Phenotypic Characterization of New Metastatic Cutaneous Squamous Cell Carcinoma Cell Lines Reveal Novel Drug Susceptibilities. Int. J. Mol. Sci. 2020, 21, 9536. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α–Selective Inhibition with Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results from the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci. Transl. Med. 2015, 7, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL Mediates Resistance to PI3Kα Inhibition by Activating the EGFR/PKC/mTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichoń, A.M.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; MacKenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The receptor tyrosine kinase Axl regulates cell–cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2013, 33, 4185–4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.; Ikram, M.; Vyas, J.; Patel, N.; Proby, C.M.; Ghali, L.; Leigh, I.M.; O’Toole, E.A.; Storey, A. Overexpression of the Axl tyrosine kinase receptor in cutaneous SCC-derived cell lines and tumours. Br. J. Cancer 2006, 94, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Badarni, M.; Prasad, M.; Balaban, N.; Zorea, J.; Yegodayev, K.M.; Ben-Zion, J.; Dinur, A.B.; Grénman, R.; Rotblat, B.; Cohen, L.; et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight 2019, 4, 125341. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, C.; O’Shaughnessy, R.F.L. Protein kinases involved in epidermal barrier formation: The AKT family and other an-imals. Exp. Dermatol. 2018, 27, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Mannella, V.; Bergamaschi, D.; Harwood, C.; Maffucci, T. Targeting selective phosphoinositide-3-kinase isoforms as a novel strategy in cutaneous squamous cell carcinoma. In Proceedings of the British Society for Investigative Dermatology Annual Meeting Norcroft Conference Centre, University of Bradford, Bradford, UK, 1–3 April 2019; Volume 180, pp. e183–e215. [Google Scholar]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannella, V.; Boehm, K.; Celik, S.; Ali, T.; Mirza, A.N.; El Hasnaouy, M.; Kaffa, A.; Lyu, Y.; Kafaei Golahmadi, D.; Leigh, I.M.; et al. Growth and Viability of Cutaneous Squamous Cell Carcinoma Cell Lines Display Different Sensitivities to Isoform-Specific Phosphoinositide 3-Kinase Inhibitors. Int. J. Mol. Sci. 2021, 22, 3567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073567
Mannella V, Boehm K, Celik S, Ali T, Mirza AN, El Hasnaouy M, Kaffa A, Lyu Y, Kafaei Golahmadi D, Leigh IM, et al. Growth and Viability of Cutaneous Squamous Cell Carcinoma Cell Lines Display Different Sensitivities to Isoform-Specific Phosphoinositide 3-Kinase Inhibitors. International Journal of Molecular Sciences. 2021; 22(7):3567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073567
Chicago/Turabian StyleMannella, Viviana, Kira Boehm, Suheyla Celik, Tasnim Ali, Amnah N. Mirza, Mariam El Hasnaouy, Andreas Kaffa, Yanshuang Lyu, Donya Kafaei Golahmadi, Irene M. Leigh, and et al. 2021. "Growth and Viability of Cutaneous Squamous Cell Carcinoma Cell Lines Display Different Sensitivities to Isoform-Specific Phosphoinositide 3-Kinase Inhibitors" International Journal of Molecular Sciences 22, no. 7: 3567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073567