
Unveiling the N-Terminal Homodimerization of BCL11B by Hybrid Solvent Replica-Exchange Simulations

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Role of BCL11B
1.2. Enhanced Molecular Dynamics Simulations
1.3. Zinc Finger Proteins in Force Field-Based Simulations
2. Results and Discussion
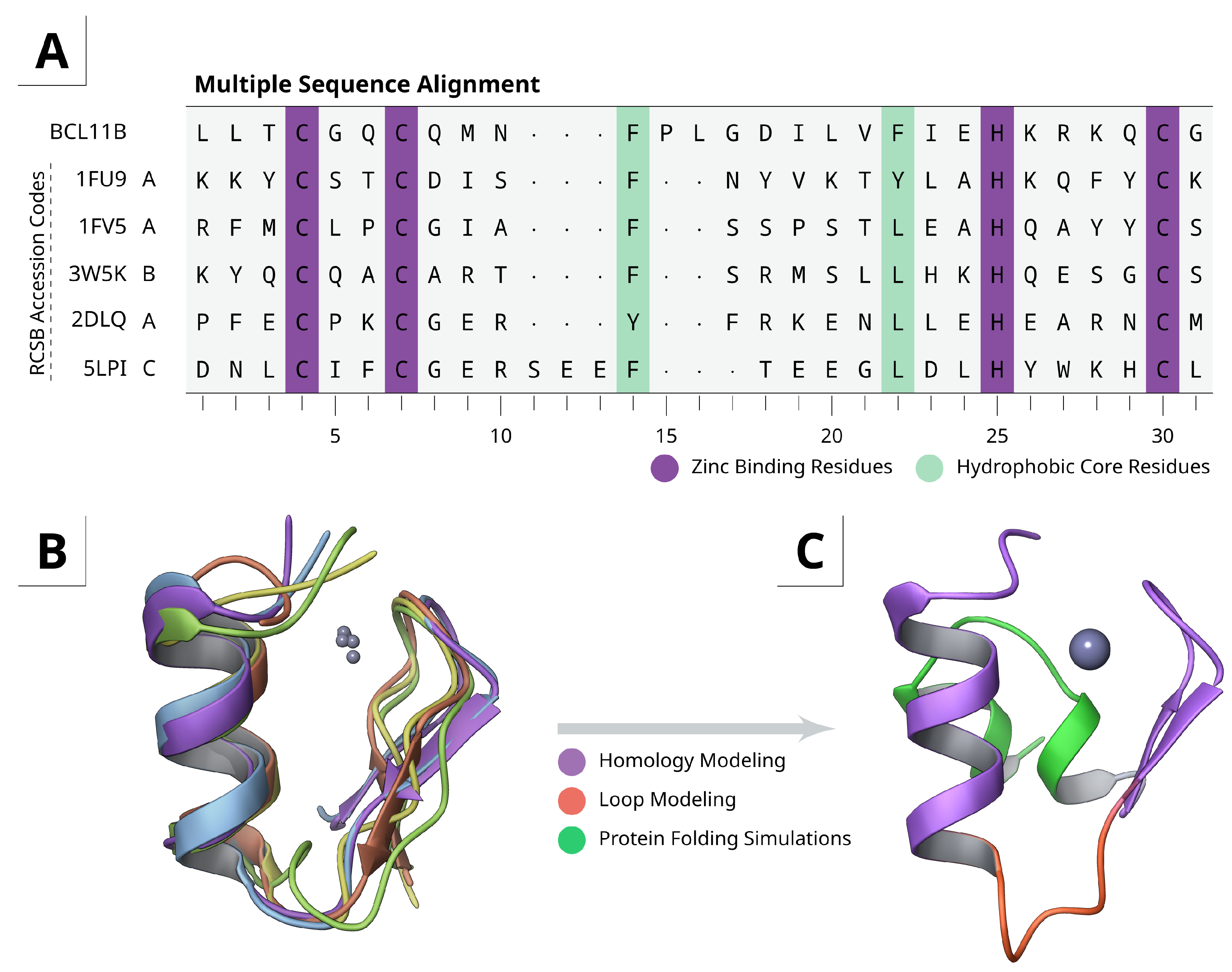
2.1. Homology and Loop Modeling
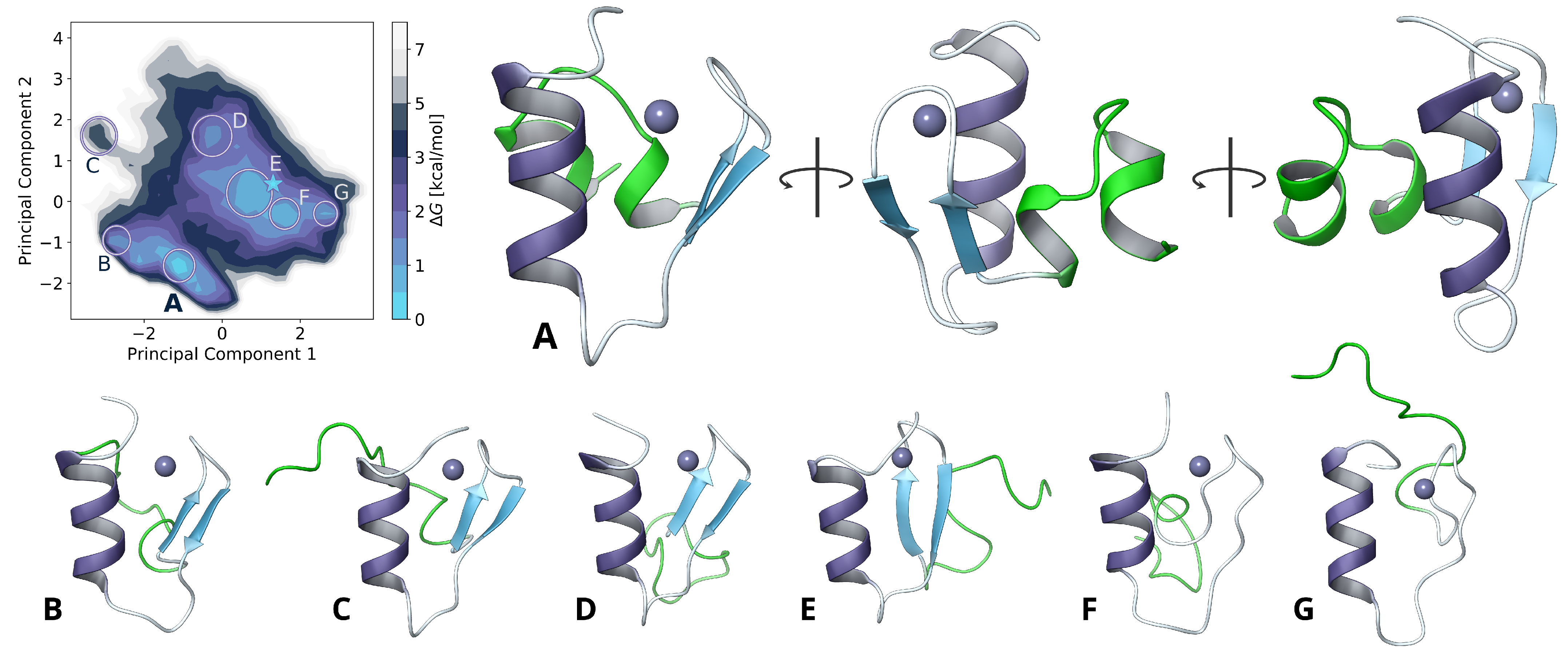
2.2. Model Extension and Monomer Conformational Sampling
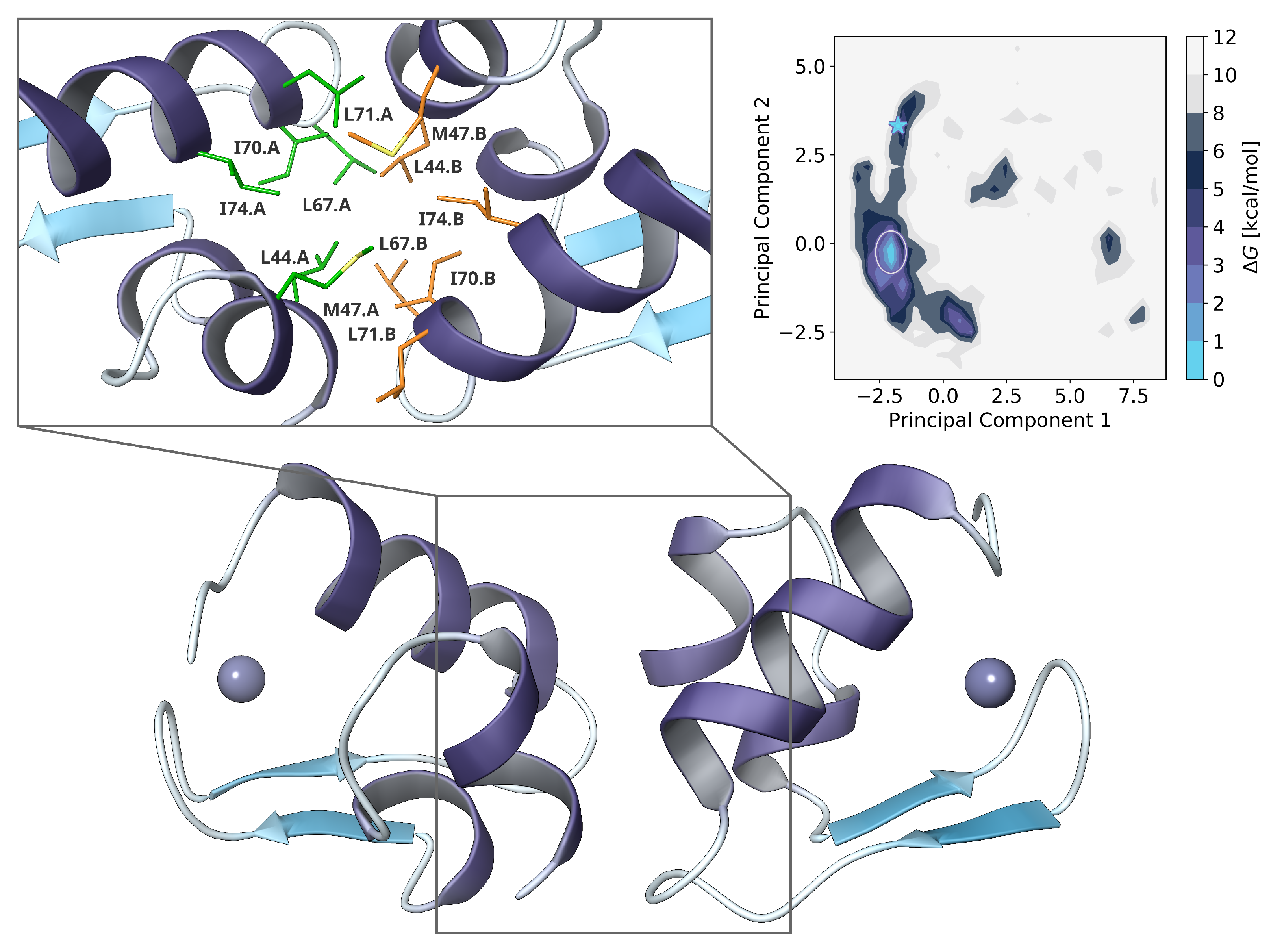
2.3. Dimer Protein-Protein-Docking and Refinement
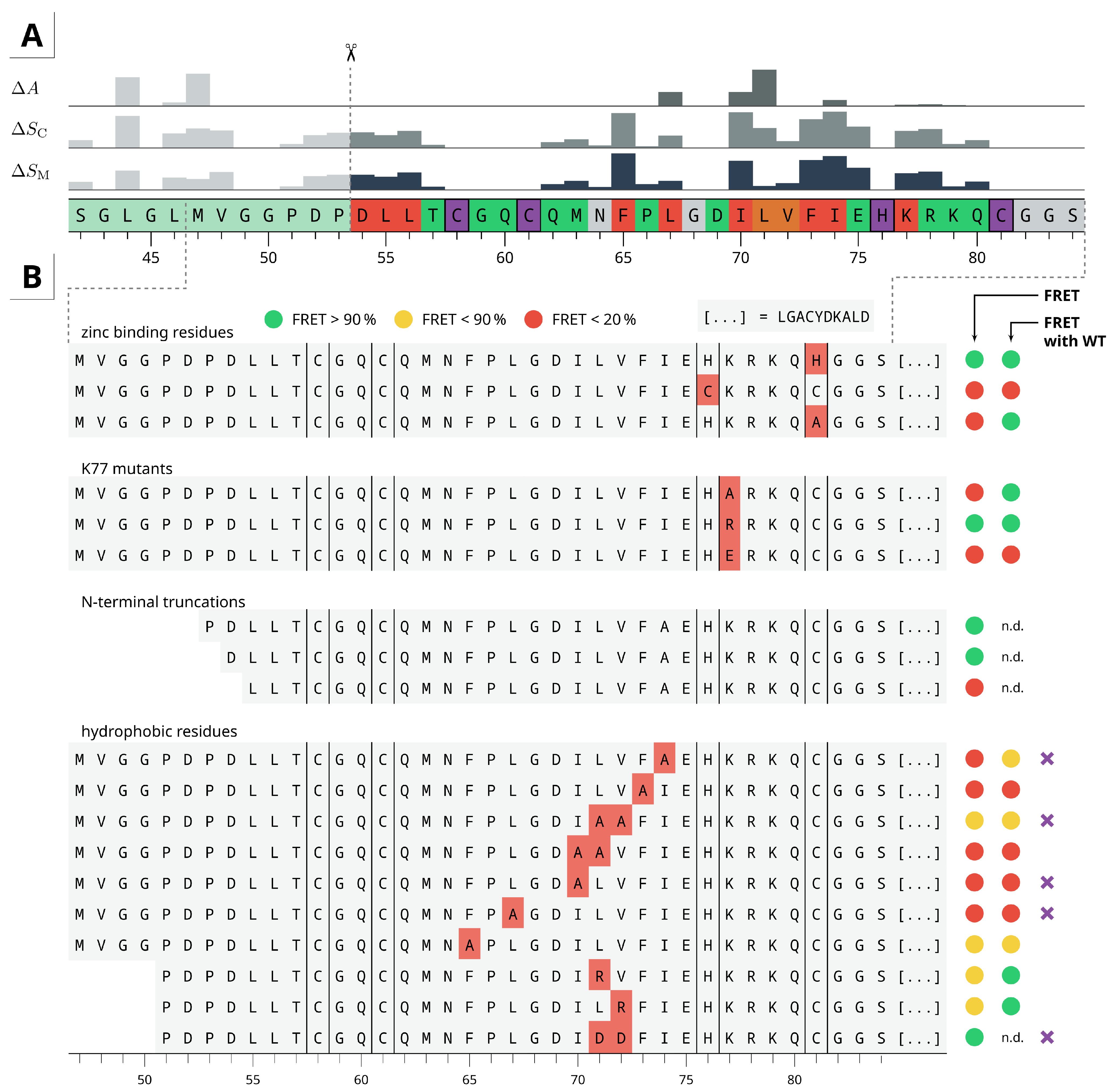
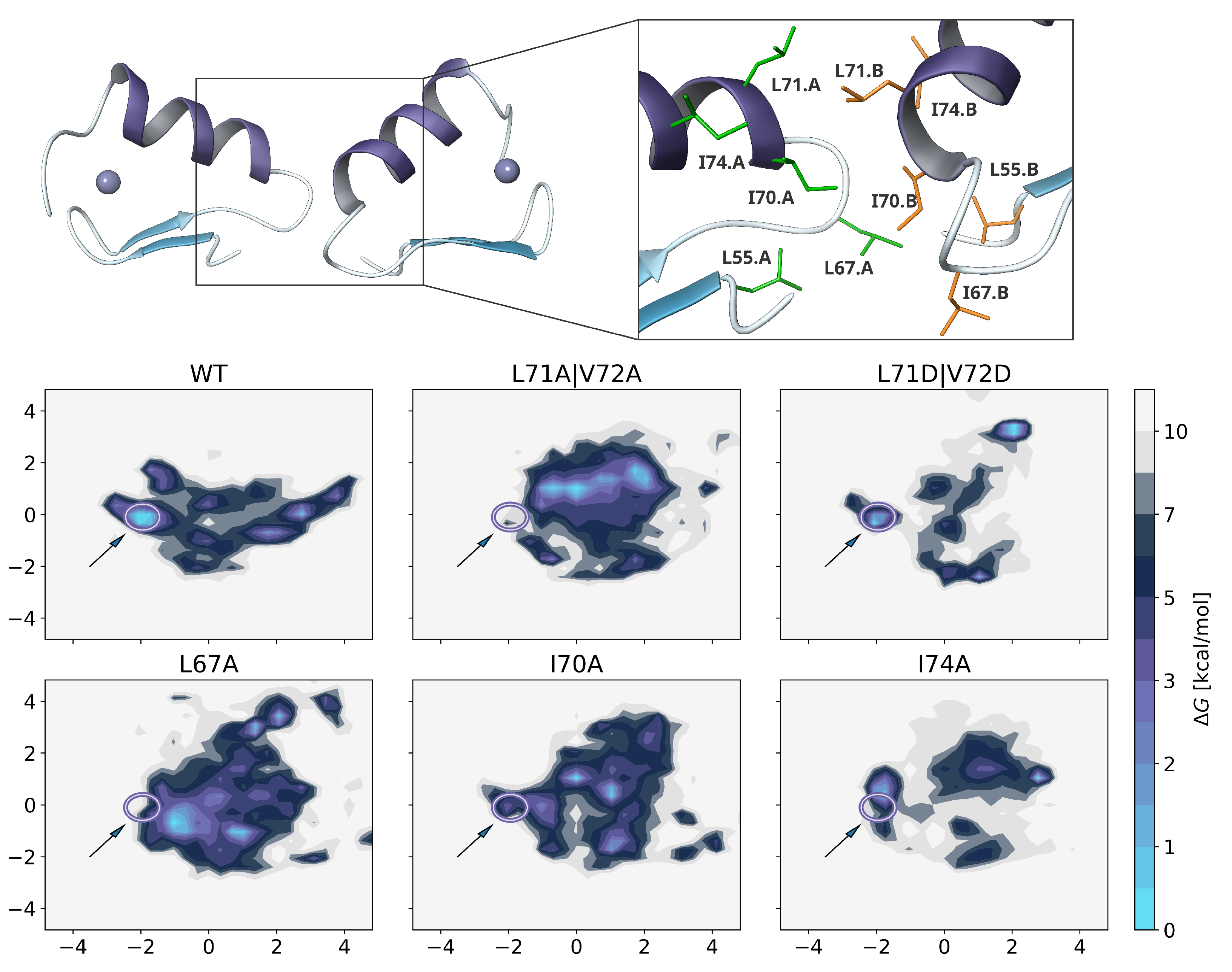
2.4. Residue Importance by FACS-FRET Measurements
2.5. Dimer Protein-Protein-Interactions of Mutants
3. Conclusions and Perspectives
4. Methods
4.1. Homology and Loop Modeling
4.2. Simulation System Preparation
4.3. General Molecular Dynamics Setup
4.4. Monomer Conformational Sampling
4.5. Dimer Protein-Protein-Docking
4.6. Mutants Protein-Protein-Docking
4.7. Alanine Scanning
4.8. FACS-FRET Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ECFP | Enhanced Cyan Fluorescent Protein. |
| EYFP | Enhanced Yellow Fluorescent Protein |
| FACS | Fluorescence-activated Cell Sorting |
| FFT | Fast Fourier Transform |
| FRET | Fluorescence Resonance Energy Transfer |
| GBSA | Generalized Born Model with Accessible Surface Area Term |
| (d)PCA | (Dihedral) Princicipal Component Analysis |
| MDS | Molecular Dynamics Simulation |
| REMD | Replica-Exchange Molecular Dynamics |
| TIGER2h(s) | Temperature Intervals with Global Exchange of Replica with Hybrid Solvent (and Shell) |
| ZF | Zinc Finger |
References
- Cismasiu, V.B.; Adamo, K.; Gecewicz, J.; Duque, J.; Lin, Q.; Avram, D. BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 2005, 24, 6753–6764. [Google Scholar] [CrossRef] [Green Version]
- Senawong, T.; Peterson, V.J.; Avram, D.; Shepherd, D.M.; Frye, R.A.; Minucci, S.; Leid, M. Involvement of the Histone Deacetylase SIRT1 in Chicken Ovalbumin Upstream Promoter Transcription Factor (COUP-TF)-interacting Protein 2-mediated Transcriptional Repression. J. Biol. Chem. 2003, 278, 43041–43050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topark-Ngarm, A.; Golonzhka, O.; Peterson, V.J.; Barrett, B.; Martinez, B.; Crofoot, K.; Filtz, T.M.; Leid, M. CTIP2 Associates with the NuRD Complex on the Promoter ofp57KIP2, a Newly Identified CTIP2 Target Gene. J. Biol. Chem. 2006, 281, 32272–32283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Lint, C.V.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherrier, T.; Suzanne, S.; Redel, L.; Calao, M.; Marban, C.; Samah, B.; Mukerjee, R.; Schwartz, C.; Gras, G.; Sawaya, B.E.; et al. p21WAF1 gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 2009, 28, 3380–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- juan Zhang, L.; Vogel, W.K.; Liu, X.; Topark-Ngarm, A.; Arbogast, B.L.; Maier, C.S.; Filtz, T.M.; Leid, M. Coordinated Regulation of Transcription Factor Bcl11b Activity in Thymocytes by the Mitogen-activated Protein Kinase (MAPK) Pathways and Protein Sumoylation. J. Biol. Chem. 2012, 287, 26971–26988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubuissez, M.; Loison, I.; Paget, S.; Vorng, H.; Ait-Yahia, S.; Rohr, O.; Tsicopoulos, A.; Leprince, D. Protein Kinase C-Mediated Phosphorylation of BCL11B at Serine 2 Negatively Regulates Its Interaction with NuRD Complexes during CD4+T-Cell Activation. Mol. Cell. Biol. 2016, 36, 1881–1898. [Google Scholar] [CrossRef] [Green Version]
- Cismasiu, V.B.; Ghanta, S.; Duque, J.; Albu, D.I.; Chen, H.M.; Kasturi, R.; Avram, D. BCL11B participates in the activation of IL2 gene expression in CD4+ T lymphocytes. Blood 2006, 108, 2695–2702. [Google Scholar] [CrossRef] [Green Version]
- Arlotta, P.; Molyneaux, B.J.; Chen, J.; Inoue, J.; Kominami, R.; Macklis, J.D. Neuronal Subtype-Specific Genes that Control Corticospinal Motor Neuron Development In Vivo. Neuron 2005, 45, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, Y.; Watanabe, H.; Inoue, J.; Takeda, N.; Sakata, J.; Mishima, Y.; Hitomi, J.; Yamamoto, T.; Utsuyama, M.; Niwa, O.; et al. Bcl11b is required for differentiation and survival of αβ-T lymphocytes. Nat. Immunol. 2003, 4, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Golonzhka, O.; Liang, X.; Messaddeq, N.; Bornert, J.M.; Campbell, A.L.; Metzger, D.; Chambon, P.; Ganguli-Indra, G.; Leid, M.; Indra, A.K. Dual Role of COUP-TF-Interacting Protein 2 in Epidermal Homeostasis and Permeability Barrier Formation. J. Investig. Dermatol. 2009, 129, 1459–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golonzhka, O.; Metzger, D.; Bornert, J.M.; Bay, B.K.; Gross, M.K.; Kioussi, C.; Leid, M. Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4278–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrylkova, K.; Kyryachenko, S.; Biehs, B.; Klein, O.; Kioussi, C.; Leid, M. BCL11B Regulates Epithelial Proliferation and Asymmetric Development of the Mouse Mandibular Incisor. PLoS ONE 2012, 7, e37670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Kalisky, T.; Sahoo, D.; Dalerba, P.; Feng, W.; Lin, Y.; Qian, D.; Kong, A.; Yu, J.; Wang, F.; et al. A Quiescent Bcl11b High Stem Cell Population Is Required for Maintenance of the Mammary Gland. Cell Stem Cell 2017, 20, 247–260.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punwani, D.; Zhang, Y.; Yu, J.; Cowan, M.J.; Rana, S.; Kwan, A.; Adhikari, A.N.; Lizama, C.O.; Mendelsohn, B.A.; Fahl, S.P.; et al. Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant BCL11B. N. Engl. J. Med. 2016, 375, 2165–2176. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Burke, S.; Wang, J.; Chen, X.; Ortiz, M.; Lee, S.C.; Lu, D.; Campos, L.; Goulding, D.; Ng, B.L.; et al. Reprogramming of T Cells to Natural Killer-Like Cells upon Bcl11b Deletion. Science 2010, 329, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, A.; Kentsis, A.; Sanda, T.; Holmfeldt, L.; Chen, S.C.; Zhang, J.; Protopopov, A.; Chin, L.; Dahlberg, S.E.; Neuberg, D.S.; et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 2011, 118, 4169–4173. [Google Scholar] [CrossRef] [Green Version]
- Ohi, H.; Mishima, Y.; Kamimura, K.; Maruyama, M.; Sasai, K.; Kominami, R. Multi-step lymphomagenesis deduced from DNA changes in thymic lymphomas and atrophic thymuses at various times after γ-irradiation. Oncogene 2007, 26, 5280–5289. [Google Scholar] [CrossRef] [Green Version]
- Roy, U.; Raghavan, S.C. Deleterious Point Mutations in T-cell Acute Lymphoblastic Leukemia: Mechanistic Insights into Leukemogenesis. Int. J. Cancer 2021. [Google Scholar] [CrossRef]
- Ganguli-Indra, G.; Wasylyk, C.; Liang, X.; Millon, R.; Leid, M.; Wasylyk, B.; Abecassis, J.; Indra, A. CTIP2 Expression in Human Head and Neck Squamous Cell Carcinoma Is Linked to Poorly Differentiated Tumor Status. PLoS ONE 2009, 4, e5367. [Google Scholar] [CrossRef]
- Wiles, E.T.; Lui-Sargent, B.; Bell, R.; Lessnick, S.L. BCL11B Is Up-Regulated by EWS/FLI and Contributes to the Transformed Phenotype in Ewing Sarcoma. PLoS ONE 2013, 8, e59369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabarczyk, P.; Przybylski, G.K.; Depke, M.; Völker, U.; Bahr, J.; Assmus, K.; Bröker, B.M.; Walther, R.; Schmidt, C.A. Inhibition of BCL11B expression leads to apoptosis of malignant but not normal mature T cells. Oncogene 2007, 26, 3797–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, K.; Mishima, Y.; Obata, M.; Endo, T.; Aoyagi, Y.; Kominami, R. Lack of Bcl11b tumor suppressor results in vulnerability to DNA replication stress and damages. Oncogene 2007, 26, 5840–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabarczyk, P.; Nähse, V.; Delin, M.; Przybylski, G.; Depke, M.; Hildebrandt, P.; Völker, U.; Schmidt, C.A. Increased Expression of Bcl11b Leads to Chemoresistance Accompanied by G1 Accumulation. PLoS ONE 2010, 5, e12532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanam, N.K.; Grabarczyk, P.; Hammer, E.; Scharf, C.; Venz, S.; Gesell-Salazar, M.; Barthlen, W.; Przybylski, G.K.; Schmidt, C.A.; Volker, U. Proteome Analysis Reveals New Mechanisms of Bcl11b-loss Driven Apoptosis. J. Proteome Res. 2010, 9, 3799–3811. [Google Scholar] [CrossRef]
- Grabarczyk, P.; Winkler, P.; Delin, M.; Sappa, P.K.; Bekeschus, S.; Hildebr, T.P.; Przybylski, G.K.; Völker, U.; Hammer, E.; Schmidt, C.A. The N-Terminal CCHC Zinc Finger Motif Mediates Homodimerization of Transcription Factor BCL11B. Mol. Cell. Biol. 2017, 38. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Rosta, E.; Hummer, G. Error and efficiency of replica exchange molecular dynamics simulations. J. Chem. Phys. 2009, 131, 165102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulke, M.; Geist, N.; Möller, D.; Langel, W. Replica-Based Protein Structure Sampling Methods: Compromising between Explicit and Implicit Solvents. J. Phys. Chem. B 2018, 122, 7295–7307. [Google Scholar] [CrossRef]
- Geist, N.; Kulke, M.; Schulig, L.; Link, A.; Langel, W. Replica-Based Protein Structure Sampling Methods II: Advanced Hybrid Solvent TIGER2hs. J. Phys. Chem. B 2019, 123, 5995–6006. [Google Scholar] [CrossRef] [PubMed]
- Sakharov, D.V.; Lim, C. Zn Protein Simulations Including Charge Transfer and Local Polarization Effects. J. Am. Chem. Soc. 2005, 127, 4921–4929. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, J.; Wang, J.; Wang, W. Metal-Coupled Folding of Cys2His2Zinc-Finger. J. Am. Chem. Soc. 2008, 130, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Jiao, X.; Hu, J.P.; Chen, Y.; Tian, X.H. Stability and Folding Behavior Analysis of Zinc-Finger Using Simple Models. Int. J. Mol. Sci. 2010, 11, 4014–4034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluska, K.; Adamczyk, J.; Krężel, A. Metal binding properties, stability and reactivity of zinc fingers. Coord. Chem. Rev. 2018, 367, 18–64. [Google Scholar] [CrossRef]
- Kulke, M.; Uhrhan, M.; Geist, N.; Brüggemann, D.; Ohler, B.; Langel, W.; Köppen, S. Phosphorylation of Fibronectin Influences the Structural Stability of the Predicted Interchain Domain. J. Chem. Inf. Model. 2019, 59, 4383–4392. [Google Scholar] [CrossRef] [PubMed]
- Malgieri, G.; Palmieri, M.; Russo, L.; Fattorusso, R.; Pedone, P.V.; Isernia, C. The prokaryotic zinc-finger: Structure, function and comparison with the eukaryotic counterpart. FEBS J. 2015, 282, 4480–4496. [Google Scholar] [CrossRef] [Green Version]
- Namuswe, F.; Berg, J.M. Secondary interactions involving zinc-bound ligands: Roles in structural stabilization and macromolecular interactions. J. Inorg. Biochem. 2012, 111, 146–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padhorny, D.; Hall, D.R.; Mirzaei, H.; Mamonov, A.B.; Moghadasi, M.; Alekseenko, A.; Beglov, D.; Kozakov, D. Protein–ligand docking using FFT based sampling: D3R case study. J. Comput.-Aided Mol. Des. 2017, 32, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.K.; Kowalski, K.; Fox, A.H.; Newton, A.; Sharpe, B.K.; Crossley, M.; Mackay, J.P. Solution Structures of Two CCHC Zinc Fingers from the FOG Family Protein U-Shaped that Mediate Protein–Protein Interactions. Structure 2000, 8, 1157–1166. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M.N.; Merz, K.M. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Case, D.A.; Zhong, E.D. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput.-Aided Mol. Des. 2016, 31, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulig, L.; Grabarczyk, P.; Geist, N.; Delin, M.; Forkel, H.; Kulke, M.; Delcea, M.; Schmidt, C.A.; Link, A. Unveiling the N-Terminal Homodimerization of BCL11B by Hybrid Solvent Replica-Exchange Simulations. Int. J. Mol. Sci. 2021, 22, 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073650
Schulig L, Grabarczyk P, Geist N, Delin M, Forkel H, Kulke M, Delcea M, Schmidt CA, Link A. Unveiling the N-Terminal Homodimerization of BCL11B by Hybrid Solvent Replica-Exchange Simulations. International Journal of Molecular Sciences. 2021; 22(7):3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073650
Chicago/Turabian StyleSchulig, Lukas, Piotr Grabarczyk, Norman Geist, Martin Delin, Hannes Forkel, Martin Kulke, Mihaela Delcea, Christian A. Schmidt, and Andreas Link. 2021. "Unveiling the N-Terminal Homodimerization of BCL11B by Hybrid Solvent Replica-Exchange Simulations" International Journal of Molecular Sciences 22, no. 7: 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073650