
Role of Rho GTPase Interacting Proteins in Subcellular Compartments of Podocytes


Abstract
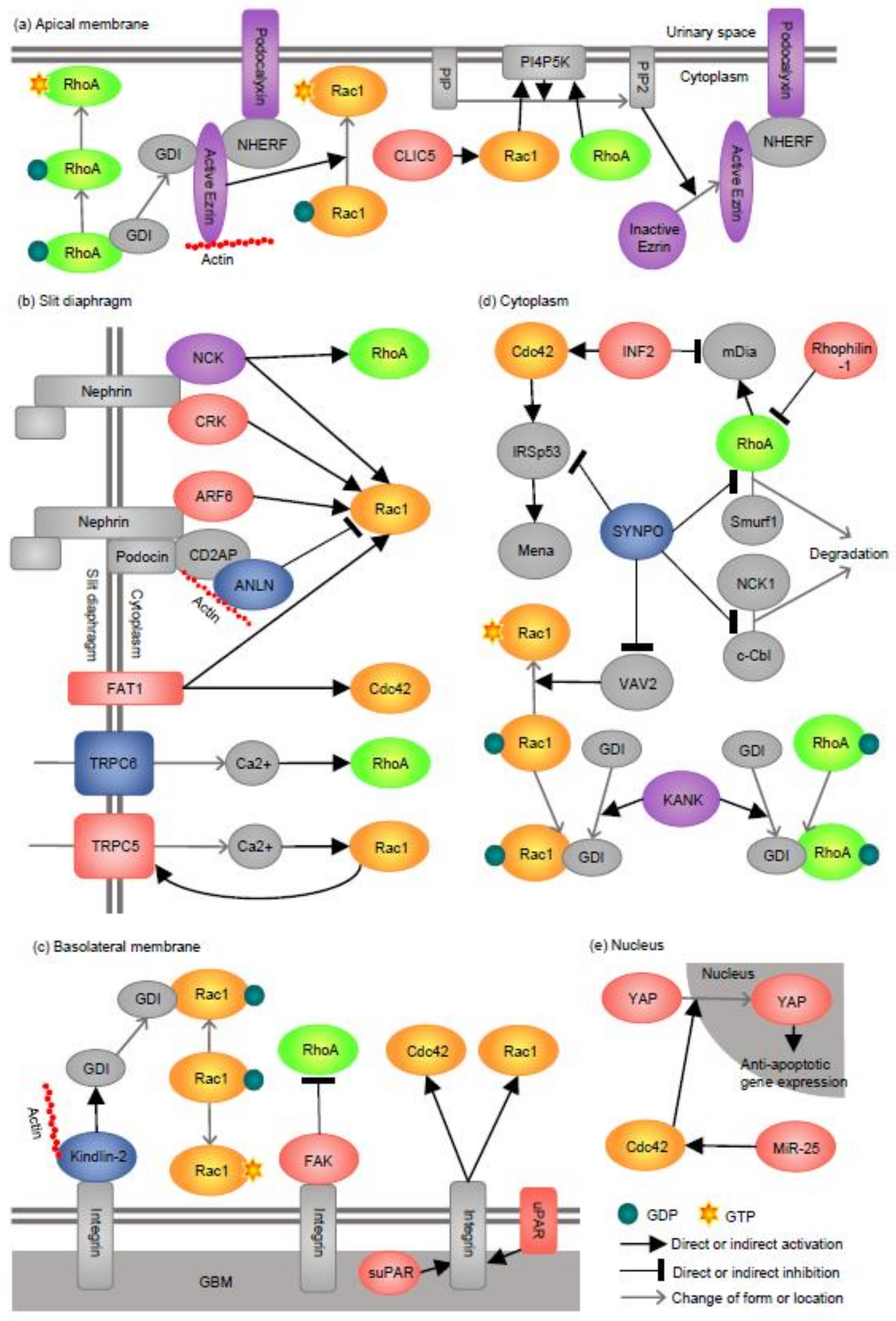
:1. Introduction
2. Apical Membrane
2.1. Podocalyxin
2.2. Ezrin
2.3. CLIC5
3. Slit Diaphragm (SD)
3.1. NCK
3.2. CRK
3.3. ARF6
3.4. ANLN
3.5. FAT1
3.6. TRPC5 and TRPC6
4. Basolateral Membrane
4.1. FAK
4.2. Kindlin-2
4.3. uPAR, suPAR, and Integrin
5. Cytoplasm (Actin Cytoskeleton)
5.1. SYNPO
5.2. INF2
5.3. KANK
5.4. Rhophilin-1
6. Nucleus
7. Rho GTPase Interacting Proteins Whose Localization Could Not Be Identified
7.1. aPKC
7.2. NMDAR1
8. Summary and Future Directions
- In the apical membrane and the SD, a certain level of RhoA and Rac1 activities are important for the normal development and basal maintenance of podocyte morphology.
- In all three membrane domains of foot processes, overactivation of Rac1/Cdc42 is pathogenic and contributes to podocyte injury.
- In the basolateral membrane and the cytoplasm, RhoA abundance/activity correlates with rapid recovery after podocyte injury.
- A certain level of Cdc42 activity is critical for podocyte survival because it is required for YAP translocation to the nucleus and subsequent expression of anti-apoptotic genes.
Author Contributions
Funding
Conflicts of Interest
References
- Garg, P. A review of podocyte biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Welsh, G.I.; Saleem, M.A. The podocyte cytoskeleton-key to a functioning glomerulus in health and disease. Nat. Rev. Nephrol. 2011, 8, 14–21. [Google Scholar] [CrossRef]
- Tian, X.; Ishibe, S. Targeting the podocyte cytoskeleton: From pathogenesis to therapy in proteinuric kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1577–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatecka-Urban, A. Endocytic trafficking at the mature podocyte slit diaphragm. Front. Pediatr. 2017, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.; Nissaisorakarn, P.; Husain, I.; Jim, B. Proteinuric kidney diseases: A podocyte’s slit diaphragm and cytoskeleton approach. Front. Med. 2018, 5, 221. [Google Scholar] [CrossRef]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol. 2013, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.P.; Hawley, S.P.; Ruston, J.; Du, J.; Brakebusch, C.; Jones, N.; Pawson, T. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Jiang, R.; Aoudjit, L.; Jones, N.; Takano, T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Cote, J.F.; Gupta, I.R.; Takano, T. Rac1 activation in podocytes induces the spectrum of nephrotic syndrome. Kidney Int. 2017, 92, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Suleiman, H.; Kim, A.; Miner, J.; Dani, A.; Shaw, A.; Akilesh, S. Rac1 Activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell. Biol. 2013, 33, 4755–4764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouawad, F.; Tsui, H.; Takano, T. Role of Rho-GTPases and their regulatory proteins in glomerular podocyte function. Can. J. Physiol. Pharmacol. 2013, 91, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.A.; Welsh, G.I. Podocyte RhoGTPases: New therapeutic targets for nephrotic syndrome? F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Matsuda, J.; Asano-Matsuda, K.; Kitzler, T.; Takano, T. Rho GTPase regulatory proteins in podocytes. Kidney Int. 2021, 99, 336–345. [Google Scholar] [CrossRef]
- Schmieder, S.; Nagai, M.; Orlando, R.A.; Takeda, T.; Farquhar, M.G. Podocalyxin activates RhoA and induces actin reorganization through NHERF1 and Ezrin in MDCK cells. J. Am. Soc. Nephrol. 2004, 15, 2289–2298. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, H.; Obayashi, H.; Schmieder, S.; Lee, J.; Ghosh, P.; Farquhar, M.G. Phosphorylation of podocalyxin (Ser415) Prevents RhoA and ezrin activation and disrupts its interaction with the actin cytoskeleton. Am. J. Pathol. 2011, 179, 2254–2265. [Google Scholar] [CrossRef]
- Matsui, T.; Yonemura, S.; Tsukita, S.; Tsukita, S. Activation of ERM proteins in vivo by Rho involves phosphatidylinositol 4-phosphate 5-kinase and not ROCK kinases. Curr. Biol. 1999, 9, 1259–1262. [Google Scholar] [CrossRef] [Green Version]
- Hatano, R.; Takeda, A.; Abe, Y.; Kawaguchi, K.; Kazama, I.; Matsubara, M.; Asano, S. Loss of ezrin expression reduced the susceptibility to the glomerular injury in mice. Sci. Rep. 2018, 8, 4512. [Google Scholar] [CrossRef] [PubMed]
- Tavasoli, M.; Li, L.; Al-Momany, A.; Zhu, L.F.; Adam, B.A.; Wang, Z.; Ballermann, B.J. The chloride intracellular channel 5A stimulates podocyte Rac1, protecting against hypertension-induced glomerular injury. Kidney Int. 2016, 89, 833–847. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Buvall, L.; Rashmi, P.; Lopez-Rivera, E.; Andreeva, S.; Weins, A.; Wallentin, H.; Greka, A.; Mundel, P. Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat. Commun. 2013, 4, 2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, B.; Fan, Q.; Dlugos, C.P.; Soofi, A.A.; Zhang, J.; Verma, R.; Park, T.J.; Wong, H.; Curran, T.; Nihalani, D.; et al. Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int. 2014, 85, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.S.; Jeon, J.S.; Fan, Q.; Wong, H.N.; Palmer, M.B.; Holzman, L.B. ARF6 mediates nephrin tyrosine phosphorylation-induced podocyte cellular dynamics. PLoS ONE 2017, 12, e0184575. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.; Lane, B.M.; Khan, K.; Pediaditakis, I.; Xiao, J.; Wu, G.; Wang, L.; Kovalik, M.E.; Chryst-Stangl, M.; Davis, E.E.; et al. The human FSGS-Causing ANLN R431C Mutation Induces Dysregulated PI3K/AKT/mTOR/Rac1 signaling in podocytes. J. Am. Soc. Nephrol. 2018, 29, 2110–2122. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Jacobo, S.; Billing, D.; Rozkalne, A.; Gage, S.; Anagnostou, T.; Pavenstädt, H.; Hsu, H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Echtermeyer, F.; Thilo, F.; Theilmeier, G.; Schmidt, A.; Schulein, R.; Jensen, B.L.; Loddenkemper, C.; Jankowski, V.; Marcussen, N.; et al. The proteoglycan syndecan 4 regulates transient receptor potential canonical 6 channels via RhoA/Rho-associated protein kinase signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaldecker, T.; Kim, S.; Tarabanis, C.; Tian, D.; Hakroush, S.; Castonguay, P.; Ahn, W.; Wallentin, H.; Heid, H.; Hopkins, C.R.; et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J. Clin. Investig. 2013, 123, 5298–5309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerides, V.J.; Ramsey, I.S.; Kotecha, S.; Greka, A.; Clapham, D.E. Rapid vesicular translocation and insertion of TRP channels. Nat. Cell Biol. 2004, 6, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Togawa, A.; Soda, K.; Zhang, J.; Lee, S.; Ma, M.; Yu, Z.; Ardito, T.; Czyzyk, J.; Diggs, L.; et al. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J. Am. Soc. Nephrol. 2010, 21, 1145–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Guo, C.; Ma, P.; Lai, Y.; Yang, F.; Cai, J.; Cheng, Z.; Zhang, K.; Liu, Z.; Tian, Y.; et al. Kindlin-2 association with Rho GDP-dissociation inhibitor alpha suppresses Rac1 activation and podocyte injury. J. Am. Soc. Nephrol. 2017, 28, 3545–3562. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Moller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2008, 14, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, T.H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 2015, 26, 133–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asanuma, K.; Yanagida-Asanuma, E.; Faul, C.; Tomino, Y.; Kim, K.; Mundel, P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell Biol. 2006, 8, 485–491. [Google Scholar] [CrossRef]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Buvall, L.; Wallentin, H.; Sieber, J.; Andreeva, S.; Choi, H.Y.; Mundel, P.; Greka, A. Synaptopodin is a coincidence detector of tyrosine versus serine/threonine phosphorylation for the modulation of Rho protein crosstalk in podocytes. J. Am. Soc. Nephrol. 2017, 28, 837–851. [Google Scholar] [CrossRef]
- Sun, H.; Schlondorff, J.; Higgs, H.N.; Pollak, M.R. Inverted formin 2 regulates actin dynamics by antagonizing Rho/diaphanous-related formin signaling. J. Am. Soc. Nephrol. 2013, 24, 917–929. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Hao, X.; Azeloglu, E.U.; Ren, H.; Wang, Z.; Ma, J.; Liu, J.; Ma, X.; Wang, W.; Pan, X.; et al. Novel mutations in the inverted formin 2 gene of Chinese families contribute to focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Zhang, F.; Ashraf, S.; Kohl, S.; Sadowski, C.E.; Vega-Warner, V.; Zhou, W.; Lovric, S.; Fang, H.; Nettleton, M.; et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Investig. 2015, 125, 2375–2384. [Google Scholar] [CrossRef] [Green Version]
- Lal, M.A.; Andersson, A.C.; Katayama, K.; Xiao, Z.; Nukui, M.; Hultenby, K.; Wernerson, A.; Tryggvason, K. Rhophilin-1 is a key regulator of the podocyte cytoskeleton and is essential for glomerular filtration. J. Am. Soc. Nephrol. 2015, 26, 647–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.S.; Pawson, T.; Wrana, J.; McNeill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, J.; Maier, M.; Aoudjit, L.; Baldwin, C.; Takano, T. ARHGEF7 (beta-PIX) is required for the maintenance of podocyte architecture and glomerular function. J. Am. Soc. Nephrol. 2020, 31, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Liu, J.; Han, P.; Li, X.; Bai, H.; Zhang, C.; Sun, X.; Teng, Y.; Zhang, Y.; et al. Variations in MicroRNA-25 expression influence the severity of diabetic kidney disease. J. Am. Soc. Nephrol. 2017, 28, 3627–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthmann, K.; Leitges, M.; Teng, B.; Sestu, M.; Tossidou, I.; Samson, T.; Haller, H.; Huber, T.B.; Schiffer, M. Def-6, a novel regulator of small GTPases in podocytes, acts downstream of atypical protein kinase C (aPKC) lambda/iota. Am. J. Pathol. 2013, 183, 1945–1959. [Google Scholar] [CrossRef]
- Shen, J.; Wang, R.; He, Z.; Huang, H.; He, X.; Zhou, J.; Yan, Y.; Shen, S.; Shao, X.; Shen, X.; et al. NMDA receptors participate in the progression of diabetic kidney disease by decreasing Cdc42-GTP activation in podocytes. J. Pathol. 2016, 240, 149–160. [Google Scholar] [CrossRef]
- Shankland, S.J. The podocyte’s response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int. 2006, 69, 2131–2147. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; McQuistan, T.; Orlando, R.A.; Farquhar, M.G. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J. Clin. Investig. 2001, 108, 289–301. [Google Scholar] [CrossRef]
- Doyonnas, R.; Kershaw, D.; Duhme, C.; Merkens, H.; Chelliah, S.; Graf, T.; McNagny, K. Anuria, omphalocele, and perinatal lethality in mice lacking the CD34-related protein podocalyxin. J. Exp. Med. 2001, 194, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Refaeli, I.; Hughes, M.R.; Wong, A.K.; Bissonnette, M.L.Z.; Roskelley, C.D.; Wayne Vogl, A.; Barbour, S.J.; Freedman, B.S.; McNagny, K.M. Distinct functional requirements for podocalyxin in immature and mature podocytes reveal mechanisms of human kidney disease. Sci. Rep. 2020, 10, 9419. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Powell, R.; Nakamura, T.; Kurihara, H.; Sakai, T.; Obara, T. Podocalyxin regulates pronephric glomerular development in zebrafish. Physiol. Rep. 2013, 1. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Sasaki, T.; Mammoto, A.; Takaishi, K.; Kameyama, T.; Tsukita, S.; Tsukita, S.; Takai, Y. Direct Interaction of the Rho GDP dissociation inhibitor with Ezrin/Radixin/Moesin initiates the activation of the Rho Small G protein. J. Biol. Chem. 1997, 272, 23371–23375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, R.; Fujii, E.; Segawa, H.; Mukaisho, K.; Matsubara, M.; Miyamoto, K.; Hattori, T.; Sugihara, H.; Asano, S. Ezrin, a membrane cytoskeletal cross-linker, is essential for the regulation of phosphate and calcium homeostasis. Kidney Int. 2013, 83, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, B.; Al-Momany, A.; Kulak, S.C.; Kozlowski, K.; Obeidat, M.; Jahroudi, N.; Paes, J.; Berryman, M.; Ballermann, B.J. CLIC5A, a component of the ezrin-podocalyxin complex in glomeruli, is a determinant of podocyte integrity. Am. J. Physiol. Renal Physiol. 2010, 298, F1492–F1503. [Google Scholar] [CrossRef] [Green Version]
- Pierchala, B.A.; Munoz, M.R.; Tsui, C.C. Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int. 2010, 78, 868–882. [Google Scholar] [CrossRef] [Green Version]
- Al-Momany, A.; Li, L.; Alexander, R.T.; Ballermann, B.J. Clustered PI(4,5)P(2) accumulation and ezrin phosphorylation in response to CLIC5A. J. Cell Sci. 2014, 127, 5164–5178. [Google Scholar] [CrossRef] [Green Version]
- Tryggvason, K. Unraveling the mechanisms of glomerular ultrafiltration: Nephrin, a key component of the slit diaphragm. J. Am. Soc. Nephrol. 1999, 10, 2440–2445. [Google Scholar]
- Putaala, H.; Sainio, K.; Sariola, H.; Tryggvason, K. Primary structure of mouse and rat nephrin cDNA and structure and expression of the mouse gene. J. Am. Soc. Nephrol. 2000, 11, 991–1001. [Google Scholar] [PubMed]
- Jones, N.; Blasutig, I.M.; Eremina, V.; Ruston, J.M.; Bladt, F.; Li, H.; Huang, H.; Larose, L.; Li, S.S.; Takano, T.; et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006, 440, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, J.; Aoudjit, L.; Latreille, M.; Kawachi, H.; Larose, L.; Takano, T. Rat nephrin modulates cell morphology via the adaptor protein Nck. Biochem. Biophys. Res. Commun. 2006, 349, 310–316. [Google Scholar] [CrossRef]
- New, L.A.; Martin, C.E.; Scott, R.P.; Platt, M.J.; Keyvani Chahi, A.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Maddox, A.S. Anillin. Curr. Biol. 2010, 20, R135–R136. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Yaoita, E.; Kurihara, H.; Shimizu, F.; Sakai, T.; Kobayashi, T.; Ohshiro, K.; Kawachi, H.; Okada, H.; Suzuki, H.; et al. FAT is a component of glomerular slit diaphragms. Kidney Int. 2001, 59, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.; Conlon, P.; Lynn, K.; Farrington, M.; Creazzo, T.; Hawkins, A.; Daskalakis, N.; Kwan, S.; Ebersviller, S.; Burchette, J.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riehle, M.; Buscher, A.K.; Gohlke, B.O.; Kassmann, M.; Kolatsi-Joannou, M.; Brasen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D loss-of-function mutation associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef] [Green Version]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.Z.; Saleem, M.A.; et al. TRPC6 binds to and activates calpain, independent of its channel activity, and regulates podocyte cytoskeleton, cell adhesion, and motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.; Schermer, B.; Müller, R.; Höhne, M.; Bartram, M.; Calixto, A.; Hagmann, H.; Reinhardt, C.; Koos, F.; Kunzelmann, K.; et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc. Natl. Acad. Sci. USA 2006, 103, 17079–17086. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Tang, J.; Chen, Z.; Trost, C.; Flockerzi, V.; Li, M.; Ramesh, V.; Zhu, M.X. Association of mammalian trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J. Biol. Chem. 2000, 275, 37559–37564. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Castonguay, P.; Sidhom, E.; Clark, A.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.; Andreeva, S.; et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durvasula, R.; Shankland, S. Podocyte injury and targeting therapy: An update. Curr. Opin. Nephrol. Hypertens. 2006, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, K.; Kim, K.; Oh, J.; Giardino, L.; Chabanis, S.; Faul, C.; Reiser, J.; Mundel, P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J. Clin. Investig. 2005, 115, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Boyer, O.; Benoit, G.; Gribouval, O.; Nevo, F.; Tete, M.J.; Dantal, J.; Gilbert-Dussardier, B.; Touchard, G.; Karras, A.; Presne, C.; et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Cong, E.; Arrondel, C.; Tête, M.; Montjean, R.; et al. INF2 mutations in Charcot–Marie–Tooth disease with glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, B.; Chun, J.; Perez-Gill, C.; Yan, P.; Stillman, I.E.; Higgs, H.N.; Alper, S.L.; Schlondorff, J.S.; Pollak, M.R. FSGS-causing INF2 mutation impairs Cleaved INF2 N-Fragment functions in podocytes. J. Am. Soc. Nephrol. 2020, 31, 374–391. [Google Scholar] [CrossRef]
- Subramanian, B.; Sun, H.; Yan, P.; Charoonratana, V.T.; Higgs, H.N.; Wang, F.; Lai, K.V.; Valenzuela, D.M.; Brown, E.J.; Schlondorff, J.S.; et al. Mice with mutant Inf2 show impaired podocyte and slit diaphragm integrity in response to protamine-induced kidney injury. Kidney Int. 2016, 90, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Madrid, R.; Aranda, J.F.; Rodriguez-Fraticelli, A.E.; Ventimiglia, L.; Andres-Delgado, L.; Shehata, M.; Fanayan, S.; Shahheydari, H.; Gomez, S.; Jimenez, A.; et al. The formin INF2 regulates basolateral-to-apical transcytosis and lumen formation in association with Cdc42 and MAL2. Dev. Cell 2010, 18, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kakinuma, N.; Zhu, Y.; Kiyama, R. Nucleo-cytoplasmic shuttling of human Kank protein accompanies intracellular translocation of beta-catenin. J. Cell Sci. 2006, 119, 4002–4010. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, G.; Saito, Y.; Madaule, P.; Ishizaki, T.; Fujisawa, K.; Morii, N.; Mukai, H.; Ono, Y.; Kakizuka, A.; Narumiya, S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 1996, 271, 645–648. [Google Scholar] [CrossRef]
- Schwartzman, M.; Reginensi, A.; Wong, J.S.; Basgen, J.M.; Meliambro, K.; Nicholas, S.B.; D’Agati, V.; McNeill, H.; Campbell, K.N. Podocyte-specific deletion of yes-associated protein causes FSGS and progressive renal failure. J. Am. Soc. Nephrol. 2016, 27, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, T.B.; Hartleben, B.; Winkelmann, K.; Schneider, L.; Becker, J.U.; Leitges, M.; Walz, G.; Haller, H.; Schiffer, M. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J. Am. Soc. Nephrol. 2009, 20, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Donnelly, M.; Merscher-Gomez, S.; Chang, Y.H.; Franz, S.; Delfgaauw, J.; Chang, J.M.; Choi, H.Y.; Campbell, K.N.; Kim, K.; et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 2008, 14, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, M.; Teng, B.; Gu, C.; Shchedrina, V.A.; Kasaikina, M.; Pham, V.A.; Hanke, N.; Rong, S.; Gueler, F.; Schroder, P.; et al. Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat. Med. 2015, 21, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Rho GTPase | ||||||
|---|---|---|---|---|---|---|
| Compartment | Interacting Protein | RhoA | Rac1 | Cdc42 | Known Modulators | Reference Number |
| Apical membrane | Podocalyxin | ✓ | Ezrin, NHERF, RhoGDI | [18,19] | ||
| Ezrin | ✓ | ✓ | RhoGDI | [20,21] | ||
| CLIC5 | ✓ | [22] | ||||
| Slit diaphragm | NCK | ✓ | ✓ | Nephrin | [23,24] | |
| CRK | ✓ | Nephrin | [25,26] | |||
| ARF6 | ✓ | Nephrin | [27] | |||
| ANLN | ✓ | CD2AP | [28] | |||
| FAT1 | ✓ | ✓ | [29] | |||
| TRPC6 | ✓ | [30,31] | ||||
| TRPC5 | ✓ | [32,33] | ||||
| Basolateral membrane | FAK | ✓ | Integrin | [34] | ||
| Kindlin-2 | ✓ | RhoGDI, Integrin | [35] | |||
| uPAR, suPAR | ✓ | ✓ | Integrin | [36,37] | ||
| Cytoplasm | SYNPO | ✓ | ✓ | ✓ | VAV2, IRSp53, Smurf1, c-Cbl | [24,38,39,40] |
| INF2 | ✓ | ✓ | mDia | [41,42] | ||
| KANK | ✓ | ✓ | RhoGDI | [43] | ||
| Rhophilin-1 | ✓ | [44] | ||||
| Nucleus | YAP | ✓ | [45,46] | |||
| MiR-25 | ✓ | [47] | ||||
| Undetermined | aPKC | ✓ | ✓ | ✓ | Def-6 | [48] |
| NMDAR1 | ✓ | [49] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano-Matsuda, K.; Ibrahim, S.; Takano, T.; Matsuda, J. Role of Rho GTPase Interacting Proteins in Subcellular Compartments of Podocytes. Int. J. Mol. Sci. 2021, 22, 3656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073656
Asano-Matsuda K, Ibrahim S, Takano T, Matsuda J. Role of Rho GTPase Interacting Proteins in Subcellular Compartments of Podocytes. International Journal of Molecular Sciences. 2021; 22(7):3656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073656
Chicago/Turabian StyleAsano-Matsuda, Kana, Sajida Ibrahim, Tomoko Takano, and Jun Matsuda. 2021. "Role of Rho GTPase Interacting Proteins in Subcellular Compartments of Podocytes" International Journal of Molecular Sciences 22, no. 7: 3656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073656