
Analysis of Missense Variants in the Human Histamine Receptor Family Reveals Increased Constitutive Activity of E4106.30×30K Variant in the Histamine H1 Receptor


Abstract
:1. Introduction
2. Results
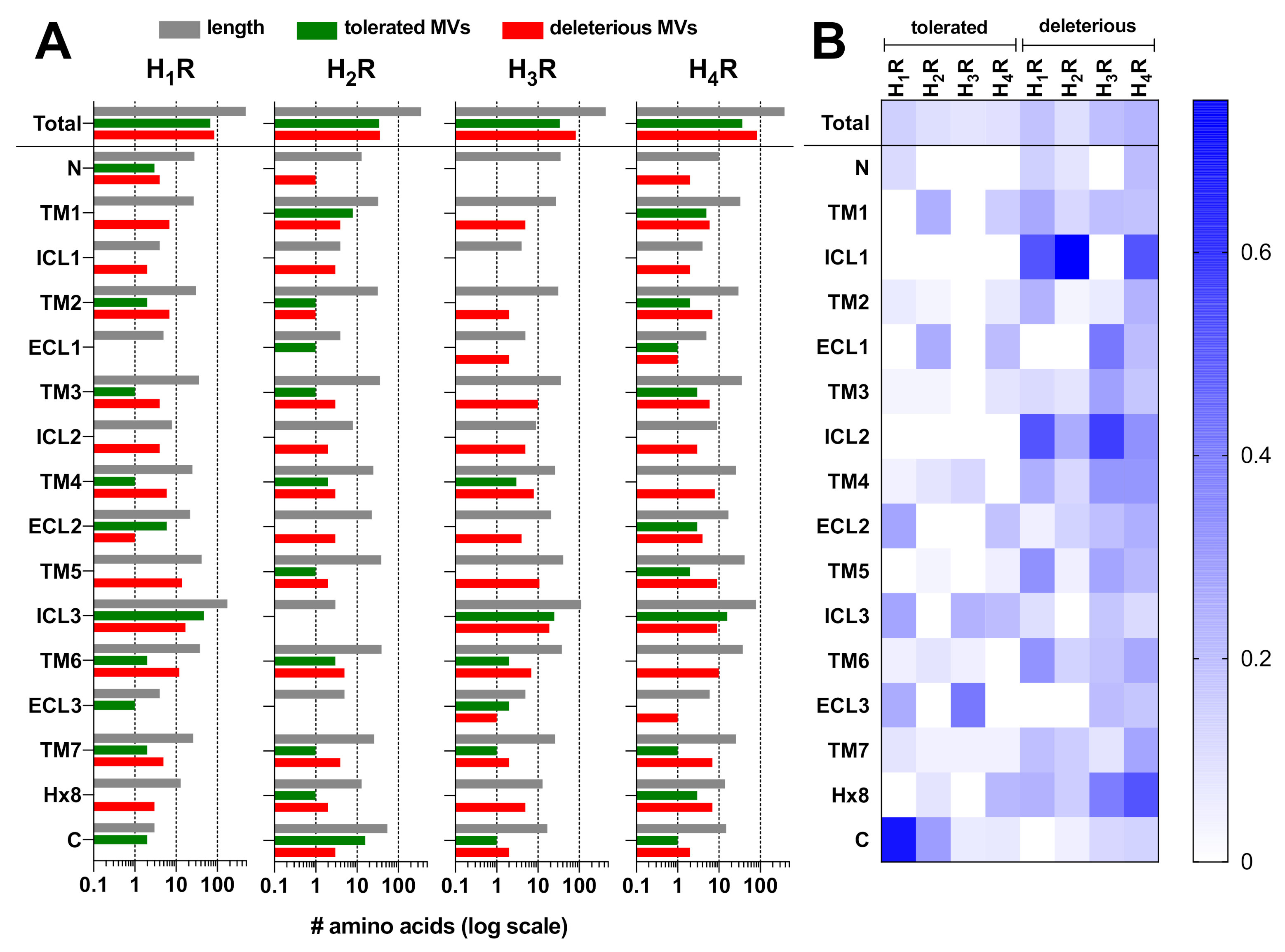
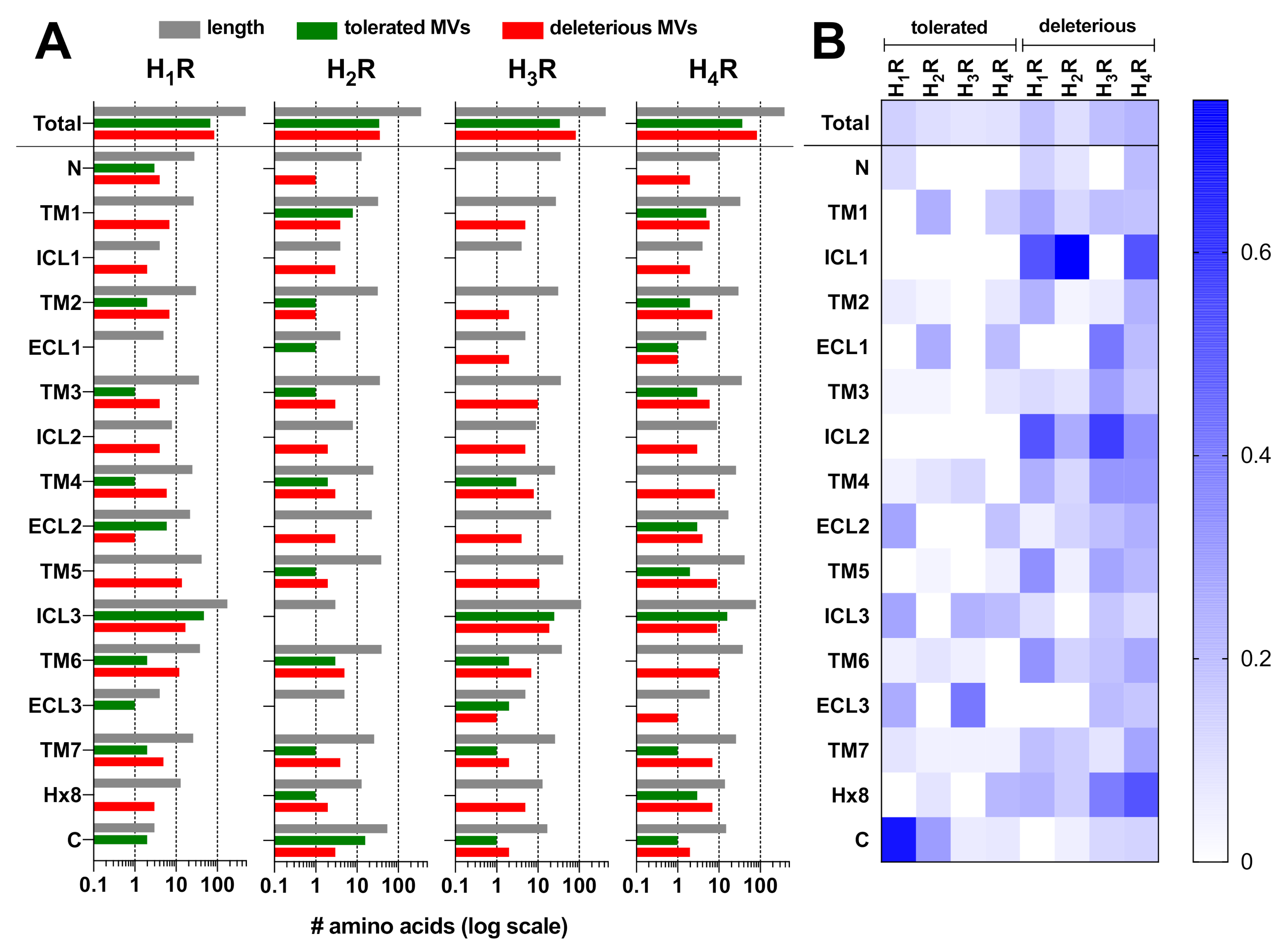
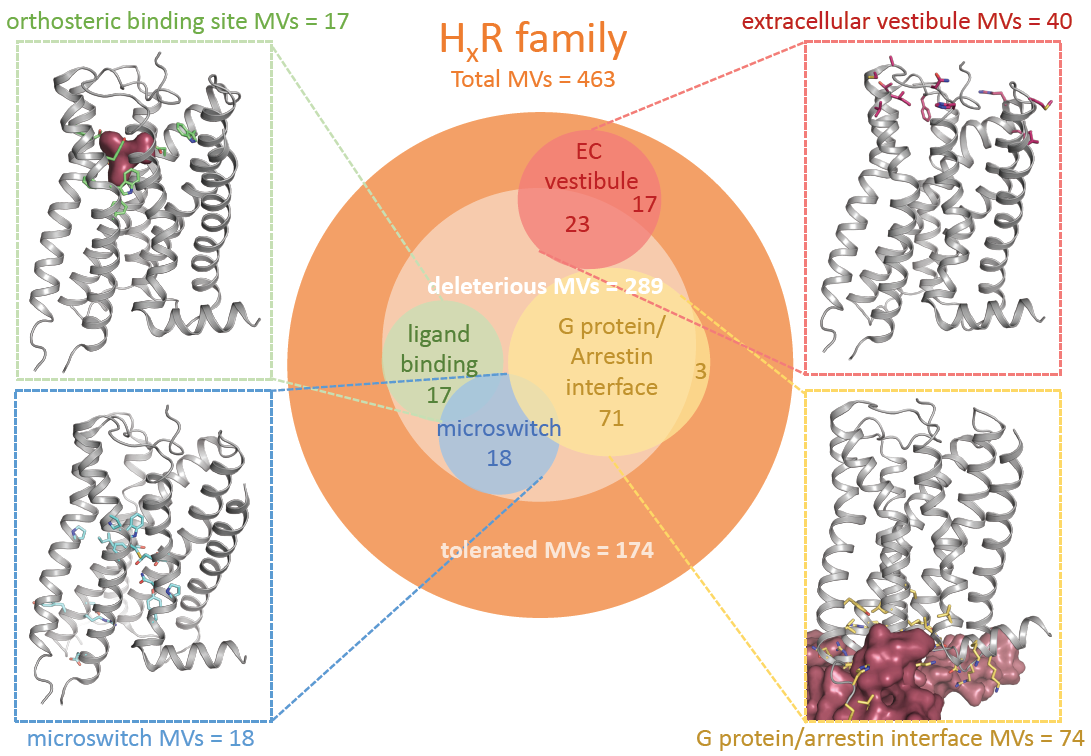
2.1. Genetic Missense Variations in the Histamine Receptor Family
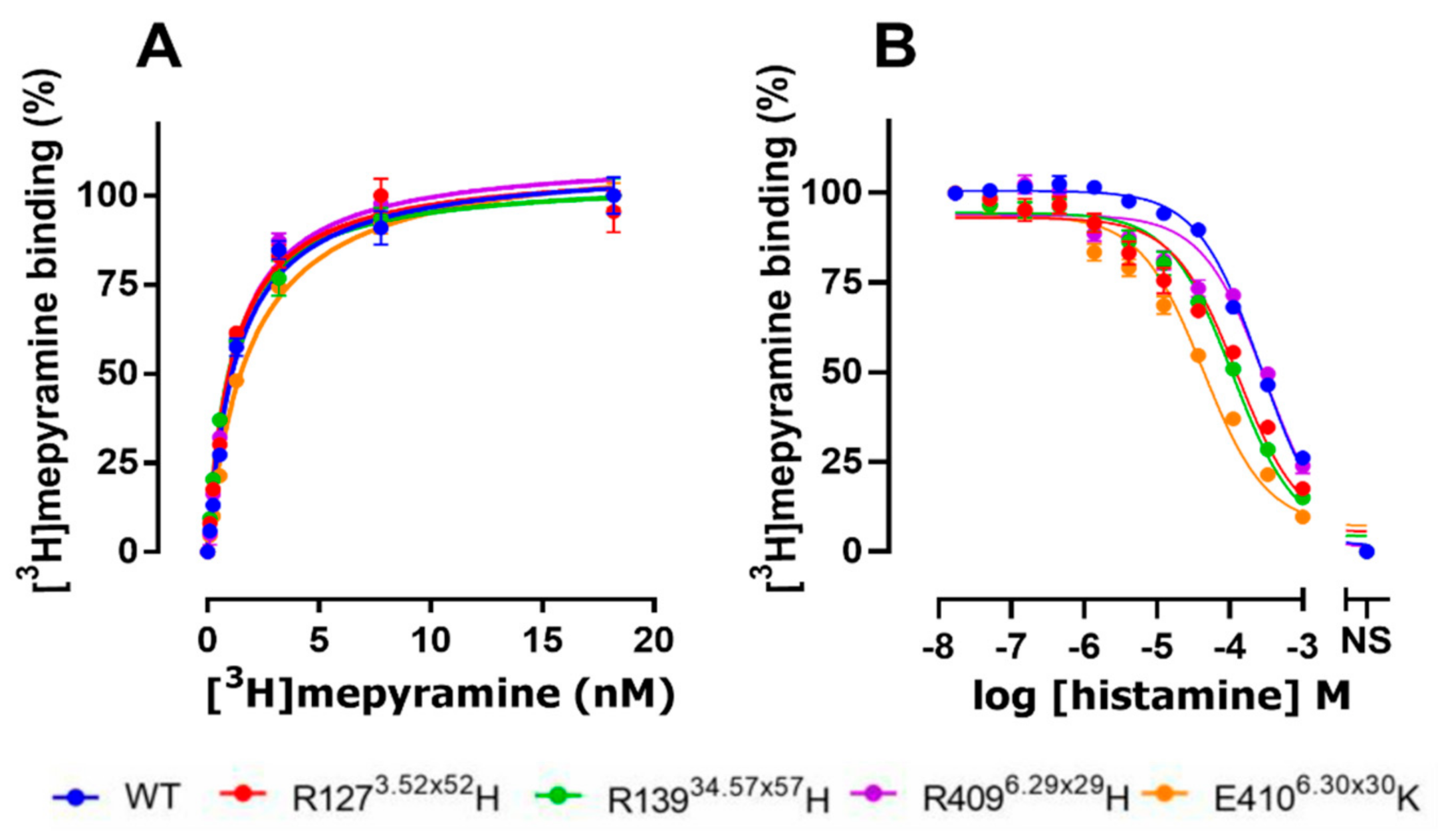
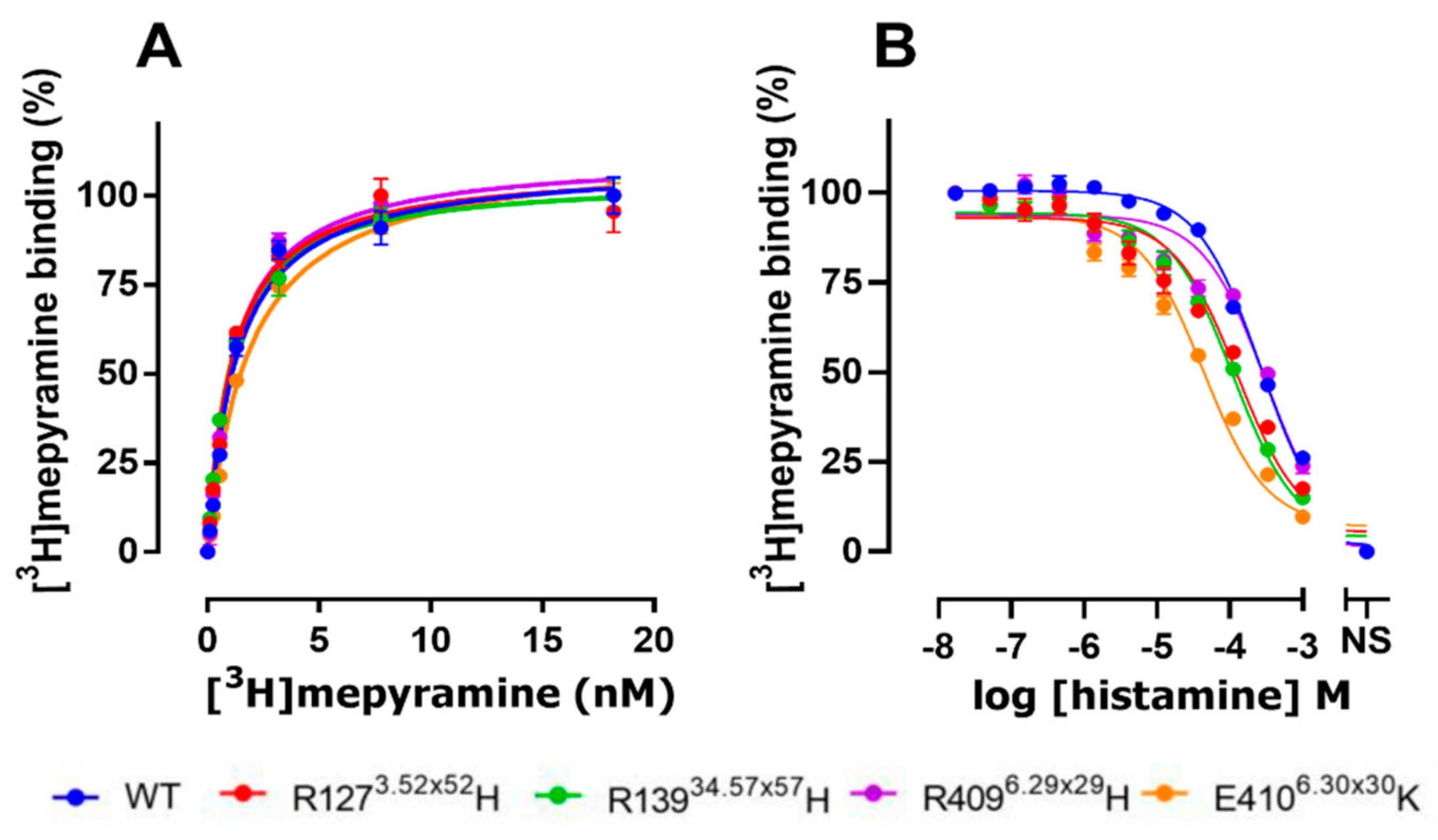
2.2. The E4106.30×30K Natural Variant Displays Increased Histamine Affinity
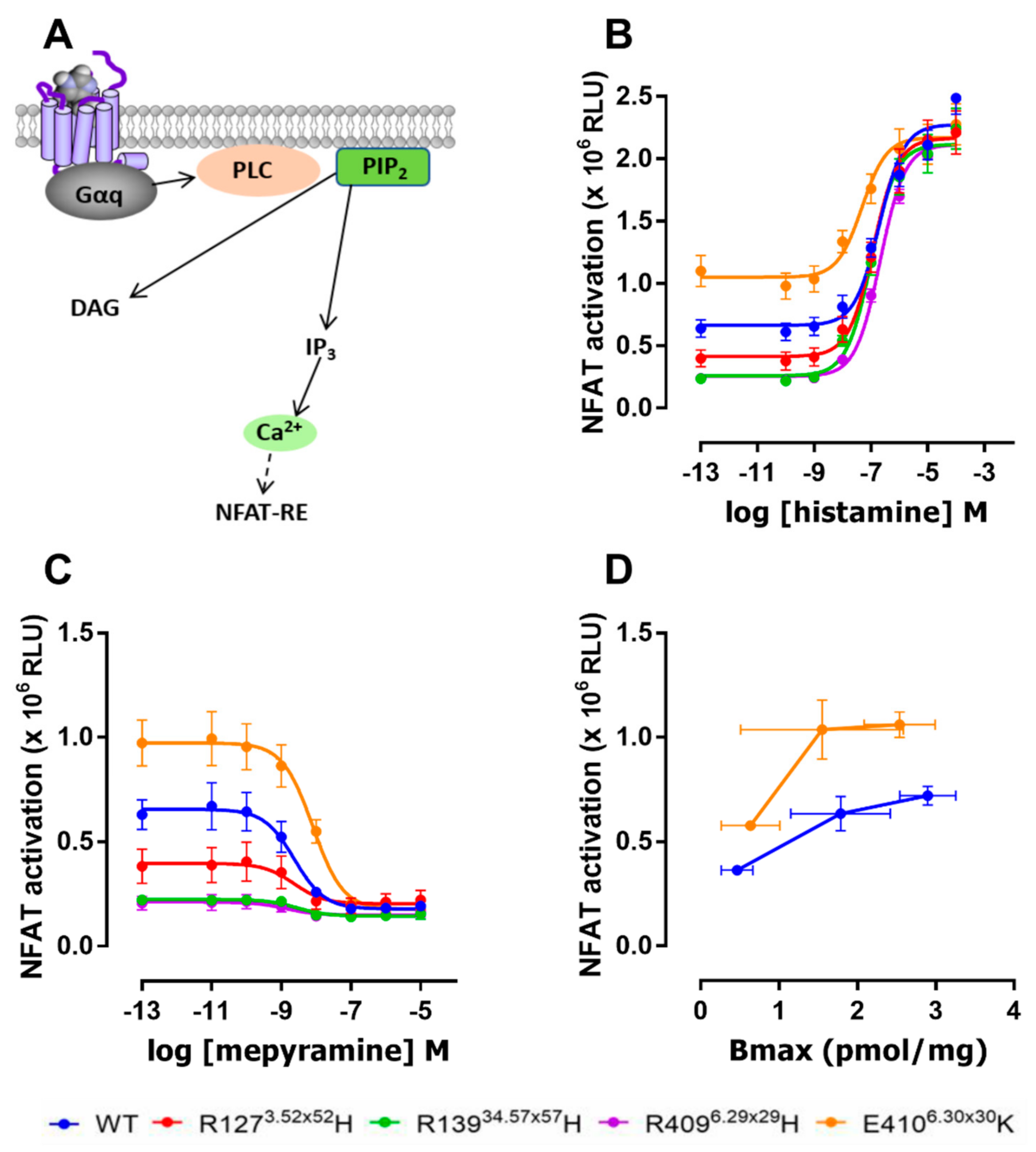
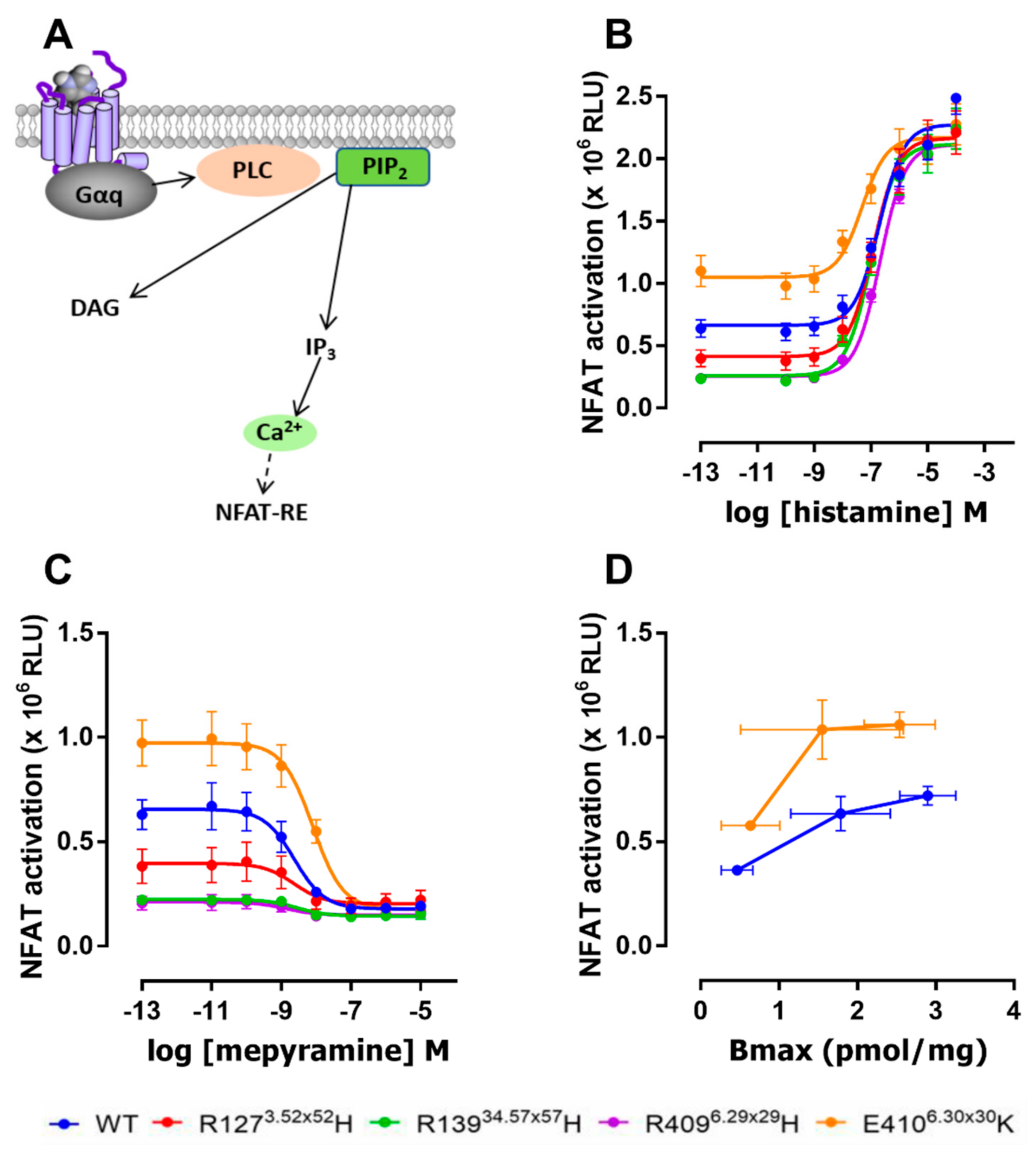
2.3. The E4106.30×30K Variant Displays Increased Constitutive Activity in G Protein Signaling
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Residue Numbering
4.3. Genetic Variation Dataset
4.4. Generation of hH1R Variants
4.5. Cell Culture and Transfection
4.6. Radioligand Binding Experiments
4.7. Nuclear Factor Activated T-Cells (NFAT)-Driven Reporter Gene Assay
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Almén, M.S.; Nordström, K.J.V.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19. [Google Scholar] [CrossRef] [Green Version]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Tiligada, E.; Ennis, M. Histamine pharmacology: From Sir Henry Dale to the 21st century. Br. J. Pharmacol. 2020, 177, 469–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Łażewska, D.; Kieć-Kononowicz, K. Progress in the development of histamine H3 receptor antagonists/inverse agonists: A patent review (2013–2017). Expert Opin. Ther. Pat. 2018, 28, 175–196. [Google Scholar] [CrossRef]
- Syed, Y.Y. Pitolisant: First Global Approval. Drugs 2016, 76, 1313–1318. [Google Scholar] [CrossRef]
- Urquhart, L. FDA new drug approvals in Q3 2019. Nat. Rev. Drug Discov. 2019, 18, 816. [Google Scholar] [CrossRef]
- Thurmond, R.L.; Venable, J.; Savall, B.; La, D.; Snook, S.; Dunford, P.J.; Edwards, J.P. Clinical Development of Histamine H4 Receptor Antagonists. Handb. Exp. Pharmacol. 2017, 241, 301–320. [Google Scholar] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.F.; Devree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β(2) adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Altenbach, C.; Thurmond, R.L.; Khorana, H.G.; Hubbell, W.L. Structure and function in rhodopsin: Rhodopsin mutants with a neutral amino acid at E134 have a partially activated conformation in the dark state. Proc. Natl. Acad. Sci. USA 1997, 94, 14273–14278. [Google Scholar] [CrossRef] [Green Version]
- Ramon, E.; Cordomi, A.; Bosch, L.; Zernii, E.Y.; Senin, I.I.; Manyosa, J.; Philippov, P.P.; Pérez, J.J.; Garriga, P. Critical role of electrostatic interactions of amino acids at the cytoplasmic region of helices 3 and 6 in rhodopsin conformational properties and activation. J. Biol. Chem. 2007, 282, 14272–14282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Jensen, A.D.; Liapakis, G.; Rasmussen, S.G.; Shi, L.; Gether, U.; Javitch, J.A. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [Google Scholar] [CrossRef] [Green Version]
- Valentin-Hansen, L.; Groenen, M.; Nygaard, R.; Frimurer, T.M.; Holliday, N.D.; Schwartz, T.W. The arginine of the DRY motif in transmembrane segment III functions as a balancing micro-switch in the activation of the β2-adrenergic receptor. J. Biol. Chem. 2012, 287, 31973–31982. [Google Scholar] [CrossRef] [Green Version]
- Alewijnse, A.E.; Timmerman, H.; Jacobs, E.H.; Smit, M.J.; Roovers, E.; Cotecchia, S.; Leurs, R. The effect of mutations in the DRY motif on the constitutive activity and structural instability of the histamine H(2) receptor. Mol. Pharmacol. 2000, 57, 890–898. [Google Scholar]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Bakker, R.A.; Wieland, K.; Timmerman, H.; Leurs, R. Constitutive activity of the histamine H(1) receptor reveals inverse agonism of histamine H(1) receptor antagonists. Eur. J. Pharmacol. 2000, 387, R5–R7. [Google Scholar] [CrossRef]
- Bakker, R.A.; Schoonus, S.B.; Smit, M.J.; Timmerman, H.; Leurs, R. Histamine H(1)-receptor activation of nuclear factor-kappa B: Roles for G beta gamma- and G alpha(q/11)-subunits in constitutive and agonist-mediated signaling. Mol. Pharmacol. 2001, 60, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Atienza, J.M.; Bernard, J.; Blanc, S.; Zhu, J.; Wang, X.; Xu, X.; Abassi, Y.A. Real-time monitoring of morphological changes in living cells by electronic cell sensor arrays: An approach to study G protein-coupled receptors. Anal. Chem. 2006, 78, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Jongejan, A.; Bruysters, M.; Ballesteros, J.A.; Haaksma, E.; Bakker, R.A.; Pardo, L.; Leurs, R. Linking agonist binding to histamine H1 receptor activation. Nat. Chem. Biol. 2005, 1, 98–103. [Google Scholar] [CrossRef]
- Bakker, R.A.; Jongejan, A.; Sansuk, K.; Hacksell, U.; Timmerman, H.; Brann, M.R.; Weiner, D.M.; Pardo, L.; Leurs, R. Constitutively active mutants of the histamine H1 receptor suggest a conserved hydrophobic asparagine-cage that constrains the activation of class A G protein-coupled receptors. Mol. Pharmacol. 2008, 73, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansuk, K.; Deupi, X.; Torrecillas, I.R.; Jongejan, A.; Nijmeijer, S.; Bakker, R.A.; Pardo, L.; Leurs, R. A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol. Pharmacol. 2011, 79, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Bosma, R.; Moritani, R.; Leurs, R.; Vischer, H.F. BRET-based β-arrestin2 recruitment to the histamine H1 receptor for investigating antihistamine binding kinetics. Pharmacol. Res. 2016, 111, 679–687. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Högger, P.; Shockley, M.S.; Lameh, J.; Sadée, W. Activating and inactivating mutations in N- and C-terminal i3 loop junctions of muscarinic acetylcholine Hm1 receptors. J. Biol. Chem. 1995, 270, 7405–7410. [Google Scholar] [CrossRef] [Green Version]
- Leppik, R.A.; Miller, R.C.; Eck, M.; Paquet, J.L. Role of acidic amino acids in the allosteric modulation by gallamine of antagonist binding at the m2 muscarinic acetylcholine receptor. Mol. Pharmacol. 1994, 45, 983–990. [Google Scholar] [PubMed]
- Ghanouni, P.; Schambye, H.; Seifert, R.; Lee, T.W.; Rasmussen, S.G.; Gether, U.; Kobilka, B.K. The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J. Biol. Chem. 2000, 275, 3121–3127. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, D.A.; Kristiansen, K.; Weiner, D.M.; Kroeze, W.K.; Roth, B.L. Evidence for a model of agonist-induced activation of 5-hydroxytryptamine 2A serotonin receptors that involves the disruption of a strong ionic interaction between helices 3 and 6. J. Biol. Chem. 2002, 277, 11441–11449. [Google Scholar] [CrossRef] [Green Version]
- Greasley, P.J.; Fanelli, F.; Rossier, O.; Abuin, L.; Cotecchia, S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol. Pharmacol. 2002, 61, 1025–1032. [Google Scholar] [CrossRef]
- Montanelli, L.; Delbaere, A.; Di Carlo, C.; Nappi, C.; Smits, G.; Vassart, G.; Costagliola, S. A mutation in the follicle-stimulating hormone receptor as a cause of familial spontaneous ovarian hyperstimulation syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 1255–1258. [Google Scholar] [CrossRef] [Green Version]
- Laue, L.; Chan, W.Y.; Hsueh, A.J.; Kudo, M.; Hsu, S.Y.; Wu, S.M.; Blomberg, L.; Cutler, G.B. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc. Natl. Acad. Sci. USA 1995, 92, 1906–1910. [Google Scholar] [CrossRef] [Green Version]
- Parma, J.; Duprez, L.; Van Sande, J.; Cochaux, P.; Gervy, C.; Mockel, J.; Dumont, J.; Vassart, G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993, 365, 649–651. [Google Scholar] [CrossRef]
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.J.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L.; et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 2015, 347, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Micallef, S.; Sasse, A. Genetic Polymorphisms in the Histamine Receptor Family. In Histamine Receptors; Blandina, P., Passani, M.B., Eds.; Preclinical and Clinical Aspects; Humana: Cham, Switzerland, 2016; Volume 28, pp. 51–74. [Google Scholar]
- García-Martín, E.; Ayuso, P.; Martínez, C.; Blanca, M.; Agúndez, J.A.G. Histamine pharmacogenomics. Pharmacogenomics 2009, 10, 867–883. [Google Scholar] [CrossRef]
- Godlewska, B.R.; Olajossy-Hilkesberger, L.; Olajossy, M.; Limon, J.; Landowski, J. Polymorphisms of the histamine receptor (H1HR) gene are not associated with olanzapine-induced weight gain. J. Clin. Psychopharmacol. 2013, 33, 436–437. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-J.; Lin, C.-H.; Yu, Y.W.-Y.; Chang, S.-C.; Wang, S.-Y.; Tsai, S.-J. Genetic variant of the histamine-1 receptor (glu349asp) and body weight change during clozapine treatment. Psychiatr. Genet. 2002, 12, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Ujike, H.; Nomura, A.; Morita, Y.; Morio, A.; Okahisa, Y.; Kotaka, T.; Kodama, M.; Ishihara, T.; Kuroda, S. Multiple genetic factors in olanzapine-induced weight gain in schizophrenia patients: A cohort study. J. Clin. Psychiatry 2008, 69, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- García-Martín, E.; Ayuso, P.; Luengo, A.; Martínez, C.; Agúndez, J.A. Genetic variability of histamine receptors in patients with Parkinson’s disease. BMC Med. Genet. 2008, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wei, X.; Shi, L.; Chen, B.; Zhao, G.; Yang, H. Integrative genomic analyses of the histamine H1 receptor and its role in cancer prediction. Int. J. Mol. Med. 2014, 33, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Millán-Guerrero, R.O.; Baltazar-Rodríguez, L.M.; Cárdenas-Rojas, M.I.; Ramírez-Flores, M.; Isais-Millán, S.; Delgado-Enciso, I.; Caballero-Hoyos, R.; Trujillo-Hernández, B. A280V polymorphism in the histamine H3 receptor as a risk factor for migraine. Arch. Med. Res. 2011, 42, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, P.; Bönisch, H.; Oerters, F.; Brüss, M. Structure of the human histamine H3 receptor gene (HRH3) and identification of naturally occurring variations. J. Neural Transm. 2002, 109, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Flores-Clemente, C.; Osorio-Espinoza, A.; Escamilla-Sánchez, J.; Leurs, R.; Arias, J.-M.; Arias-Montaño, J.-A. A single-point mutation (Ala280Val) in the third intracellular loop alters the signalling properties of the human histamine H3 receptor stably expressed in CHO-K1 cells. Br. J. Pharmacol. 2013, 170, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Schöneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef]
- Smit, M.J.; Vischer, H.F.; Bakker, R.A.; Jongejan, A.; Timmerman, H.; Pardo, L.; Leurs, R. Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 53–87. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, L.A.; Vernall, A.J.; Bouzo-Lorenzo, M.; Bosma, R.; Kooistra, A.J.; de Graaf, C.; Vischer, H.F.; Leurs, R.; Briddon, S.J.; Kellam, B.; et al. Development of novel fluorescent histamine H1-receptor antagonists to study ligand-binding kinetics in living cells. Sci. Rep. 2018, 8, 1572. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, C.; Foata, N.; Engkvist, O.; Rognan, D. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins 2008, 71, 599–620. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs—Theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural features for functional selectivity at serotonin receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Heydenreich, F.M.; Flock, T.; Miljus, T.; Balaji, S.; Bouvier, M.; Veprintsev, D.B.; Tate, C.G.; et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 2016, 536, 484–487. [Google Scholar] [CrossRef]
- Zarzycka, B.; Zaidi, S.A.; Roth, B.L.; Katritch, V. Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev. 2019, 71, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020, 159, 1022–1025. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.-L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; et al. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 2019, 29, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Blomenröhr, M.; Vischer, H.F.; Bogerd, J. Receptor mutagenesis strategies for examination of structure-function relationships. Methods Mol. Biol. 2004, 259, 307–322. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Missense Variations | Average Density 1 | Total Rare MVs 2 | Total Common MVs 3 | Predicted Deleterious Mutations 4 | Total Loss-of-Function Variants 5 | |
|---|---|---|---|---|---|---|
| All GPCRs 6 | 65,539 | 0.27 | 155 7 | 4.8 7 | 4066 | |
| Class A GPCRs 8 | 36,340 | 0.27 | 35,323 | 1017 | 24,207 | 2305 |
| Histamine receptors | 463 | 0.24 | 453 | 10 | 289 | 30 |
| H1R | 154 | 0.32 | 149 | 5 | 86 | 6 |
| H2R | 71 | 0.20 | 70 | 1 | 36 | 1 |
| H3R | 117 | 0.26 | 117 | 0 | 83 | 2 |
| H4R | 121 | 0.31 | 117 | 4 | 84 | 21 |
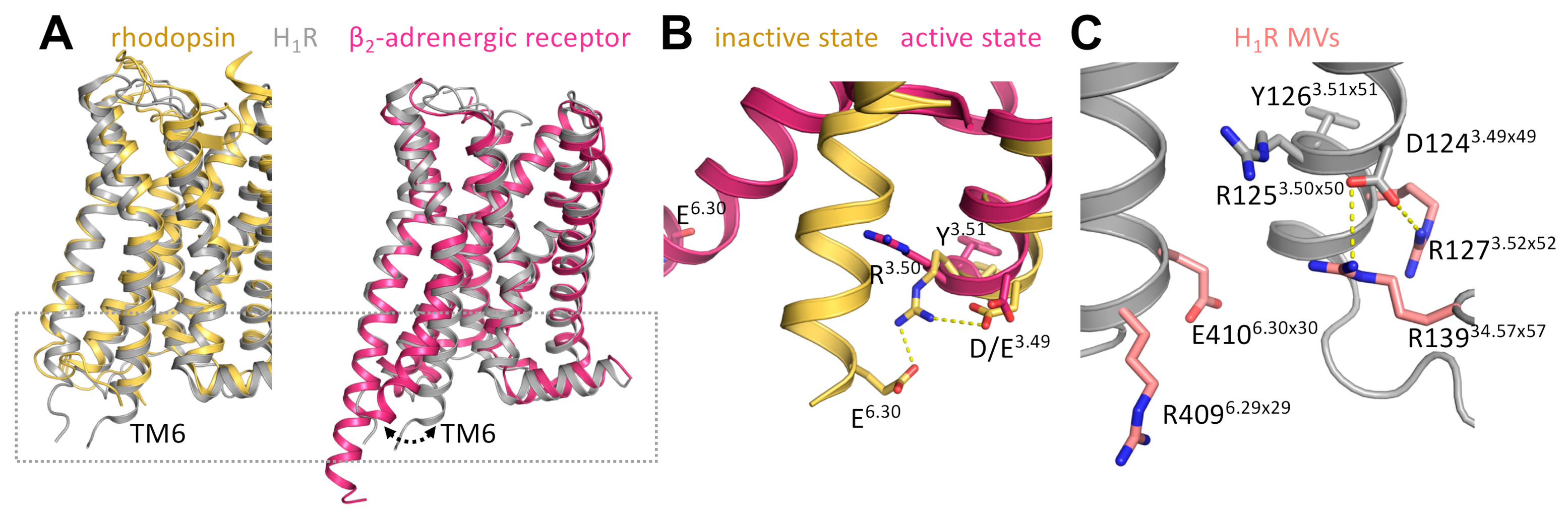
| Position | GPCRdb | Segment | Variant | Allele Count 2 | Allele Number 3 | Allele Frequency 4 | Homozygotes 5 | SIFT 6 | PolyPhen 7 |
|---|---|---|---|---|---|---|---|---|---|
| 127 | 3.52 × 52 | TM3 | R => H | 3 | 121266 | 2.47 × 10−5 | 0 | 0.02 | 0.909 |
| 139 | 34.57 × 57 | ICL2 | R => H | 7 | 121276 | 5.77 × 10−5 | 1 | 0 | 1 |
| 409 | 6.29 × 29 | TM6 | R => H | 2 | 121228 | 1.65 × 10−5 | 0 | 0 | 0.996 |
| 410 | 6.30 × 30 | TM6 | E => K | 4 | 121242 | 3.30 × 10−5 | 0 | 0 | 0.996 |
| hH1R Variants | pKd | Bmax (pmol/mg) |
|---|---|---|
| WT | 8.9 ± 0.10 | 42.5 ± 12.07 |
| R1273.52×52H | 8.9 ± 0.13 | 35.1 ± 12.01 |
| R13934.57×57H | 9.0 ± 0.06 | 38.4 ± 8.42 |
| R4096.29×29H | 8.9 ± 0.03 | 48.4 ± 12.33 |
| E4106.30×30K | 8.7 ± 0.05 | 27.8 ± 7.39 |
| hH1R Variants | Histamine | Mepyramine | Levocetirizine | Doxepin |
|---|---|---|---|---|
| WT | 4.3 ± 0.05 | 8.8 ± 0.06 | 7.7 ± 0.17 | 9.7 ± 0.19 |
| R1273.52×52H | 4.7 ± 0.02 * | 8.9 ± 0.09 | 7.6 ± 0.08 | 9.8 ± 0.17 |
| R13934.57×57H | 4.8 ± 0.03 * | 8.8 ± 0.09 | 7.8 ± 0.09 | 9.6 ± 0.17 |
| R4096.29×29H | 4.3 ± 0.09 | 9.0 ± 0.33 | 7.8 ± 0.17 | 9.9 ± 0.29 |
| E4106.30×30K | 5.1 ± 0.10 * | 8.7 ± 0.16 | 7.8 ± 0.17 | 9.7 ± 0.06 |
| hH1R Variants | Histamine | Mepyramine |
|---|---|---|
| WT | 6.7 ± 0.05 | 8.7 ± 0.23 |
| R1273.52×52H | 6.9 ± 0.09 | 8.6 ± 0.20 |
| R13934.57×57H | 7.0 ± 0.06 | 8.6 ± 0.13 |
| R4096.29×29H | 6.7 ± 0.09 | 8.8 ± 0.27 |
| E4106.30×30K | 7.3 ± 0.23 * | 8.1 ± 0.22 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Segura, M.A.; Zarzycka, B.; Vischer, H.F.; Leurs, R. Analysis of Missense Variants in the Human Histamine Receptor Family Reveals Increased Constitutive Activity of E4106.30×30K Variant in the Histamine H1 Receptor. Int. J. Mol. Sci. 2021, 22, 3702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073702
Ma X, Segura MA, Zarzycka B, Vischer HF, Leurs R. Analysis of Missense Variants in the Human Histamine Receptor Family Reveals Increased Constitutive Activity of E4106.30×30K Variant in the Histamine H1 Receptor. International Journal of Molecular Sciences. 2021; 22(7):3702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073702
Chicago/Turabian StyleMa, Xiaoyuan, Marta Arimont Segura, Barbara Zarzycka, Henry F. Vischer, and Rob Leurs. 2021. "Analysis of Missense Variants in the Human Histamine Receptor Family Reveals Increased Constitutive Activity of E4106.30×30K Variant in the Histamine H1 Receptor" International Journal of Molecular Sciences 22, no. 7: 3702. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073702