
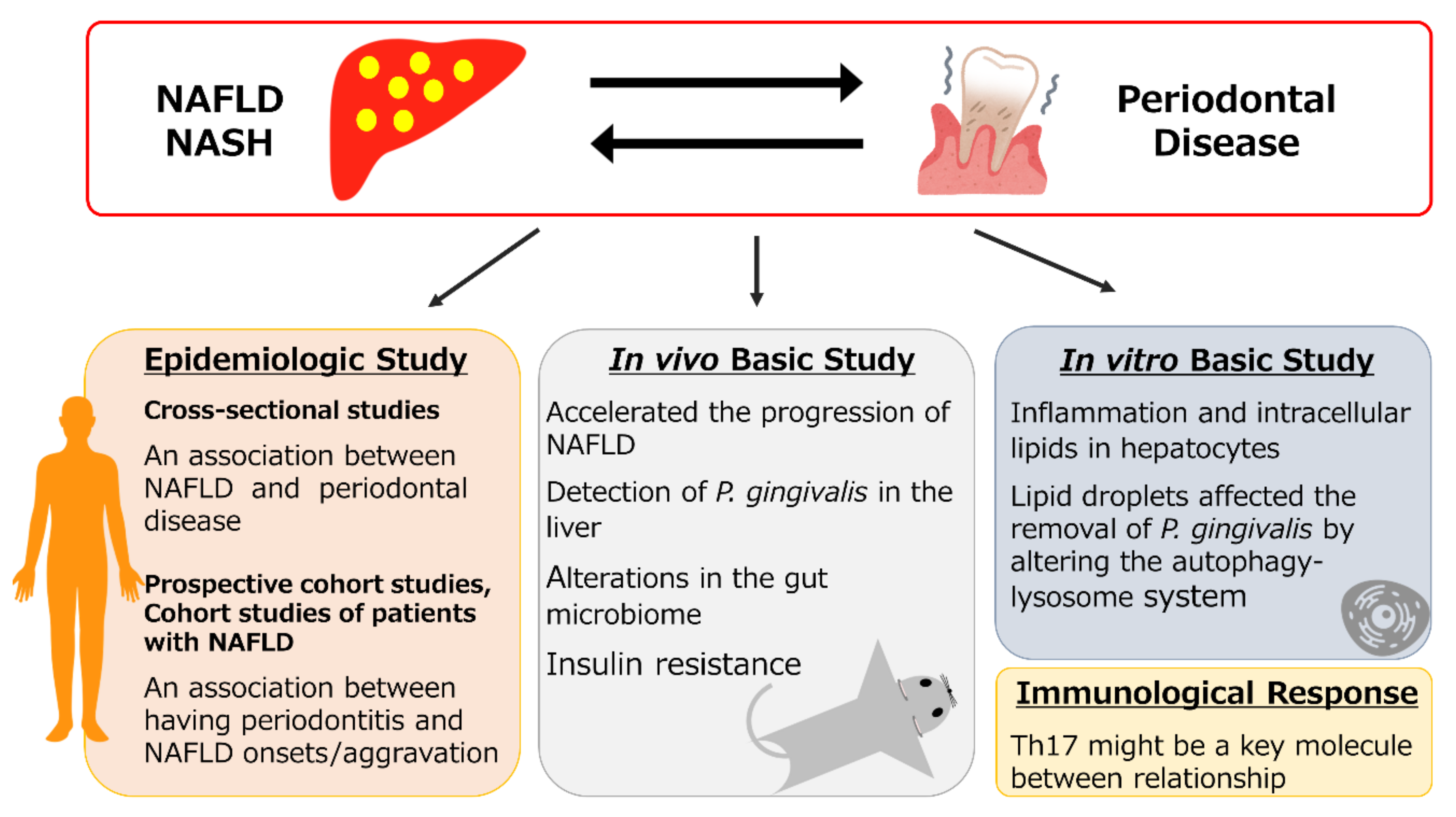
Relationship between NAFLD and Periodontal Disease from the View of Clinical and Basic Research, and Immunological Response

 , , and
, , and
Abstract
:1. Introduction
2. NAFLD
2.1. NAFLD and Nonalcoholic Steatohepatitis (NASH)
2.2. From NAFLD to Metabolic Associated Fatty Liver Disease (MAFLD)
3. Human Epidemiologic Studies
3.1. Cross-Sectional Studies Regarding the Relationship between NAFLD and Periodontal Disease
3.2. Prospective Cohort Studies Regarding the Relationship between NAFLD and Periodontal Disease
3.3. Meta-Analysis Regarding the Relationship between NAFLD and Periodontal Disease
3.4. Reports Regarding Periodontal Disease in Patients with NAFLD
4. In Vivo Basic Research Regarding the Relationship between NAFLD and Periodontal Disease
4.1. P. gingivalis Lipopolysaccharide (LPS) Injection in Gingiva Model
4.2. Pulp Chamber Model
4.3. Intravenous Injection of the P. gingivalis Model
4.4. Oral Administration Model
4.5. Ligature-Induced Periodontitis Model

{kind=link}
| Ref. No. | Study | Animals | High-Fat Diet | Model | Major Findings |
|---|---|---|---|---|---|
| [8] | Yoneda et al. | Mice | + | Intravenous injection of P. gingivalis | Increase in the body weight acceleration in the progression of NAFLD |
| [9] | Komazaki et al. | Mice | + | Oral administration of A. actinomycetemcomitans | Increased liver steatosis the enriched glucagon-signaling pathway, adipocytokine signaling pathway, insulin resistance in the liver decrease in the genus Turicibacter in the gut. |
| [54] | Nakahara et al. | Mice | + | Pulp chamber model | Fatty acid metabolism was disrupted, and expression levels of SCD1 and ELOVL6 were reduced. |
| [55] | Furusho et al. | Mice | + | Pulp chamber model | P. gingivalis was detected in Kupffer cells and hepatocytes |
| [56] | Fujita et al. | Rats | + | P. gingivalis LPS injection in gingiva | Large fat droplets Ballooning degeneration Infiltration of inflammatory cells |
| [57] | Varela-López et al. | Rabbits | + | P. gingivalis LPS injection in gingiva | High score of acinar inflammation Increase in the blood triglyceride and phospholipid levels |
| [58] | Nagasaki et al. | Mice | + | Pulp chamber model | Ipregulation of the immunoexpression of phosphorylated Smad2 and Galectin-3 |
| [60] | Sasaki et al. | Mice | + | Intravenous injection of P. gingivalis | Impaired glucose tolerance, insulin resistance, and liver steatosis Alteration of the gut microbiome |
| [61] | Arimatsu et al. | Mice | - | Oral administration of P. gingivalis | Increase in insulin resistance and systemic inflammation Increase in the order Bacteroidales in the gut |
| [68] | Vasconcelos et al. | Rats | - | Ligature-induced periodontitis model | Decrease of GSH and increase of MDA concentrations |
| [69] | Kuraji et al. | Rats | + | Ligature-induced periodontitis model | Perivenular lipid deposition, including large fatty drops, ballooning degeneration, and focal necrosis with inflammation. |
5. In Vitro Basic Research Regarding the Relationship between NAFLD and Periodontal Disease
6. Immunological Responses in Periodontitis and NAFLD
6.1. Role of T Cells in Periodontal Disease
6.2. Innate Immune Response in NAFLD
6.3. Role of T Cells in NAFLD
6.4. Interrelationship between NAFLD and Periodontal Disease
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Benakanakere, M.; Kinane, D.F. Innate cellular responses to the periodontal biofilm. Front. Oral Biol. 2012, 15, 41–55. [Google Scholar]
- Genco, R.J.; Borgnakke, W.S. Diabetes as a potential risk for periodontitis: Association studies. Periodontology 2000 2020, 83, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Polak, D.; Sanui, T.; Nishimura, F.; Shapira, L. Diabetes as a risk factor for periodontal disease-plausible mechanisms. Periodontology 2000 2020, 83, 46–58. [Google Scholar] [CrossRef]
- Orlandi, M.; Graziani, F.; D’Aiuto, F. Periodontal therapy and cardiovascular risk. Periodontology 2000 2020, 83, 107–124. [Google Scholar] [CrossRef]
- Schenkein, H.A.; Papapanou, P.N.; Genco, R.; Sanz, M. Mechanisms underlying the association between periodontitis and atherosclerotic disease. Periodontology 2000 2020, 83, 90–106. [Google Scholar] [CrossRef]
- Figuero, E.; Han, Y.W.; Furuichi, Y. Periodontal diseases and adverse pregnancy outcomes: Mechanisms. Periodontology 2000 2020, 83, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Naka, S.; Nakano, K.; Wada, K.; Endo, H.; Mawatari, H.; Imajo, K.; Nomura, R.; Hokamura, K.; Ono, M.; et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Komazaki, R.; Katagiri, S.; Takahashi, H.; Maekawa, S.; Shiba, T.; Takeuchi, Y.; Kitajima, Y.; Ohtsu, A.; Udagawa, S.; Sasaki, N.; et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci. Rep. 2017, 7, 13950. [Google Scholar] [CrossRef]
- Kamata, Y.; Kessoku, T.; Shimizu, T.; Kobayashi, T.; Kurihashi, T.; Sato, S.; Kuraji, S.; Aoyama, N.; Iwasaki, T.; Takashiba, S.; et al. Efficacy and safety of PERIOdontal treatment versus usual care for Nonalcoholic liver disease: Protocol of the PERION multicenter, two-arm, open-label, randomized trial. Trials 2020, 21, 291. [Google Scholar] [CrossRef] [Green Version]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015, 86, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slade, G.D.; Offenbacher, S.; Beck, J.D.; Heiss, G.; Pankow, J.S. Acute-phase inflammatory response to periodontal disease in the US population. J. Dent. Res. 2000, 79, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Trevisan, M.; Genco, R.J.; Falkner, K.L.; Dorn, J.P.; Sempos, C.T. Examination of the relation between periodontal health status and cardiovascular risk factors: Serum total and high density lipoprotein cholesterol, C-reactive protein, and plasma fibrinogen. Am. J. Epidemiol. 2000, 151, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, S.; Nitta, H.; Nagasawa, T.; Uchimura, I.; Izumiyama, H.; Inagaki, K.; Kikuchi, T.; Noguchi, T.; Kanazawa, M.; Matsuo, A.; et al. Multi-center intervention study on glycohemoglobin (HbA1c) and serum, high-sensitivity CRP (hs-CRP) after local anti-infectious periodontal treatment in type 2 diabetic patients with periodontal disease. Diabetes Res. Clin. Pract. 2009, 83, 308–315. [Google Scholar] [CrossRef]
- Page, R.C. The pathobiology of periodontal diseases may affect systemic diseases: Inversion of a paradigm. Ann. Periodontol. 1998, 3, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Von Troil-Linden, B.; Torkko, H.; Alaluusua, S.; Jousimies-Somer, H.; Asikainen, S. Salivary levels of suspected periodontal pathogens in relation to periodontal status and treatment. J. Dent. Res. 1995, 74, 1789–1795. [Google Scholar] [CrossRef]
- Katagiri, S.; Shiba, T.; Tohara, H.; Yamaguchi, K.; Hara, K.; Nakagawa, K.; Komatsu, K.; Watanabe, K.; Ohsugi, Y.; Maekawa, S.; et al. Re-initiation of Oral Food Intake Following Enteral Nutrition Alters Oral and Gut Microbiota Communities. Front. Cell Infect. Microbiol. 2019, 9, 434. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [Green Version]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Drew, L. Drug development: Sprint finish. Nature 2017, 551, S86–S89. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Schaffner, F.; Thaler, H. Nonalcoholic fatty liver disease. Prog. Liver Dis. 1986, 8, 283–298. [Google Scholar]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Nibali, L.; Tatarakis, N.; Needleman, I.; Tu, Y.K.; D’Aiuto, F.; Rizzo, M.; Donos, N. Clinical review: Association between metabolic syndrome and periodontitis: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2013, 98, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Nagao, Y.; Sata, M. Dental problems delaying the initiation of interferon therapy for HCV-infected patients. Virol. J. 2010, 7, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef]
- Furuta, M.; Ekuni, D.; Yamamoto, T.; Irie, K.; Koyama, R.; Sanbe, T.; Yamanaka, R.; Morita, M.; Kuroki, K.; Tobe, K. Relationship between periodontitis and hepatic abnormalities in young adults. Acta Odontol. Scand. 2010, 68, 27–33. [Google Scholar] [CrossRef]
- Ahmad, A.; Furuta, M.; Shinagawa, T.; Takeuchi, K.; Takeshita, T.; Shimazaki, Y.; Yamashita, Y. Association of periodontal status with liver abnormalities and metabolic syndrome. J. Oral Sci. 2015, 57, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, T.; Yamazaki, Y.; Fujiharu, C.; Ishii, T.; Seto, M.; Nishinoue, N.; Sasaki, Y.; Kawato, T.; Motohashi, M.; Maeno, M. Serum γ-glutamyltransferase level is associated with periodontal disease independent of drinking habits in Japanese adults. Med. Sci. Monit. 2014, 20, 2109–2116. [Google Scholar]
- Iwasaki, T.; Hirose, A.; Azuma, T.; Ohashi, T.; Watanabe, K.; Obora, A.; Deguchi, F.; Kojima, T.; Isozaki, A.; Tomofuji, T. Correlation between ultrasound-diagnosed non-alcoholic fatty liver and periodontal condition in a cross-sectional study in Japan. Sci. Rep. 2018, 8, 7496. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, G.N.; Song, H.C.; Park, Y.M.; Ahn, Y.B.; Han, K.; Ko, S.H. Association between Fatty Liver Index and Periodontitis: The Korea National Health and Nutrition Examination Survey. Sci. Rep. 2020, 10, 3805. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.S. Association between periodontal status and non-alcoholic fatty liver disease in a Korean adult population: A nationwide cross-sectional study. J. Periodontol. 2020, 91, 524–532. [Google Scholar] [CrossRef]
- Qiao, F.; Fu, K.; Zhang, Q.; Liu, L.; Meng, G.; Wu, H.; Xia, Y.; Bao, X.; Gu, Y.; Shi, H.; et al. The association between missing teeth and non-alcoholic fatty liver disease in adults. J. Clin. Periodontol. 2018, 45, 941–951. [Google Scholar] [CrossRef]
- Weintraub, J.A.; Lopez Mitnik, G.; Dye, B.A. Oral Diseases Associated with Nonalcoholic Fatty Liver Disease in the United States. J. Dent. Res. 2019, 98, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Alazawi, W.; Bernabe, E.; Tai, D.; Janicki, T.; Kemos, P.; Samsuddin, S.; Syn, W.K.; Gillam, D.; Turner, W. Periodontitis is associated with significant hepatic fibrosis in patients with non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0185902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinkugbe, A.A.; Barritt, A.S.; Cai, J.; Offenbacher, S.; Thyagarajan, B.; Khambaty, T.; Singer, R.; Kallwitz, E.; Heiss, G.; Slade, G.D. Periodontitis and prevalence of elevated aminotransferases in the Hispanic Community Health Study/Study of Latinos. J. Periodontol. 2018, 89, 949–958. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Peloso, G.M.; Pencina, M.J.; Hoffmann, U.; Fox, C.S.; Cupples, L.A.; Levy, D.; D’Agostino, R.B.; Hwang, S.J.; O’Donnell, C.J. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: The Framingham Heart Study. Circ. Cardiovasc. Genet. 2012, 5, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Liu, Z.J.; Tao, S.; Sun, Y.M.; Jiang, D.; Li, H.L.; Chen, H.; Liu, X.; Lapin, B.; Wang, C.H.; et al. Risk prediction for sporadic Alzheimer’s disease using genetic risk score in the Han Chinese population. Oncotarget 2015, 6, 36955–36964. [Google Scholar] [CrossRef] [Green Version]
- Akinkugbe, A.A.; Avery, C.L.; Barritt, A.S.; Cole, S.R.; Lerch, M.; Mayerle, J.; Offenbacher, S.; Petersmann, A.; Nauck, M.; Völzke, H.; et al. Do Genetic Markers of Inflammation Modify the Relationship between Periodontitis and Nonalcoholic Fatty Liver Disease? Findings from the SHIP Study. J. Dent. Res. 2017, 96, 1392–1399. [Google Scholar] [CrossRef]
- Kuroe, K.; Furuta, M.; Takeuchi, K.; Takeshita, T.; Suma, S.; Shinagawa, T.; Shimazaki, Y.; Yamashita, Y. Association between periodontitis and fibrotic progression of non-alcoholic fatty liver among Japanese adults. J. Clin. Periodontol. 2021, 48, 368–377. [Google Scholar] [CrossRef]
- Akinkugbe, A.A.; Slade, G.D.; Barritt, A.S.; Cole, S.R.; Offenbacher, S.; Petersmann, A.; Kocher, T.; Lerch, M.M.; Mayerle, J.; Völzke, H.; et al. Periodontitis and Non-alcoholic Fatty Liver Disease, a population-based cohort investigation in the Study of Health in Pomerania. J. Clin. Periodontol. 2017, 44, 1077–1087. [Google Scholar] [CrossRef]
- Helenius-Hietala, J.; Suominen, A.L.; Ruokonen, H.; Knuuttila, M.; Puukka, P.; Jula, A.; Meurman, J.H.; Åberg, F. Periodontitis is associated with incident chronic liver disease-A population-based cohort study. Liver Int. 2019, 39, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, Y.C.; Zhu, B.L.; Wu, C.C.; Lin, R.F.; Zhang, X. Association between periodontal disease, tooth loss and liver diseases risk. J. Clin. Periodontol. 2020, 47, 1053–1063. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Panjawatanan, P.; Cheungpasitporn, W.; Lukens, F.J.; Harnois, D.M.; Pungpapong, S.; Ungprasert, P. The Association between Periodontitis and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. J. Gastrointest. Liver Dis. 2020, 29, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Amano, A.; Nakagawa, I.; Kataoka, K.; Morisaki, I.; Hamada, S. Distribution of Porphyromonas gingivalis strains with fimA genotypes in periodontitis patients. J. Clin. Microbiol. 1999, 37, 1426–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, I.; Amano, A.; Kuboniwa, M.; Nakamura, T.; Kawabata, S.; Hamada, S. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect. Immun. 2002, 70, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, T.; Hyogo, H.; Ono, A.; Nagaoki, Y.; Kawaoka, T.; Miki, D.; Tsuge, M.; Hiraga, N.; Hayes, C.N.; Hiramatsu, A.; et al. Involvement of Porphyromonas gingivalis in the progression of non-alcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Furusho, H.; Miyauchi, M.; Hyogo, H.; Inubushi, T.; Ao, M.; Ouhara, K.; Hisatune, J.; Kurihara, H.; Sugai, M.; Hayes, C.N.; et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J. Gastroenterol. 2013, 48, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kuraji, R.; Ito, H.; Hashimoto, S.; Toen, T.; Fukada, T.; Numabe, Y. Histological effects and pharmacokinetics of lipopolysaccharide derived from Porphyromonas gingivalis on rat maxilla and liver concerning with progression into non-alcoholic steatohepatitis. J. Periodontol. 2018, 89, 1101–1111. [Google Scholar] [CrossRef]
- Varela-López, A.; Bullón, P.; Ramírez-Tortosa, C.L.; Navarro-Hortal, M.D.; Robles-Almazán, M.; Bullón, B.; Cordero, M.D.; Battino, M.; Quiles, J.L. A Diet Rich in Saturated Fat and Cholesterol Aggravates the Effect of Bacterial Lipopolysaccharide on Alveolar Bone Loss in a Rabbit Model of Periodontal Disease. Nutrients 2020, 12, 1405. [Google Scholar] [CrossRef]
- Nagasaki, A.; Sakamoto, S.; Chea, C.; Ishida, E.; Furusho, H.; Fujii, M.; Takata, T.; Miyauchi, M. Odontogenic infection by Porphyromonas gingivalis exacerbates fibrosis in NASH via hepatic stellate cell activation. Sci. Rep. 2020, 10, 4134. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Zheng, R.D.; Sun, X.Q.; Ding, W.J.; Wang, X.Y.; Fan, J.G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Sasaki, N.; Katagiri, S.; Komazaki, R.; Watanabe, K.; Maekawa, S.; Shiba, T.; Udagawa, S.; Takeuchi, Y.; Ohtsu, A.; Kohda, T.; et al. Endotoxemia by Porphyromonas gingivalis Injection Aggravates Non-alcoholic Fatty Liver Disease, Disrupts Glucose/Lipid Metabolism, and Alters Gut Microbiota in Mice. Front. Microbiol. 2018, 9, 2470. [Google Scholar] [CrossRef] [Green Version]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Nyman, M.; Fåk, F. Modulation of gut microbiota in rats fed high-fat diets by processing whole-grain barley to barley malt. Mol. Nutr. Food Res. 2015, 59, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Hajishengallis, G. Optimization of the ligature-induced periodontitis model in mice. J. Immunol. Methods 2013, 394, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, S.; Katagiri, S.; Takeuchi, Y.; Komazaki, R.; Ohtsu, A.; Udagawa, S.; Izumi, Y. Bone metabolic microarray analysis of ligature-induced periodontitis in streptozotocin-induced diabetic mice. J. Periodontal. Res. 2017, 52, 233–245. [Google Scholar] [CrossRef]
- Maekawa, S.; Onizuka, S.; Katagiri, S.; Hatasa, M.; Ohsugi, Y.; Sasaki, N.; Watanabe, K.; Ohtsu, A.; Komazaki, R.; Ogura, K.; et al. RNA sequencing for ligature induced periodontitis in mice revealed important role of S100A8 and S100A9 for periodontal destruction. Sci. Rep. 2019, 9, 14663. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, D.F.; Pereira da Silva, F.R.; Pinto, M.E.; Santana, L.A.; Souza, I.G.; Miranda de Souza, L.K.; Oliveira, N.C.; Ventura, C.A.; Novaes, P.D.; Barbosa, A.L.; et al. Decrease of Pericytes is Associated With Liver Disease Caused by Ligature-Induced Periodontitis in Rats. J. Periodontol. 2017, 88, e49–e57. [Google Scholar] [CrossRef]
- Kuraji, R.; Ito, H.; Fujita, M.; Ishiguro, H.; Hashimoto, S.; Numabe, Y. Porphyromonas gingivalis induced periodontitis exacerbates progression of non-alcoholic steatohepatitis in rats. Clin. Exp. Dent. Res. 2016, 2, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.Y.; Liang, L.Z.; Zhao, Y.X.; Yang, Y.N.; Liu, F.; Ding, Q.R.; Luo, L.J. Porphyromonas gingivalis-derived lipopolysaccharide causes excessive hepatic lipid accumulation via activating NF-κB and JNK signaling pathways. Oral Dis. 2019, 25, 1789–1797. [Google Scholar] [CrossRef]
- Zaitsu, Y.; Iwatake, M.; Sato, K.; Tsukuba, T. Lipid droplets affect elimination of Porphyromonas gingivalis in HepG2 cells by altering the autophagy-lysosome system. Microbes Infect. 2016, 18, 565–571. [Google Scholar] [CrossRef]
- Page, R.C.; Schroeder, H.E. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab. Investig. 1976, 34, 235–249. [Google Scholar] [PubMed]
- Baker, P.J.; Howe, L.; Garneau, J.; Roopenian, D.C. T cell knockout mice have diminished alveolar bone loss after oral infection with Porphyromonas gingivalis. FEMS Immunol. Med. Microbiol. 2002, 34, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.T.; Nguyen, H.; Gao, X.; Kong, Y.Y.; Gorczynski, R.M.; Singh, B.; Ellen, R.P.; Penninger, J.M. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J. Clin. Investig. 2000, 106, R59–R67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.J.; Dixon, M.; Evans, R.T.; Dufour, L.; Johnson, E.; Roopenian, D.C. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect. Immun. 1999, 67, 2804–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlet, G.P.; Cardoso, C.R.; Campanelli, A.P.; Garlet, T.P.; Avila-Campos, M.J.; Cunha, F.Q.; Silva, J.S. The essential role of IFN-gamma in the control of lethal Aggregatibacter actinomycetemcomitans infection in mice. Microbes Infect. 2008, 10, 489–496. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Gemmell, E.; Seymour, G.J. Immunoregulatory control of Th1/Th2 cytokine profiles in periodontal disease. Periodontology 2000 2004, 35, 21–41. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Hajishengallis, G. A new inflammatory cytokine on the block: Re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J. Dent. Res. 2008, 87, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Kolls, J.K.; Lindén, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional specialization of interleukin-17 family members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.O.; Chang, S.H.; Park, H.; Nurieva, R.; Shah, B.; Acero, L.; Wang, Y.H.; Schluns, K.S.; Broaddus, R.R.; Zhu, Z.; et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 2008, 205, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Tanaka, Y.; Araki, H.; Kohda, A.; Sanematsu, F.; Arasaki, T.; Duan, X.; Miura, F.; Katagiri, T.; Shindo, R.; et al. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci. Rep. 2017, 7, 17402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, P.; Rodriguez, F.H.; Kanaly, S.; Stocking, K.L.; Schurr, J.; Schwarzenberger, P.; Oliver, P.; Huang, W.; Zhang, P.; Zhang, J.; et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 2001, 194, 519–527. [Google Scholar] [CrossRef]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.H.; et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Adibrad, M.; Deyhimi, P.; Ganjalikhani Hakemi, M.; Behfarnia, P.; Shahabuei, M.; Rafiee, L. Signs of the presence of Th17 cells in chronic periodontal disease. J. Periodontal. Res. 2012, 47, 525–531. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Allam, J.P.; Duan, Y.; Heinemann, F.; Winter, J.; Götz, W.; Deschner, J.; Wenghoefer, M.; Bieber, T.; Jepsen, S.; Novak, N. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J. Clin. Periodontol. 2011, 38, 879–886. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Vonghia, L.; Michielsen, P.; Francque, S. Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int. J. Mol. Sci. 2013, 14, 19867–19890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.; Surette, F.A.; Hahn, Y.S. The Immune Landscape in Nonalcoholic Steatohepatitis. Immune Netw. 2016, 16, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambineri, E.; Torgerson, T.R.; Ochs, H.D. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 2003, 15, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, F.; Ziegler, S.F. FOXP3 and scurfy: How it all began. Nat. Rev. Immunol. 2014, 14, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Yamazaki, S.; Fukui, Y.; Aoki, K.; Yagita, H.; Nishina, T.; Mikami, T.; Katagiri, S.; Shiraishi, A.; Kimura, S.; et al. JunB plays a crucial role in development of regulatory T cells by promoting IL-2 signaling. Mucosal Immunol. 2019, 12, 1104–1117. [Google Scholar] [CrossRef]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Kremer, M.; Hines, I.N.; Milton, R.J.; Wheeler, M.D. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology 2006, 44, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Folco, E.J.; Sukhova, G.; Shimizu, K.; Gotsman, I.; Vernon, A.H.; Libby, P. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: A role for adaptive immunity in obesity. Circ. Res. 2008, 103, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.Y.; Jeong, H.J.; Kim, H.M. The role of T-bet in obesity: Lack of T-bet causes obesity in male mice. J. Nutr. Biochem. 2013, 24, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Eljaafari, A.; Robert, M.; Chehimi, M.; Chanon, S.; Durand, C.; Vial, G.; Bendridi, N.; Madec, A.M.; Disse, E.; Laville, M.; et al. Adipose Tissue-Derived Stem Cells From Obese Subjects Contribute to Inflammation and Reduced Insulin Response in Adipocytes Through Differential Regulation of the Th1/Th17 Balance and Monocyte Activation. Diabetes 2015, 64, 2477–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Cella, M.; McCartney, S.A.; Fuchs, A.; Abumrad, N.A.; Pietka, T.A.; Chen, Z.; Finck, B.N.; Han, D.H.; Magkos, F.; et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 2013, 145, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolla, S.; Alchera, E.; Imarisio, C.; Bardina, V.; Valente, G.; Cappello, P.; Mombello, C.; Follenzi, A.; Novelli, F.; Carini, R. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin. Sci. 2016, 130, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xu, B.C.; Yang, X.G.; Peng, D.; Wang, Y.; Liu, X.B.; Cui, C.R.; Jiang, Y.F. A High Frequency of Peripheral Blood IL-22(+) CD4(+) T Cells in Patients With New Onset Type 2 Diabetes Mellitus. J. Clin. Lab. Anal. 2016, 30, 95–102. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Huppert, S.S.; Iwakura, Y.; Dong, C.; Shanmukhappa, S.K.; Divanovic, S. Regulation of Inflammation by IL-17A and IL-17F Modulates Non-Alcoholic Fatty Liver Disease Pathogenesis. PLoS ONE 2016, 11, e0149783. [Google Scholar]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Zhang, X.W.; Mi, S.; Li, Z.; Zhou, J.C.; Xie, J.; Hua, F.; Li, K.; Cui, B.; Lv, X.X.; Yu, J.J.; et al. Antagonism of Interleukin-17A ameliorates experimental hepatic fibrosis by restoring the IL-10/STAT3-suppressed autophagy in hepatocytes. Oncotarget 2017, 8, 9922–9934. [Google Scholar] [CrossRef] [PubMed]
- Eustace, G.J.; Melmed, G.Y. Therapy for Crohn’s Disease: A Review of Recent Developments. Curr. Gastroenterol. Rep. 2018, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G., 3rd; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef] [PubMed]
- Engebretson, S.; Kocher, T. Evidence that periodontal treatment improves diabetes outcomes: A systematic review and meta-analysis. J. Periodontol. 2013, 84 (Suppl. 4), S153–S169. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.C.; Weldon, J.C.; Worthington, H.V.; Needleman, I.; Wild, S.H.; Moles, D.R.; Stevenson, B.; Furness, S.; Iheozor-Ejiofor, Z. Treatment of periodontal disease for glycaemic control in people with diabetes mellitus. Cochrane Database Syst. Rev. 2015, CD004714. [Google Scholar] [CrossRef] [PubMed]

| Ref. No. | Study Country Year Sample Size | Parameters Evaluated for the Diagnosis of NAFLD | Parameters Evaluated for the Diagnosis of Periodontitis | Major Findings |
|---|---|---|---|---|
| [34] | Furuta et al. Japan 2010 n = 2225 | Serum ALT level | PD, BOP | An association between periodontitis and serum ALT in male without smoking (OR = 2.3, 95% CI = 1.0–5.2) |
| [35] | Ahmad et al. Japan 2017 n = 5683 | Serum ALT level | PD, CAL | An association among deep periodontal pockets and combination of increased serum ALT and symptoms of metabolic syndrome in male with low alcohol consumption |
| [36] | Morita et al. Japan 2014 n = 1510 | Serum GGT, ALT, AST level | CPI | An association between elevation of alglutamyltranspeptidase and having deep periodontal pockets (OR = 1.48, 95% CI = 1.16–1.90) |
| [37] | Iwasaki et al. Japan 2018 n = 1226 | Ultrasonography | PD, BOP | An association between periodontitis and NAFLD (OR = 1.881, 95% CI = 1.184–2.987) |
| [38] | Kim et al. Korea 2020 n = 4272 | FLI | CPI | An association between periodontal disease and FLI (OR = 1.63; 95% CI = 1.23–2.16) |
| [39] | Shin et al. Korea 2019 n = 4061 | FLI, HSI | CPI | An association between periodontal disease and FLI, HSI in women (OR = 1.77, 95% CI = 1.05–2.98) |
| [40] | Qiao et al. China 2018 n = 24,470 | Ultrasonography | The number of missing teeth | An association between the missing teeth and NAFLD in men (among those who with more than 6 missing teeth, OR = 1.40, 95% CI = 1.09–1.81) |
| [41] | Weintraub et al. USA 2019 n = 5421 | Ultrasonography, Fibrosis Score, FLI, US-FLI | PD, BOP, CAL the number of missing teeth | An association between periodontitis, tooth loss and all of the parameters for NAFLD |
| [42] | Alazawi et al. USA 2017 n = 8172 | Ultrasonography | PD, CAL Serum antibody titers against 19 oral bacteria | Significant associations among steatosis and bleeding on probing, PD ≥ 4 mm (%), mean PD, CAL ≥ 3 mm, and mean CAL (%) After adjusting for sociodemographic factors, only BOP and mean PD showed a significant association with steatosis (BOP: OR = 1.07, 95% CI = 1.00–1.14, mean PD: OR = 1.08, 95% CI = 1.00–1.17) |
| [43] | Akinkugbe et al. USA 2017 n = 11,914 | Ultrasonography, serum ALT level | PD, CAL | Periodontitis was associated with serum ALT and AST levels (≥ 30% of sites with PD ≥ 4 mm: OR = 1.39, 95% CI = 1.02–1.90), however, the significance was not observed after adjustment of age and sex |
| [46] | Akinkugbe et al. Germany 2017 n = 2481 | Ultrasonography | PD, CAL | A significant correlation between periodontitis and NAFLD among subjects with less than 1 mg/L serum CRP levels and/or with lower than the median weighted genetic CRP score Serum CRP modified the interaction between periodontitis and NAFLD |
| Ref. No. | Study Country Year Sample Size | Evaluation Criteria for Liver | Parameters Evaluated for the Diagnosis of Periodontitis | Observation Period | Major Findings |
|---|---|---|---|---|---|
| [47] | Kuroe et al. Japan 2020 n = 341 | NAFL (ultrasonography, NAFLD fibrosis score) | PD, CAL | 5 years | CAL and liver fibrosis were significantly associated in obese NAFL patients (OR = 2.87, 95% CI = 1.23–6.69) |
| [48] | Akinkugbe et al. Germany 2017 n = 2623 | NAFLD (ultrasonography) | PD, CAL | median 7.7 years | NAFLD incidence was elevated in participants with >30% of ≥3 mm CAL (multivariable-adjusted incidence rate ratio: 1.60, 95% CI = 1.05–2.43) |
| [49] | Helenius-Hietala et al. Finland 2017 n = 1801 | The incidence of severe liver disease (a first hospitalization owing to liver disease or liver-related death, a diagnosis of (primary) liver cancer) | PD | 13 years | The incidence of severe liver disease was increased for the patients with advanced periodontitis and NAFLD (hazard ratio = 6.94, 95% CI = 1.43–33.6) |
| Ref. No. | Study | The Number of Primary Studies | Study Designs of Primary Studies | Statistical Analysis | Results |
|---|---|---|---|---|---|
| [50] | Chen et al. 2020 n = 118,408 | 12 | Cross-sectional (4), case-control (1), cohort (7) | Generalized least-squares regressions | An association between periodontitis and NAFLD (OR = 1.19, 95% CI =1.06–1.33), and an association periodontitis and cirrhosis (OR = 2.28, 95% CI = 1.50–3.48) was reported. |
| [51] | Wijarnpreecha et al. 2020 n = 27,703 | 5 | Cross-sectional (4), cohort (1) | PRISMA, The random-effect model | NAFLD was associated with periodontitis (OR = 1.48, 95% CI = 1.13–1.89), however, significant correlation was lost after the adjusted results of the primary studies were applied. |
| Ref. No. | Study Country Year Sample Size | Parameters Evaluated for the Diagnosis of NAFLD | Parameters Evaluated for the Diagnosis of Periodontitis | Major Findings |
|---|---|---|---|---|
| [8] | Yoneda et al. Japan 2012 n = 150 | Liver biopsy | Detection of P. gingivalis in saliva by PCR | The detection of P. gingivalis was higher in NAFLD patients compared to non-NAFLD control subjects (46.7% vs. 21.7%, OR = 3.16, 95% CI = 1.58–6.33). Serum AST and ALT levels of 10 patients with NAFLD were significantly decreased after receiving periodontal treatment for 3 months. |
| [9] | Komazaki et al. Japan 2017 n = 52 | Total fat area, visceral fat area and the liver/spleen ratio evaluated by abdominal computed tomography, fasting blood insulin level, HOMA-IR, Serum AST, ALT, and γ-GTP | Serum antibody titers against A. actinomycetemcomitans, F. nucleatum, P. gingivalis | Anti-A. actinomycetemcomitans antibody titers and anti-F. Nucleatum antibody titers were slightly associated with fat area. Anti-A. actinomycetemcomitans antibody titers showed a positive correlation with fasting plasma insulin, the homeostasis model of assessment of insulin resistance, and AST, and a negative correlation with the liver/spleen ratio. |
| [42] | Alazawi et al. UK 2017 n = 69 | Ultrasonography | PD, CAL | An association among periodontitis and the severity of NAFLD (NASH vs. NAFL) and the presence of diabetes was reported. |
| [54] | Nakahara et al. Japan 2018 n = 200 | Liver biopsy | Serum antibody titers against P. gingivalis | A significant monotonic trend between the fibrosis stage and antibody titers against P. gingivalis fim A type 1 and 4 was observed. An association between antibody titers against P. gingivalis fim A type 4 and advanced fibrosis was reported (OR = 2.081, 95% CI = 1.098–3.943). |
| [55] | Furusho et al. Japan 2013 n = 40 | Liver biopsy | Detection of P. gingivalis by immunohistochemistry in hepatocytes | P. gingivalis-positive patients showed progression of hepatic fibrosis compared to patients without P. gingivalis. |
| Ref. No. | Study | Cells | Major Findings |
|---|---|---|---|
| [55] | Furusho et al. | Palmitic acid-induced Hc3716-hTERT cells | Upregulation of TLR2 expression Increase in the mRNA levels of inflammasomes and proinflammatory cytokines |
| [58] | Nagasaki et al. | Palmitic acid-induced LX-2 and Hc3716-hTERT cells | Induction of TGF-β1 and Galectin-3 production |
| [70] | Ding et al. | oleic acid-induced HepG2 cells | Accumulation of intracellular lipids Enhancement in the phosphorylation of p65 and JNK |
| [71] | Zaitsu et al. | oleic acid-induced HepG2 cells | Lipid droplets affected the removal of P. gingivalis by altering the autophagy-lysosome system |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatasa, M.; Yoshida, S.; Takahashi, H.; Tanaka, K.; Kubotsu, Y.; Ohsugi, Y.; Katagiri, T.; Iwata, T.; Katagiri, S. Relationship between NAFLD and Periodontal Disease from the View of Clinical and Basic Research, and Immunological Response. Int. J. Mol. Sci. 2021, 22, 3728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073728
Hatasa M, Yoshida S, Takahashi H, Tanaka K, Kubotsu Y, Ohsugi Y, Katagiri T, Iwata T, Katagiri S. Relationship between NAFLD and Periodontal Disease from the View of Clinical and Basic Research, and Immunological Response. International Journal of Molecular Sciences. 2021; 22(7):3728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073728
Chicago/Turabian StyleHatasa, Masahiro, Sumiko Yoshida, Hirokazu Takahashi, Kenichi Tanaka, Yoshihito Kubotsu, Yujin Ohsugi, Takaharu Katagiri, Takanori Iwata, and Sayaka Katagiri. 2021. "Relationship between NAFLD and Periodontal Disease from the View of Clinical and Basic Research, and Immunological Response" International Journal of Molecular Sciences 22, no. 7: 3728. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073728