
Genome and Pangenome Analysis of Lactobacillus hilgardii FLUB—A New Strain Isolated from Mead

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
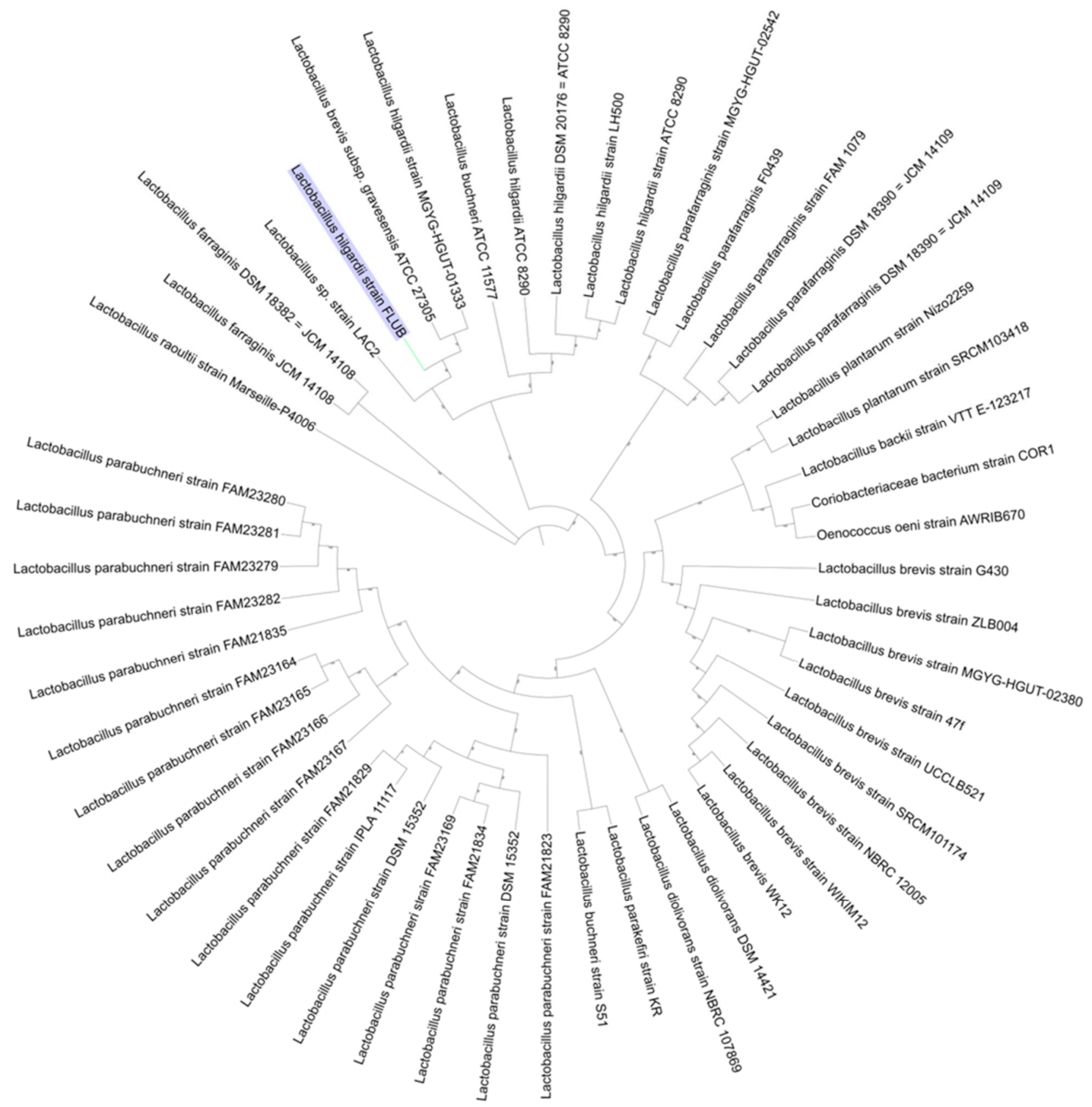
2.1. Isolation and Identification
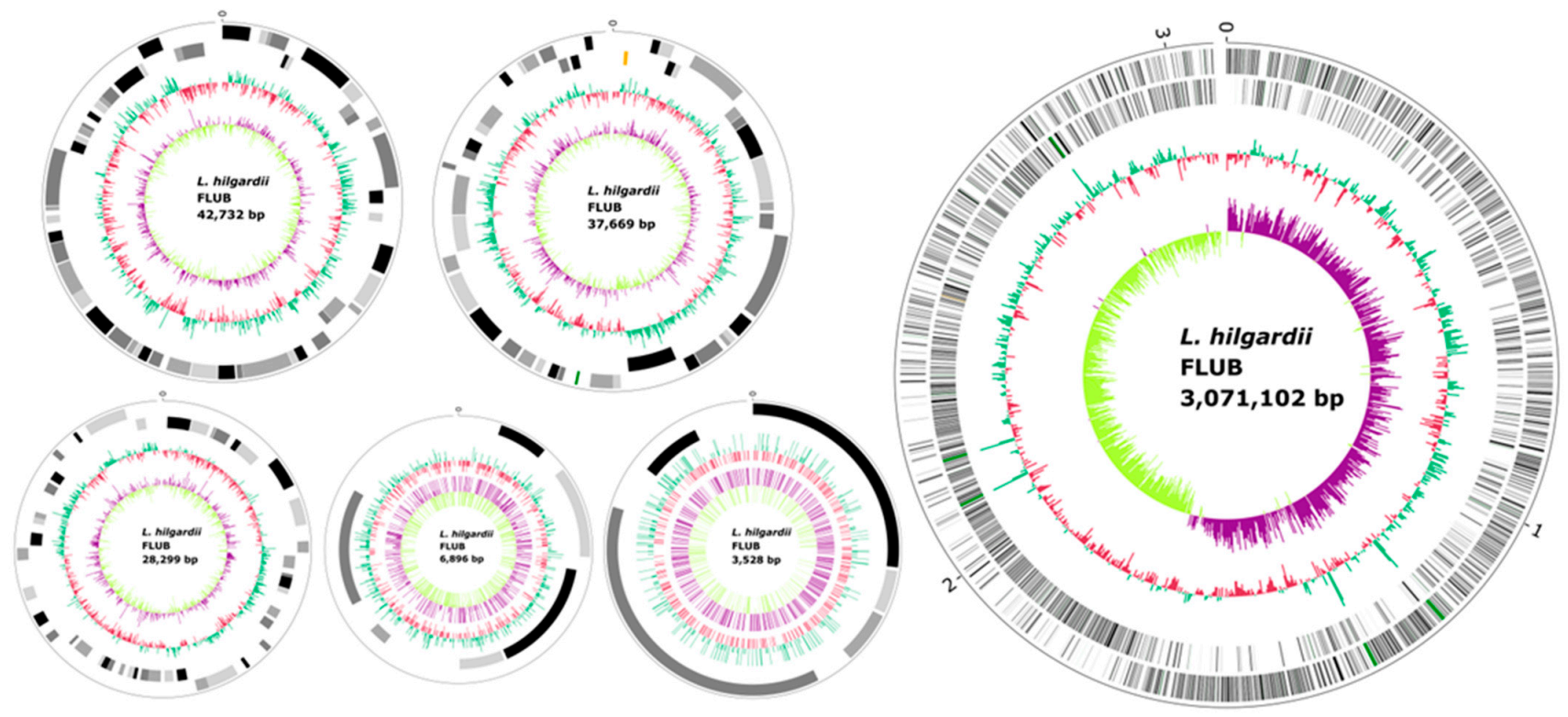
2.2. General Genome Features
2.3. Plasmids
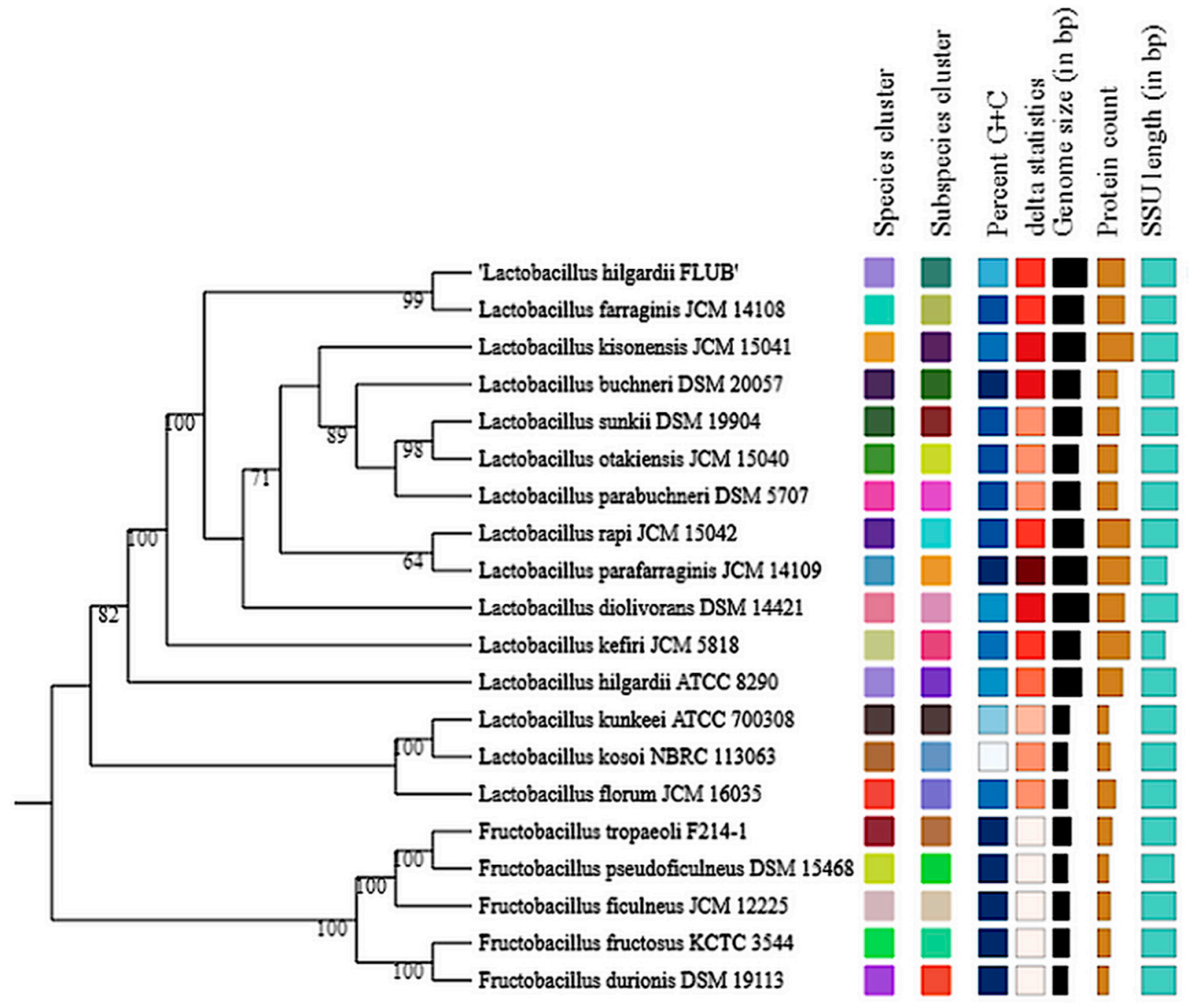
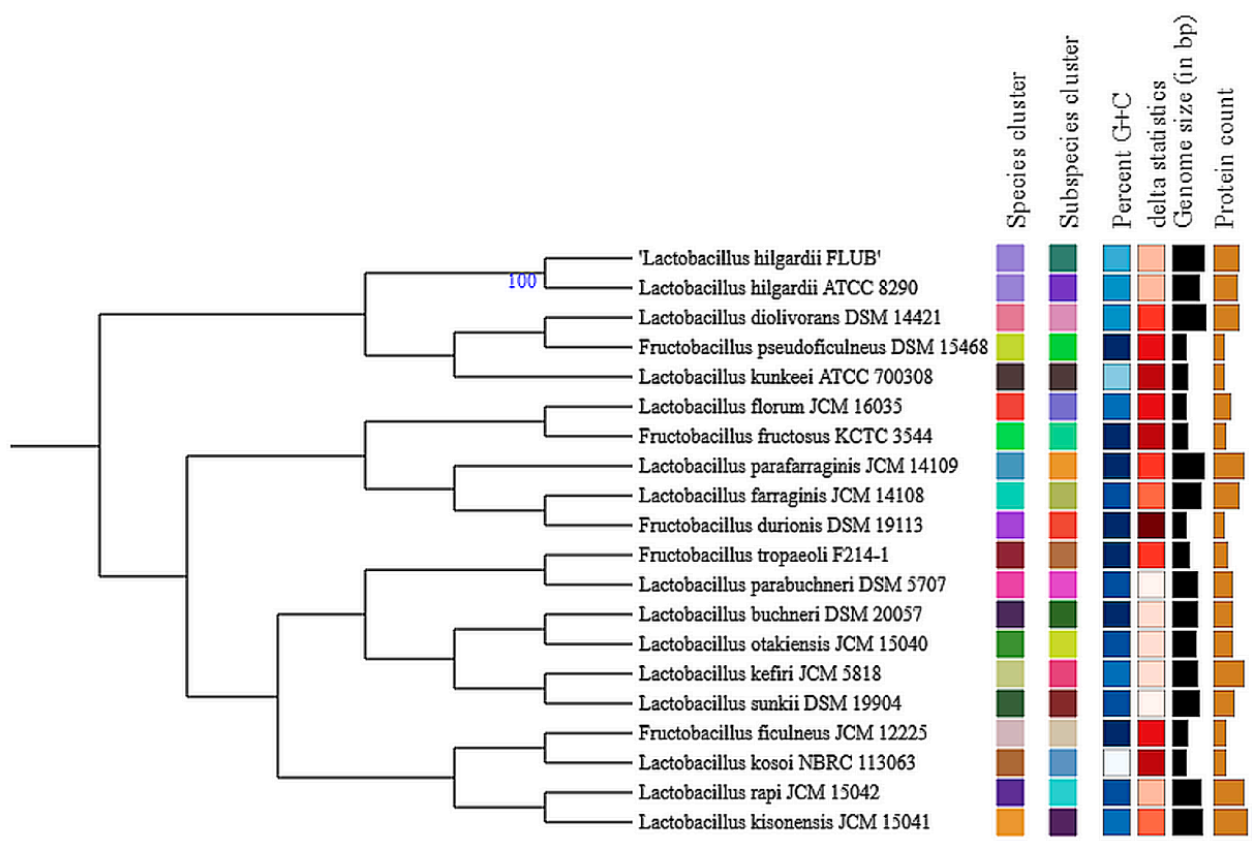
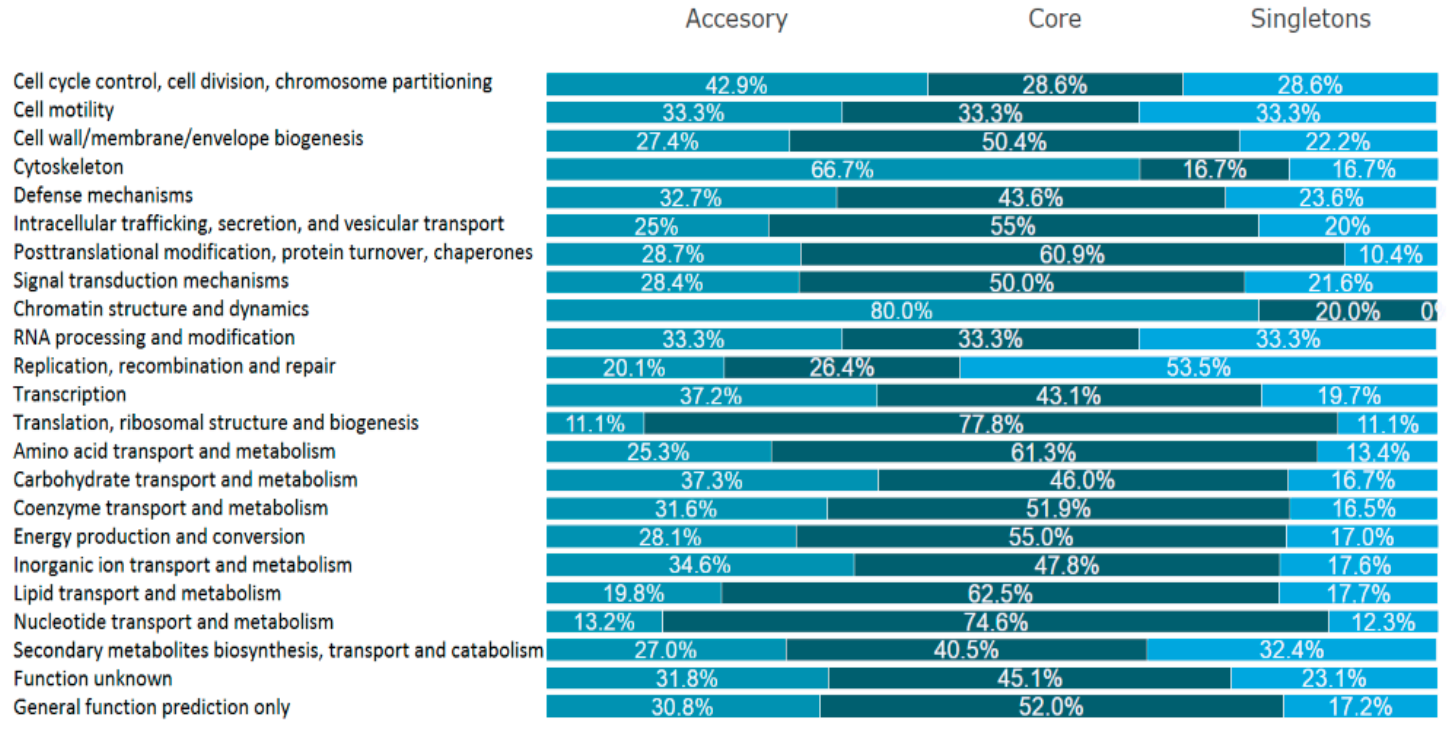
2.4. Pangenome of L. hilgardii Species
2.5. Singletons of L. hilgardii FLUB
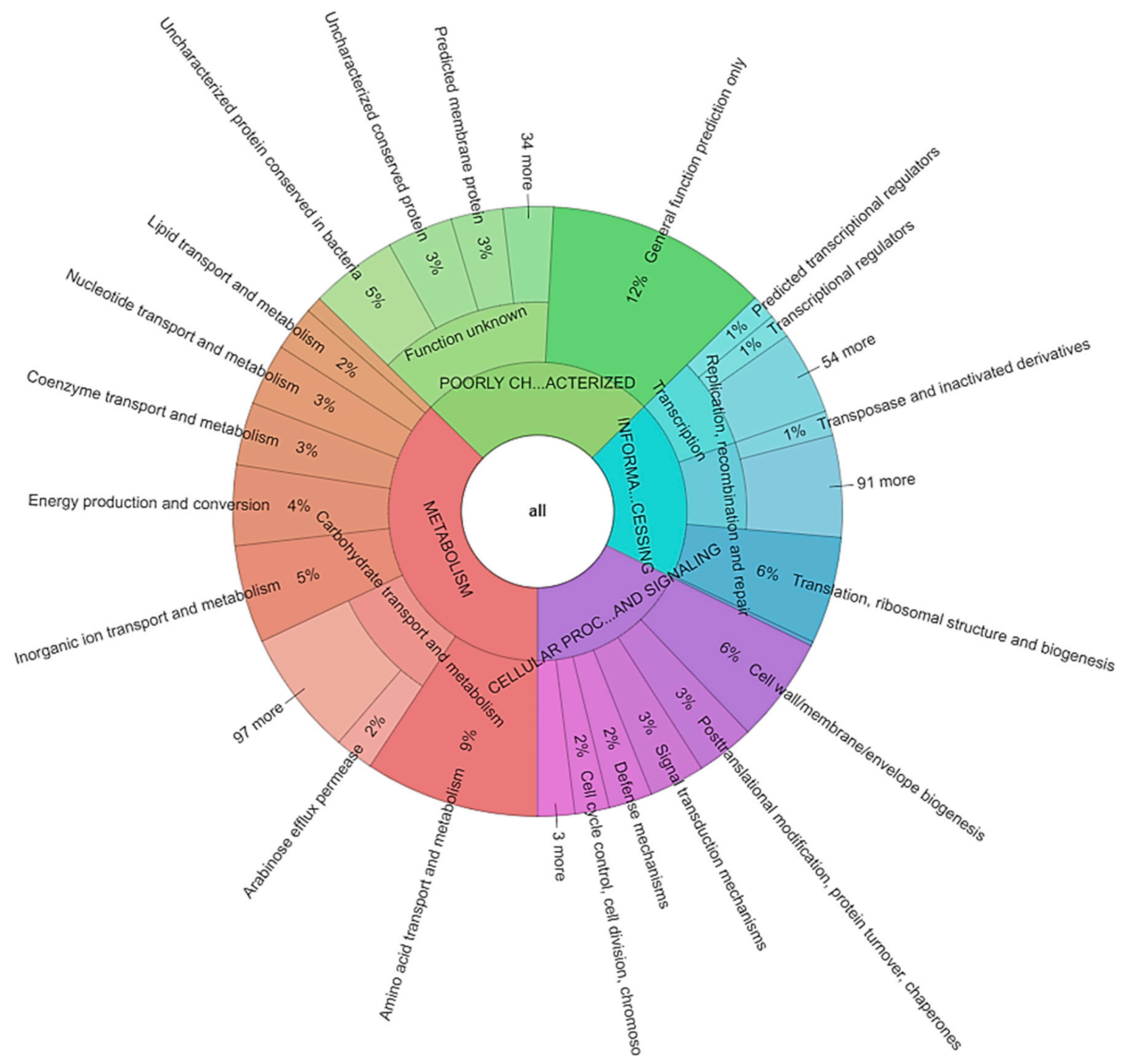
2.6. Carbohydrate Metabolism
2.7. Antimicrobial Resistance Genes
2.8. Osmoregulation, Detoxification, and Stress Response
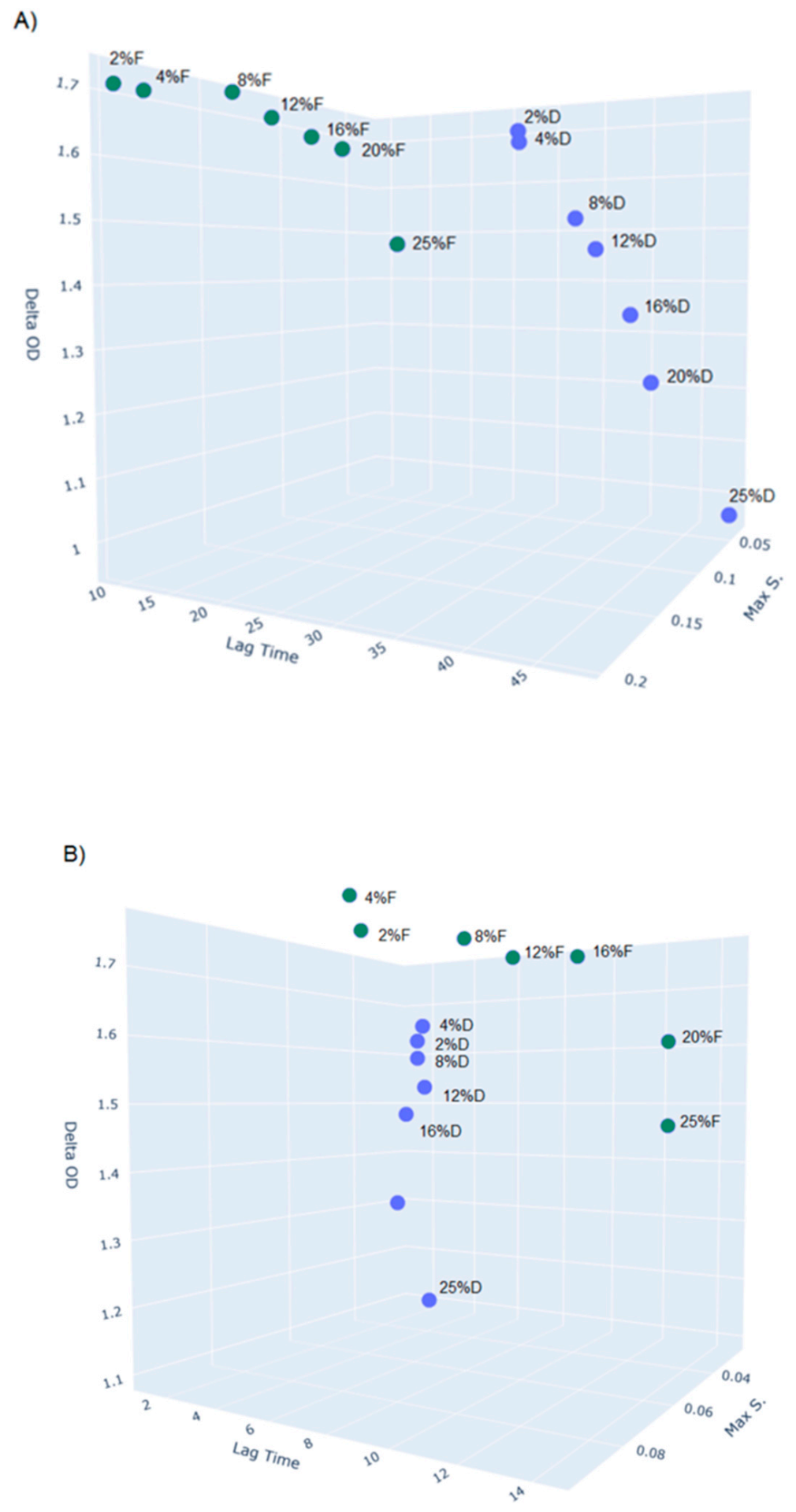
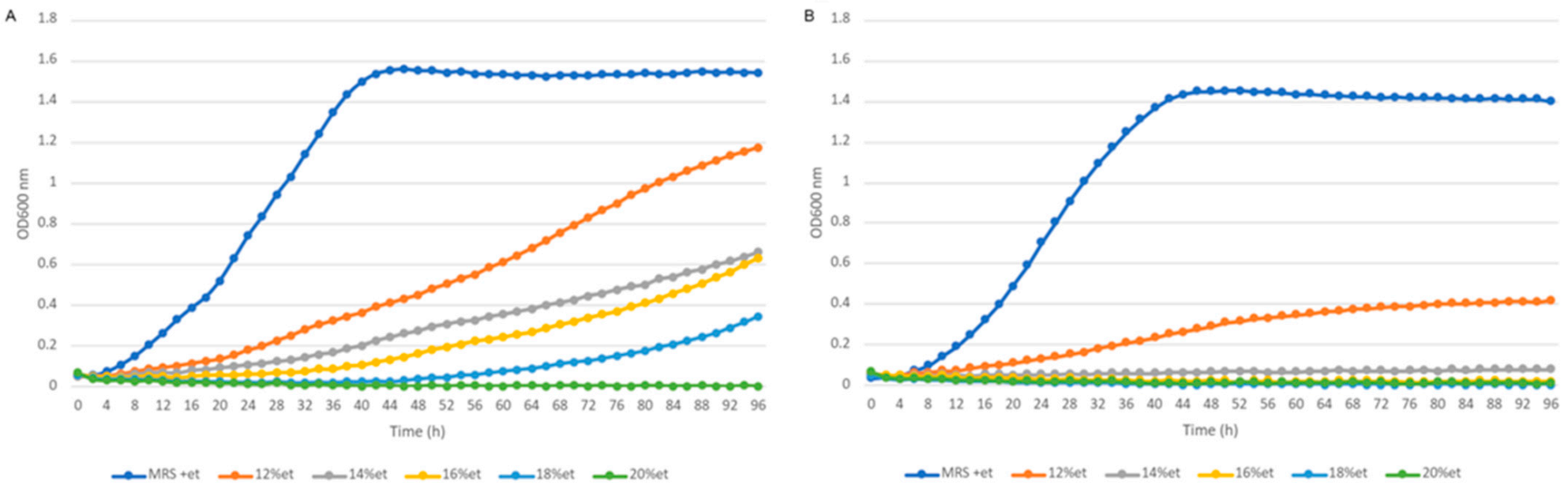
2.9. Phenotypic Properties (Growth Characteristics, API-ZYM, SDS-PAGE)
3. Materials and Methods
3.1. Isolation and Identification
3.2. Whole Genome Sequencing
3.3. Annotation and Functional Categorization
3.4. Phylogenetic Analysis
3.5. L. hilgardii Pan-Core Genome
3.6. Phenotypic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moe, D.; Oeggl, K. Palynological evidence of mead: A prehistoric drink dating back to the 3rd millennium b.c. Veg. Hist. Archaeobot. 2014, 23, 515–526. [Google Scholar] [CrossRef]
- Escalante, A.; López Soto, D.R.; Velázquez Gutiérrez, J.E.; Giles-Gómez, M.; Bolívar, F.; López-Munguía, A. Pulque, a Traditional Mexican Alcoholic Fermented Beverage: Historical, Microbiological, and Technical Aspects. Front. Microbiol. 2016, 7, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahoun, D.; Řezková, S.; Královský, J. Effect of heat treatment and storage conditions on mead composition. Food Chem. 2017, 219, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, M.; Szwengiel, A. Distinguishing between saturated and unsaturated meads based on their chemical characteristics. LWT 2020, 133, 109962. [Google Scholar] [CrossRef]
- Bednarek, M.; Szwengiel, A.; Flórez, A.B.; Czarnecki, Z.; Mayo, B. Effect of different starter cultures on chemical and microbial parameters of buckwheat honey fermentation. Food Microbiol. 2019, 82, 294–302. [Google Scholar] [CrossRef]
- Pereira, A.P.; Dias, T.; Andrade, J.; Ramalhosa, E.; Estevinho, L.M. Mead production: Selection and characterization assays of Saccharomyces cerevisiae strains. Food Chem. Toxicol. 2009, 47, 2057–2063. [Google Scholar] [CrossRef]
- Sroka, P.; Satora, P.; Tarko, T.; Duda-Chodak, A. The influence of yeast immobilization on selected parameters of young meads. J. Inst. Brew. 2017, 123, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Mandal, M.D.; Mandal, S. Honey: Its medicinal property and antibacterial activity. Asian Pac. J. Trop. Biomed. 2011, 1, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Ramalhosa, E.; Gomes, T.; Pereira, A.P.; Dias, T.; Estevinho, L.M. Mead Production: Tradition Versus Modernity. Adv. Food Nutr. Res. 2011, 63, 101–118. [Google Scholar]
- Balzan, S.; Carraro, L.; Merlanti, R.; Lucatello, L.; Capolongo, F.; Fontana, F.; Novelli, E.; Larini, I.; Vitulo, N.; Cardazzo, B. Microbial metabarcoding highlights different bacterial and fungal populations in honey samples from local beekeepers and market in north-eastern Italy. Int. J. Food Microbiol. 2020, 334, 108806. [Google Scholar] [CrossRef]
- Couto, J.A.; Hogg, T.A. Diversity of ethanol-tolerant lactobacilli isolated from Douro fortified wine: Clustering and identification by numerical analysis of electrophoretic protein profiles. J. Appl. Bacteriol. 1994, 76, 487–491. [Google Scholar] [CrossRef]
- Dias, R.; Vilas-Boas, E.; Campos, F.M.; Hogg, T.; Ant, J.E.; Couto, O. Activity of lysozyme on Lactobacillus hilgardii strains isolated from Port wine. Food Microbiol. 2015, 49, 6–11. [Google Scholar] [CrossRef]
- Figueiredo, A.R.; Campos, F.; de Freitas, V.; Hogg, T.; Couto, J.A. Effect of phenolic aldehydes and flavonoids on growth and inactivation of Oenococcus oeni and Lactobacillus hilgardii. Food Microbiol. 2008, 25, 105–112. [Google Scholar] [CrossRef]
- Reboredo-Rodríguez, P.; González-Barreiro, C.; Rial-Otero, R.; Cancho-Grande, B.; Simal-Gándara, J.; Simal, J. Effects of Sugar Concentration Processes in Grapes and Wine Aging on Aroma Compounds of Sweet Wines-A Review. Crit. Rev. Food Sci. Nutr. 2015, 55, 1053–1073. [Google Scholar] [CrossRef]
- Estevinho, L.; Pereira, A.P.; Moreira, L.; Dias, L.G.; Pereira, E. Antioxidant and antimicrobial effects of phenolic compounds extracts of Northeast Portugal honey. Food Chem. Toxicol. 2008, 46, 3774–3779. [Google Scholar] [CrossRef]
- Holland, B.R.; Huber, K.T.; Dress, A.; Moulton, V. δ plots: A tool for analyzing phylogenetic distance data. Mol. Biol. Evol. 2002, 19, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Okada, S. Lactobacillus farraginis sp. nov. and Lactobacillus parafarraginis sp. nov., heterofermentative lactobacilli isolated from a compost of distilled shochu residue. Int. J. Syst. Evol. Microbiol. 2007, 57, 708–712. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Anne, C.-D. Die Grabfunde von Manching und die latènezeitlichen Flachgräber in Südbayern. Rev. Belg. Philol. D’histoire 1989, 67, 233–234. [Google Scholar]
- Szczerba, H.; Komoń-Janczara, E.; Krawczyk, M.; Dudziak, K.; Nowak, A.; Kuzdraliński, A.; Waśko, A.; Targoński, Z. Genome analysis of a wild rumen bacterium Enterobacter aerogenes LU2—A novel bio-based succinic acid producer. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Szczerba, H.; Dudziak, K.; Krawczyk, M.; Targoński, Z. A genomic perspective on the potential of wild-type rumen bacterium enterobacter sp. Lu1 as an industrial platform for bio-based succinate production. Int. J. Mol. Sci. 2020, 21, 4835. [Google Scholar] [CrossRef]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; Oun, M.A.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.E.; Raoult, D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, D.; Mishra, A.K.; Lagier, J.C.; Padhmanabhan, R.; Rossi, M.; Sentausa, E.; Raoult, D.; Fournier, P.E. A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int. J. Syst. Evol. Microbiol. 2014, 64, 384–391. [Google Scholar] [CrossRef]
- Di Mattia, E.; Grego, S.; Cacciari, I. Eco-physiological characterization of soil bacterial populations in different states of growth. Microb. Ecol. 2002, 43, 34–43. [Google Scholar] [PubMed]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, 431–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein Families for the Microbial Genomes in the PATRIC Database. Front. Microbiol. 2016, 7, 118. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Davalos, L.M. The nature and dynamics of bacterial genomes. Science 2006, 311, 1730–1733. [Google Scholar] [CrossRef] [Green Version]
- Jarocki, P.; Komoń-Janczara, E.; Podleśny, M.; Kholiavskyi, O.; Pytka, M.; Kordowska-Wiater, M. Genomic and proteomic characterization of bacteriophage BH1 spontaneously released from probiotic Lactobacillus rhamnosus pen. Viruses 2019, 11, 1163. [Google Scholar] [CrossRef] [Green Version]
- Jarocki, P.; Podleśny, M.; Krawczyk, M.; Glibowska, A.; Pawelec, J.; Komoń-Janczara, E.; Kholiavskyi, O.; Dworniczak, M.; Targoński, Z. Complete genome sequence of Lactobacillus rhamnosus Pen, a probiotic component of a medicine used in prevention of antibiotic-associated diarrhoea in children. Gut Pathog. 2018, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Kouwen, T.R.H.M.; Trip, E.N.; Denham, E.L.; Sibbald, M.J.J.B.; Dubois, J.Y.F.; Van Dijl, J.M. The large mechanosensitive channel MscL determines bacterial susceptibility to the bacteriocin sublancin 168. Antimicrob. Agents Chemother. 2009, 53, 4702–4711. [Google Scholar] [CrossRef] [Green Version]
- Marczak, M.; Mazur, A.; Koper, P.; Żebracki, K.; Skorupska, A. Synthesis of rhizobial exopolysaccharides and their importance for symbiosis with legume plants. Genes 2017, 8, 360. [Google Scholar] [CrossRef] [Green Version]
- Lucas, P.M.; Wolken, W.A.M.; Claisse, O.; Lolkema, J.S.; Lonvaud-Funel, A. Histamine-producing pathway encoded on an unstable plasmid in Lactobacillus hilgardii 0006. Appl. Environ. Microbiol. 2005, 71, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Josson, K.; Soetaert, P.; Michiels, F.; Joos, H.; Mahillon, J. Lactobacillus hilgardii plasmid pLAB1000 consist of two functional cassettes commonly found in other gram-positive organisms. J. Bacteriol. 1990, 172, 3089–3099. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, K.; Alegría, Á.; Bron, P.A.; de Angelis, M.; Gobbetti, M.; Kleerebezem, M.; Lemos, J.A.; Linares, D.M.; Ross, P.; Stanton, C.; et al. Stress Physiology of Lactic Acid Bacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 837–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rensing, C.; Fan, B.; Sharma, R.; Mitra, B.; Rosen, B.P.; Kaback, H.R. CopA: An. Escherichia coli Cu(I)-translocating P-type ATPase. Proc. Natl. Acad. Sci. USA 2000, 97, 652–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.Y.; Lee, S.H.; Lee, M.; Song, J.H.; Chang, J.Y. Lactobacillus allii sp. Nov. isolated from scallion kimchi. Int. J. Syst. Evol. Microbiol. 2017, 67, 4936–4942. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Rosen, B.P. Dual mode of energy coupling by the oxyanion-translocating ArsB protein. J. Bacteriol. 1995, 177, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Chen, X.; Du, S.; Deng, Z.; Wang, L.; Chen, S. Genetic mechanisms of arsenic detoxification and metabolism in bacteria. Curr. Genet. 2019, 65, 329–338. [Google Scholar] [CrossRef]
- Tisa, L.S.; Rosen, B.P. Molecular characterization of an anion pump. The ArsB protein is the membrane anchor for the ArsA protein. J. Biol. Chem. 1990, 265, 190–194. [Google Scholar] [CrossRef]
- Yang, J.; Rawat, S.; Stemmler, T.L.; Rosen, B.P. Arsenic Binding and Trasfer by the ArsD As(III). Biochemistry 2010, 49, 3658–3666. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Walmsley, A.R.; Rosen, B.P. An arsenic metallochaperone for an arsenic detoxification pump. Proc. Natl. Acad. Sci. USA 2006, 103, 15617–15622. [Google Scholar] [CrossRef] [Green Version]
- Ji, G.; Silver, S. Reduction of arsenate to arsenite by the ArsC protein of the arsenic resistance operon of Staphylococcus aureus plasmid pI258. Proc. Natl. Acad. Sci. USA 1992, 89, 9474–9478. [Google Scholar] [CrossRef] [Green Version]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Tettelin, H.; Medini, D. The Pangenome; Diversity, Dynamics and Evolution of Genomes; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–307. [Google Scholar]
- Reis, C.B.; De Oliveira, A.; Santos, B.; Ferreira Carvalho, R.; Freitas, S.; Luiza, C.; Ávila, S. Wild Lactobacillus hilgardii (CCMA 0170) strain modifies the fermentation profile and aerobic stability of corn silage. J. Appl. Anim. Res. 2018, 46, 632–638. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Endo, A. Taxonomic Status of Lactic Acid Bacteria in Wine and Key Characteristics to Differentiate Species. S. Afr. J. Enol. Vitic. 2008, 30, 72–90. [Google Scholar] [CrossRef] [Green Version]
- Spano, G.; Massa, S. Environmental stress response in wine lactic acid bacteria: Beyond Bacillus subtilis. Crit. Rev. Microbiol. 2006, 32, 77–86. [Google Scholar] [CrossRef]
- Singh, P.; Dutta, S.R.; Passi, D.; Bharti, J. Benefits of alcohol on arsenic toxicity in rats. J. Clin. Diagn. Res. 2017, 11, BF01–BF06. [Google Scholar] [CrossRef]
- Ussery, D.W.; Kiil, K.; Lagesen, K.; Sicheritz-Pontén, T.; Bohlin, J.; Wassenaar, T.M. The Genus Burkholderia: Analysis of 56 Genomic Sequences; Karger Publishers: Berlin, Germany, 2009; Volume 6. [Google Scholar]
- Mellroth, P.; Daniels, R.; Eberhardt, A.; Rönnlund, D.; Blom, H.; Widengren, J.; Normark, S.; Henriques-Normark, B. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J. Biol. Chem. 2012, 287, 11018–11029. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.F.; Zhang, L.J.; Pang, X.H.; Gu, X.X.; Abdelazez, A.; Liang, Y.; Sun, S.R.; Meng, X.C. Complete genome sequence of bacteriocin-producing Lactobacillus plantarum KLDS1.0391, a probiotic strain with gastrointestinal tract resistance and adhesion to the intestinal epithelial cells. Genomics 2017, 109, 432–437. [Google Scholar] [CrossRef]
- Schmid, M.; Muri, J.; Melidis, D.; Varadarajan, A.R.; Somerville, V.; Wicki, A.; Moser, A.; Bourqui, M.; Wenzel, C.; Eugster-Meier, E.; et al. Comparative genomics of completely sequenced lactobacillus helveticus genomes provides insights into strain-specific genes and resolves metagenomics data down to the strain level. Front. Microbiol. 2018, 9, 63. [Google Scholar] [CrossRef]
- Saier, M.H.; Beatty, J.T.; Goffeau, A.; Harley, K.T.; Heijne, W.H.M.; Huang, S.C.; Jack, D.L.; Jähn, P.S.; Lew, K.; Liu, J.; et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1999, 1, 257–279. [Google Scholar]
- Antonopoulos, D.A.; Assaf, R.; Aziz, R.K.; Brettin, T.; Bun, C.; Conrad, N.; Davis, J.J.; Dietrich, E.M.; Disz, T.; Gerdes, S.; et al. PATRIC as a unique resource for studying antimicrobial resistance. Brief. Bioinform. 2018, 20, 1094–1102. [Google Scholar] [CrossRef]
- Lebars, I.; Yoshizawa, S.; Stenholm, A.R.; Guittet, E.; Douthwaite, S.; Fourmy, D. Structure of 23s rRNA hairpin 35 and its interaction with the tylosin-resistance methyltransferase RlmAII. EMBO J. 2003, 22, 183–192. [Google Scholar] [CrossRef]
- Okamoto, S.; Tamaru, A.; Nakajima, C.; Nishimura, K.; Tanaka, Y.; Tokuyama, S.; Suzuki, Y.; Ochi, K. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol. Microbiol. 2007, 63, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Al-Madboly, L.A.; Khedr, E.G.; Ali, S.M. Optimization of reduced glutathione production by a Lactobacillus plantarum isolate using Plackett-Burman and Box-Behnken designs. Front. Microbiol. 2017, 8, 772. [Google Scholar] [CrossRef] [PubMed]
- Hoeflinger, J.L.; Hoeflinger, D.E.; Miller, M.J. A dynamic regression analysis tool for quantitative assessment of bacterial growth written in Python. J. Microbiol. Methods 2017, 132, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Švecová, B.S.; Bordovská, M.; Kalvachová, D.; Hájek, T. Analysis of Czech meads: Sugar content, organic acids content and selected phenolic compounds content. J. Food Compos. Anal. 2015, 38, 80–88. [Google Scholar] [CrossRef]
- Cavia, M.M.; Fernández-Muio, M.A.; Gömez-Alonso, E.; Montes-Pérez, M.J.; Huidobro, J.F.; Sancho, M.T. Evolution of fructose and glucose in honey over one year: Influence of induced granulation. Food Chem. 2002, 78, 157–161. [Google Scholar] [CrossRef]
- Endo, A.; Okada, S. Reclassification of the genus Leuconostoc and proposals of Fructobacillus fructosus gen. nov., comb. nov., Fructobacillus durionis comb. nov., Fructobacillus ficulneus comb. nov. and Fructobacillus pseudoficulneus comb. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 2195–2205. [Google Scholar] [CrossRef] [Green Version]
- Chiou, T.Y.; Suda, W.; Oshima, K.; Hattori, M.; Matsuzaki, C.; Yamamoto, K.; Takahashi, T. Lactobacillus kosoi sp. nov., a fructophilic species isolated from kôso, a Japanese sugar-vegetable fermented beverage. Antonie Leeuwenhoek 2018, 111, 1149–1156. [Google Scholar] [CrossRef]
- Endo, A. Fructophilic lactic acid bacteria inhabit fructose-rich niches in nature. Microb. Ecol. Health Dis. 2012, 23. [Google Scholar] [CrossRef]
- Viesser, J.A.; Pereira, G.V.D.M.; Pedro, D.; Neto, D.C.; Vandenberghe, L.P.D.S.; Azevedo, V.; Brenig, B.; Góes-neto, A.; Soccol, C.R. Exploring the contribution of fructophilic lactic acid bacteria to cocoa beans fermentation: Isolation, selection and evaluation. Food Res. Int. 2020, 136, 109478. [Google Scholar] [CrossRef]
- Tyler, C.A.; Kopit, L.; Doyle, C.; Yu, A.O.; Hugenholtz, J.; Marco, M.L. Polyol production during heterofermentative growth of the plant isolate Lactobacillus florum 2F. J. Appl. Microbiol. 2016, 120, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Behare, P.V.; Ali, S.A.; McAuliffe, O. Draft Genome Sequences of Fructobacillus fructosus DPC 7238 and Leuconostoc mesenteroides DPC 7261, Mannitol-Producing Organisms Isolated from Fructose-Rich Honeybee-Resident Flowers on an Irish Farm. Microbiol. Resour. Announc. 2020, 9, 10–11. [Google Scholar] [CrossRef]
- Maeno, S.; Dicks, L.; Nakagawa, J.; Endo, A. Lactobacillus apinorum belongs to the fructophilic lactic acid bacteria 2 3. Biosci. Microbita Food Health 2017, 36, 147–149. [Google Scholar] [CrossRef] [Green Version]
- Maeno, S.; Kajikawa, A.; Dicks, L.; Endo, A. Introduction of bifunctional alcohol/acetaldehyde dehydrogenase gene (adhE) in Fructobacillus fructosus settled its fructophilic characteristics. Res. Microbiol. 2019, 170, 35–42. [Google Scholar] [CrossRef]
- Lee, S.H.; Jung, M.Y.; Song, J.H.; Lee, M.; Chang, J.Y. Complete genome sequence of Lactobacillus curvatus strain WiKim38 isolated from kimchi. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Dohm, N.; Petri, A.; Schlander, M.; Schlott, B.; König, H.; Claus, H. Molecular and biochemical properties of the S-layer protein from the wine bacterium Lactobacillus hilgardii B706. Arch. Microbiol. 2011, 193, 251–261. [Google Scholar] [CrossRef]
- Gustaw, K.; Michalak, M.; Polak-Berecka, M.; Waśko, A. Isolation and characterization of a new fructophilic Lactobacillus plantarum FPL strain from honeydew. Ann. Microbiol. 2018, 68, 459–470. [Google Scholar] [CrossRef]
- Szczerba, H.; Komoń-Janczara, E.; Dudziak, K.; Waśko, A.; Targoński, Z. A novel biocatalyst, Enterobacter aerogenes LU2, for efficient production of succinic acid using whey permeate as a cost-effective carbon source. Biotechnol. Biofuels 2020, 13, 1–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Tanizawa, Y.; Fujisawa, T.; Kaminuma, E.; Nakamura, Y.; Arita, M. DFAST and DAGA: Web-based integrated genome annotation tools and resources. Biosci. Microbiota Food Health 2016, 35, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Waśko, A.; Polak-Berecka, M.; Kuzdraliński, A.; Skrzypek, T. Variability of S-layer proteins in Lactobacillus helveticus strains. Anaerobe 2014, 25, 53–60. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Plasmid 1 | Plasmid 2 | Plasmid 3 | Plasmid 4 | Plasmid 5 | Total | |
|---|---|---|---|---|---|---|---|
| Total Sequence Length (bp): | 3,071,102 | 42,732 | 37,669 | 28,299 | 6896 | 3528 | 3,190,226 |
| Number of Sequences: | 1 | 1 | 1 | 1 | 1 | 1 | 6 |
| Longest Sequences (bp): | 3,071,102 | 42,732 | 37,669 | 28,299 | 6896 | 3528 | 3,071,102 |
| N50 (bp): | 3,071,102 | 42,732 | 37,669 | 28,299 | 6896 | 3528 | 3,071,102 |
| Gap Ratio (%): | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
| CG Content (%): | 40.12 | 39.44 | 41.56 | 37.09 | 35.64 | 37.39 | 40.1 |
| Number of CDSs: | 2858 | 50 | 41 | 30 | 6 | 4 | 2997 |
| Average Protein Length: | 303.2 | 225.3 | 226.5 | 156.9 | 162.3 | 206.8 | 298.4 |
| Coding Ratio (%): | 84.7 | 79.1 | 74.0 | 49.9 | 42.4 | 70.3 | 84.1 |
| Pseudogenes | 61 | 9 | 5 | 13 | 4 | 1 | 93 |
| Number of rRNAs: | 15 | 0 | 0 | 0 | 0 | 0 | 15 |
| Number of tRNAs: | 61 | 0 | 0 | 0 | 0 | 0 | 61 |
| Number of CRISPRs: | 1 | 0 | 1 | 0 | 1 | 0 | 3 |
| No. | Organism Name | Strain | BioSample | BioProject | Assembly | Assembly Level | Size (Mbp) | GC % | Scaffold | CDS | Number of Genes | Pseudogenes | rRNA | tRNA | Other RNA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | L. hilgardii | FLUB | SAMN13567894 | PRJNA595831 | GCA_009832765.1 | Complete | 3.19 | 40.07 | 6 | 2871 | 3043 | 93 | 15 | 61 | 3 |
| 2 | L. hilgardii | LMG 07934 | SAMN14262734 | PRJNA609644 | GCA_011765585.1 | Complete | 2.77 | 39.7 | 1 | 2540 | 2739 | 12 | 15 | 61 | 3 |
| 3 | L. hilgardii | LH500 | SAMN12777270 | PRJNA566016 | GCA_008694025.1 | Complete | 2.65 | 39.8 | 1 | 2368 | 2603 | 157 | 15 | 60 | 3 |
| 4 | L. hilgardii | MGYG-HGUT-01333 | SAMEA5850835 | PRJEB33885 | GCA_902374015.1 | Scaffold | 3.14 | 40.2 | 106 | 2768 | 2930 | 99 | 3 | 57 | 3 |
| 5 | L. hilgardii | DSM 20176 | SAMN02369502 | PRJNA222257 | GCA_001434655.1 | Contig | 2.6 | 39.6 | 125 | 2387 | 2593 | 143 | 7 | 53 | 3 |
| ATCC 8290 | SAMN00001467 | PRJNA31489 | GCA_000159315.1 | Scaffold | 2.72 | 39.9 | 113 | 2395 | 2615 | 158 | 3 | 56 | 3 | ||
| ATCC 8290 | SAMN08557741 | PRJNA434413 | GCA_004354795.1 | Scaffold | 2.77 | 39.9 | 92 | 2523 | 2746 | 146 | 16 | 58 | 3 |
| AMR Mechanism | Genes |
|---|---|
| Antibiotic target in susceptible species | Alr, Ddl, EF-G, EF-Tu, folA, Dfr, folP, gyrA, gyrB, inhA, fabI, Iso-tRNA, kasA, MurA, rpoB, rpoC, S10p, S12p |
| Antibiotic target modifying enzyme | RlmA(II) |
| Gene conferring resistance via absence | gidB |
| Protein altering cell wall charge conferring antibiotic resistance | GdpD, MprF, PgsA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustaw, K.; Koper, P.; Polak-Berecka, M.; Rachwał, K.; Skrzypczak, K.; Waśko, A. Genome and Pangenome Analysis of Lactobacillus hilgardii FLUB—A New Strain Isolated from Mead. Int. J. Mol. Sci. 2021, 22, 3780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073780
Gustaw K, Koper P, Polak-Berecka M, Rachwał K, Skrzypczak K, Waśko A. Genome and Pangenome Analysis of Lactobacillus hilgardii FLUB—A New Strain Isolated from Mead. International Journal of Molecular Sciences. 2021; 22(7):3780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073780
Chicago/Turabian StyleGustaw, Klaudia, Piotr Koper, Magdalena Polak-Berecka, Kamila Rachwał, Katarzyna Skrzypczak, and Adam Waśko. 2021. "Genome and Pangenome Analysis of Lactobacillus hilgardii FLUB—A New Strain Isolated from Mead" International Journal of Molecular Sciences 22, no. 7: 3780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073780