
Novel and Potent Small Molecules against Melanoma Harboring BRAF Class I/II/III Mutants for Overcoming Drug Resistance

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
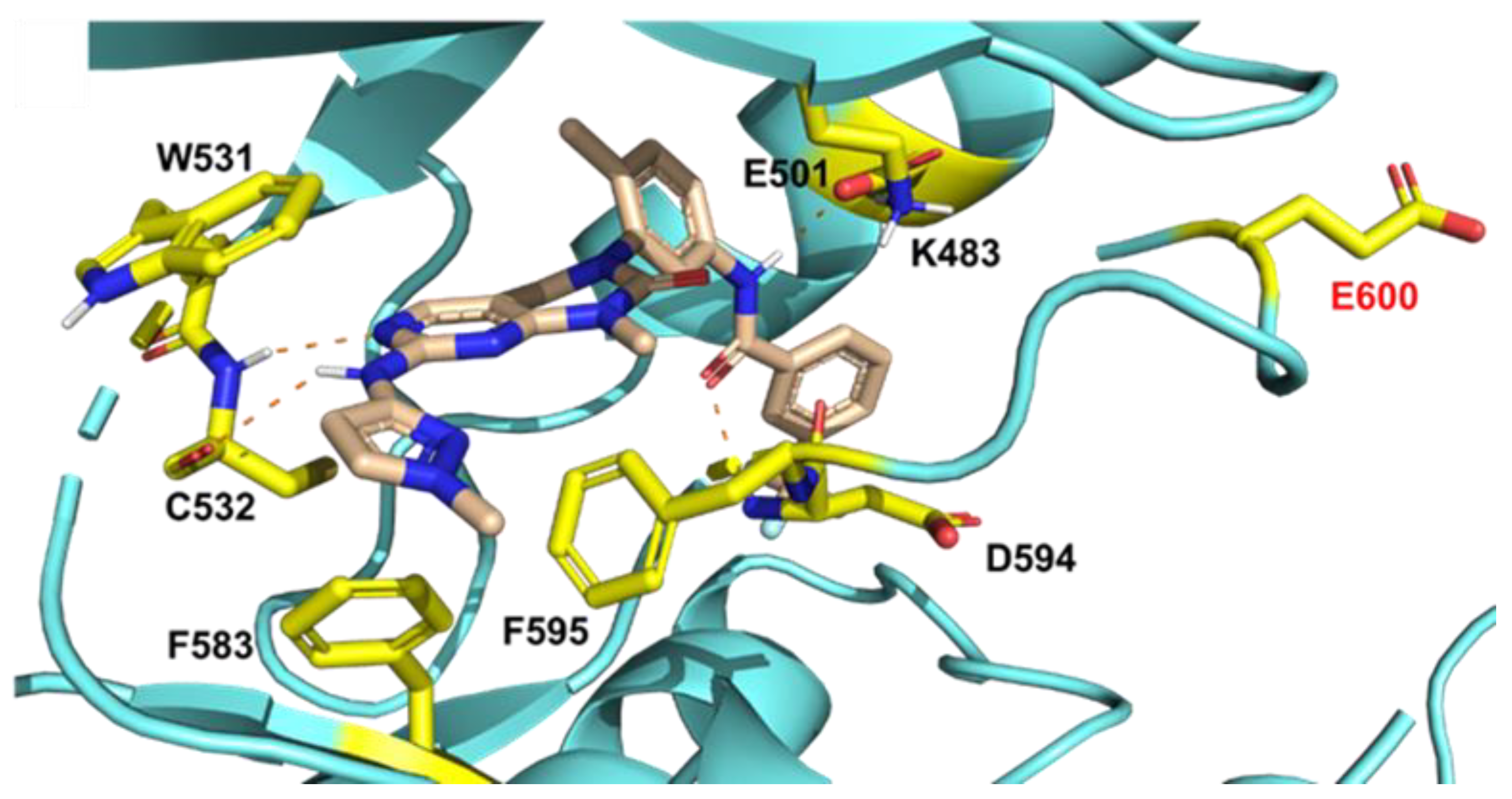
2.1. Molecular Docking Study of SIJ1777 with BRAF V600E Mutant
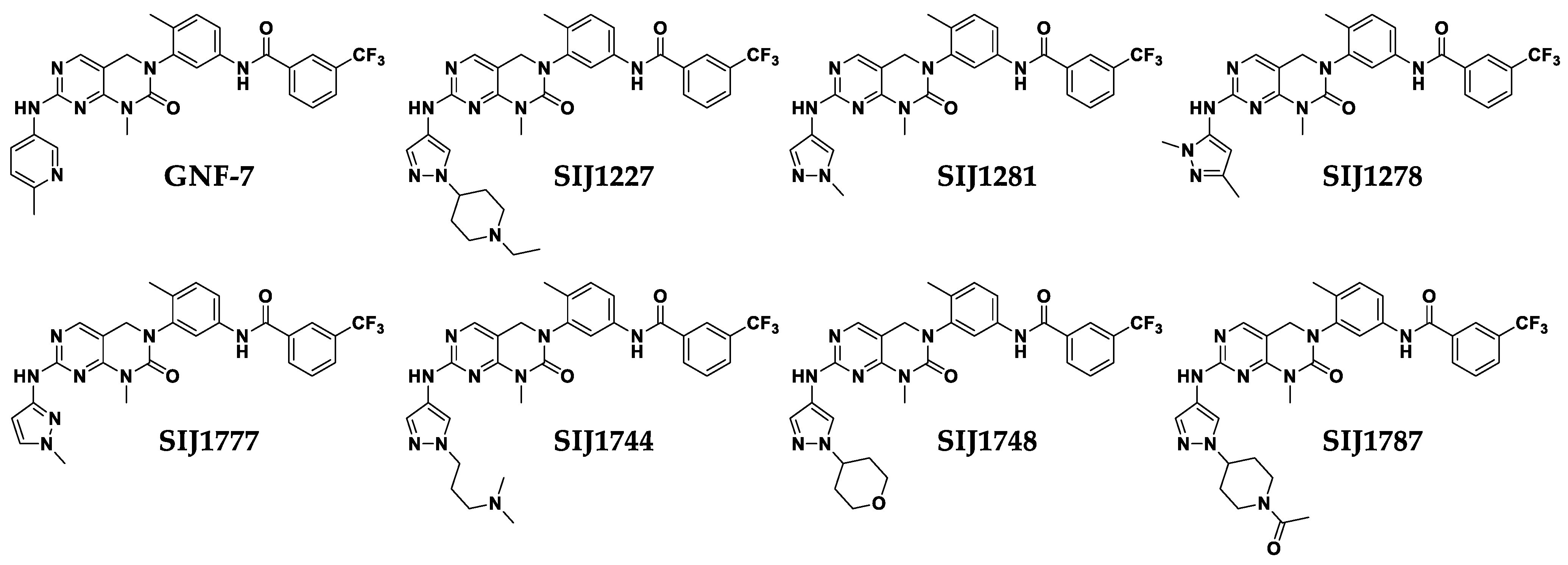
2.2. Six Derivatives Strongly Suppress Proliferation of Melanoma Cells Harboring Class I/II/III BRAF Mutations
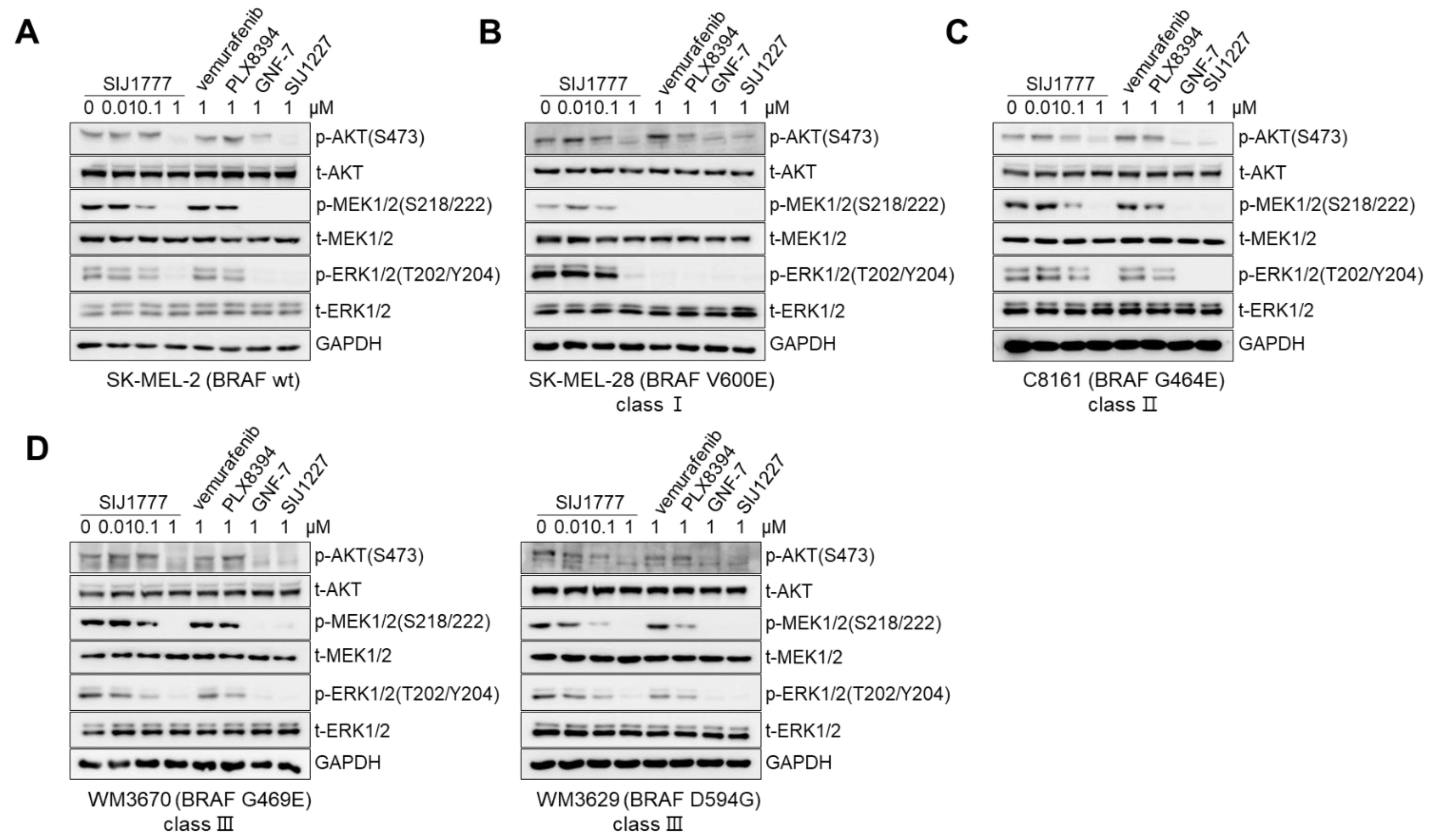
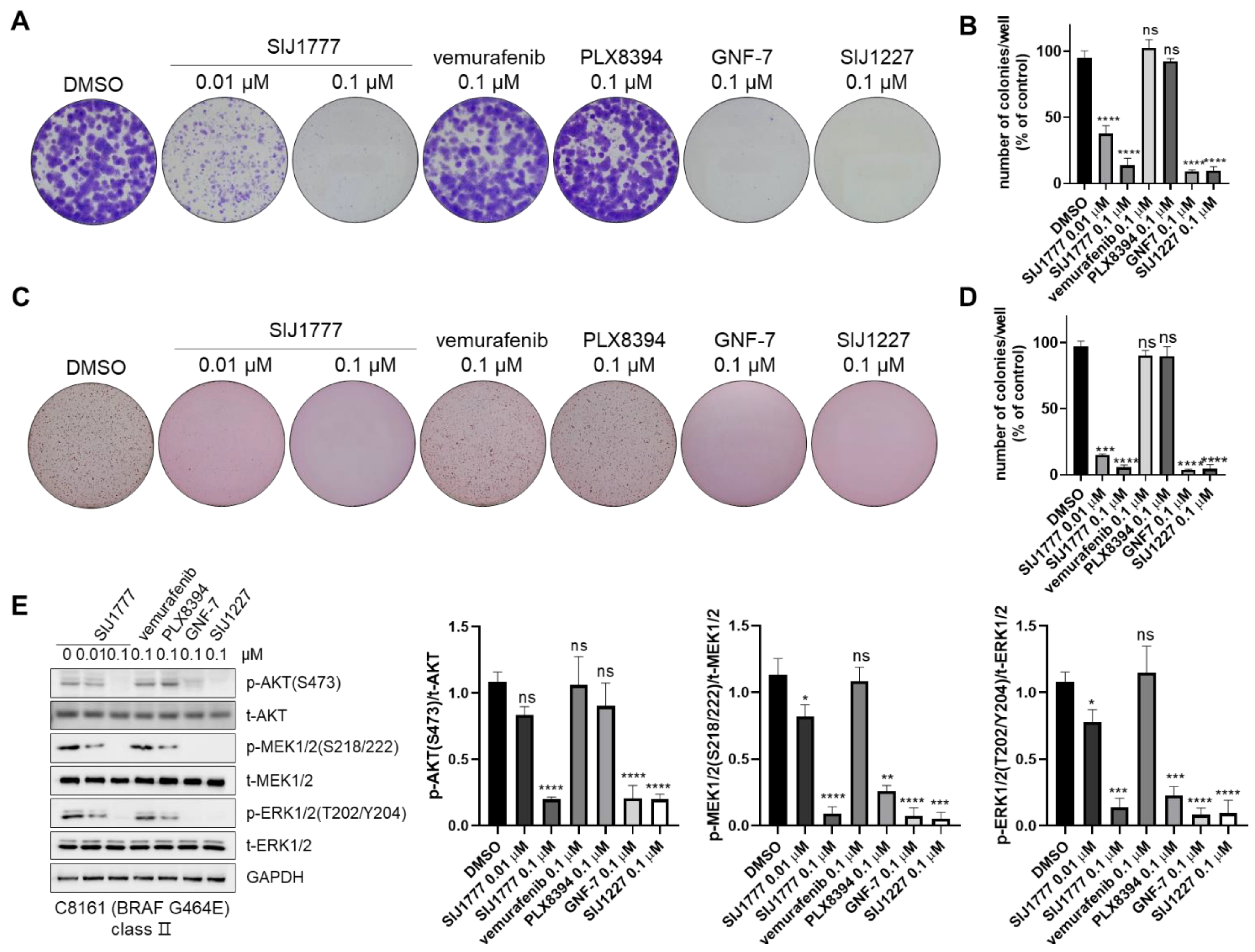
2.3. Effects of SIJ1777 on MAPK/AKT Signaling against Melanoma Cells Harboring BRAF wt or Class I/II/III Mutations
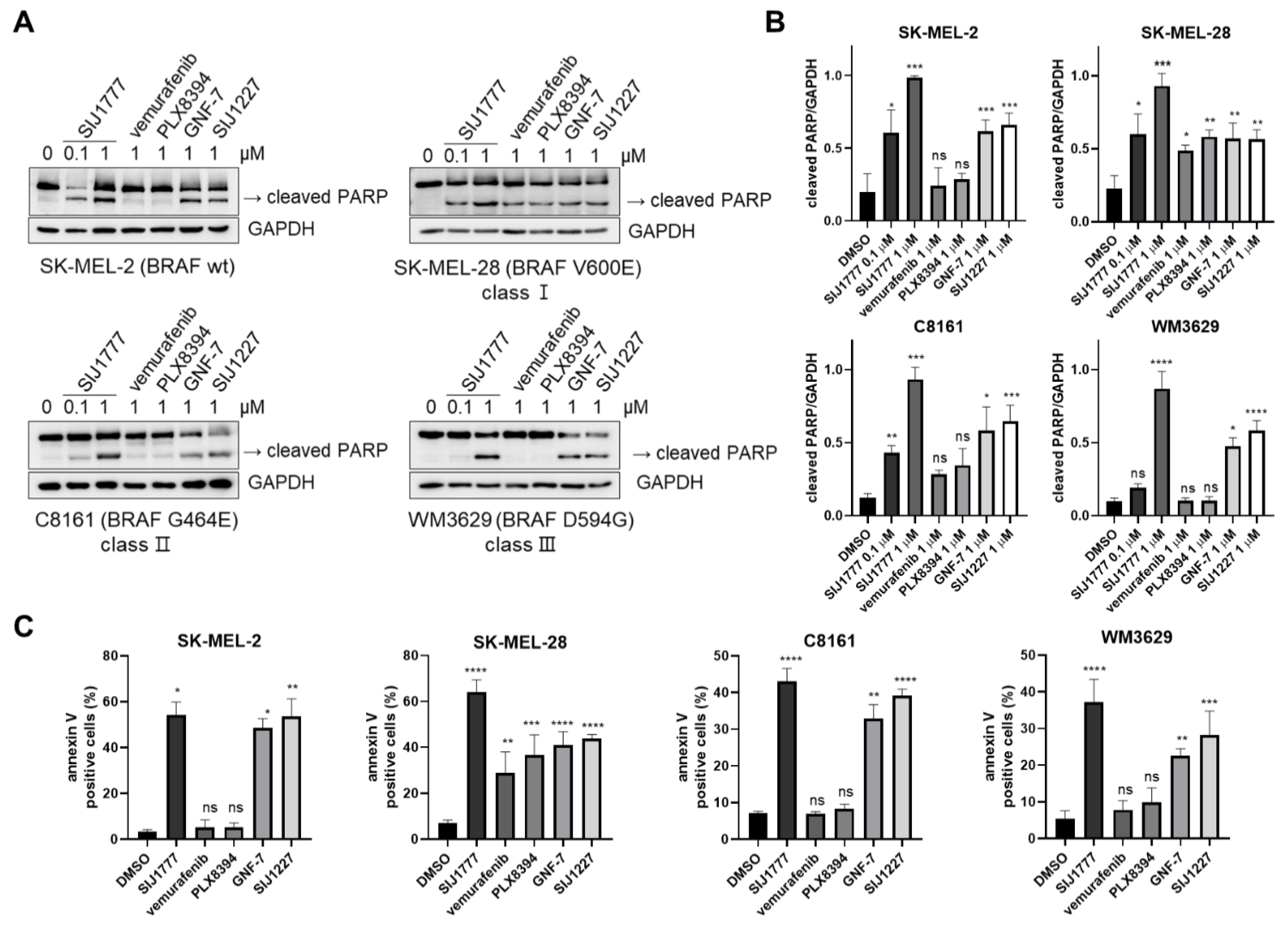
2.4. Effects of SIJ1777 on Apoptosis Induction in Melanoma Cell Lines
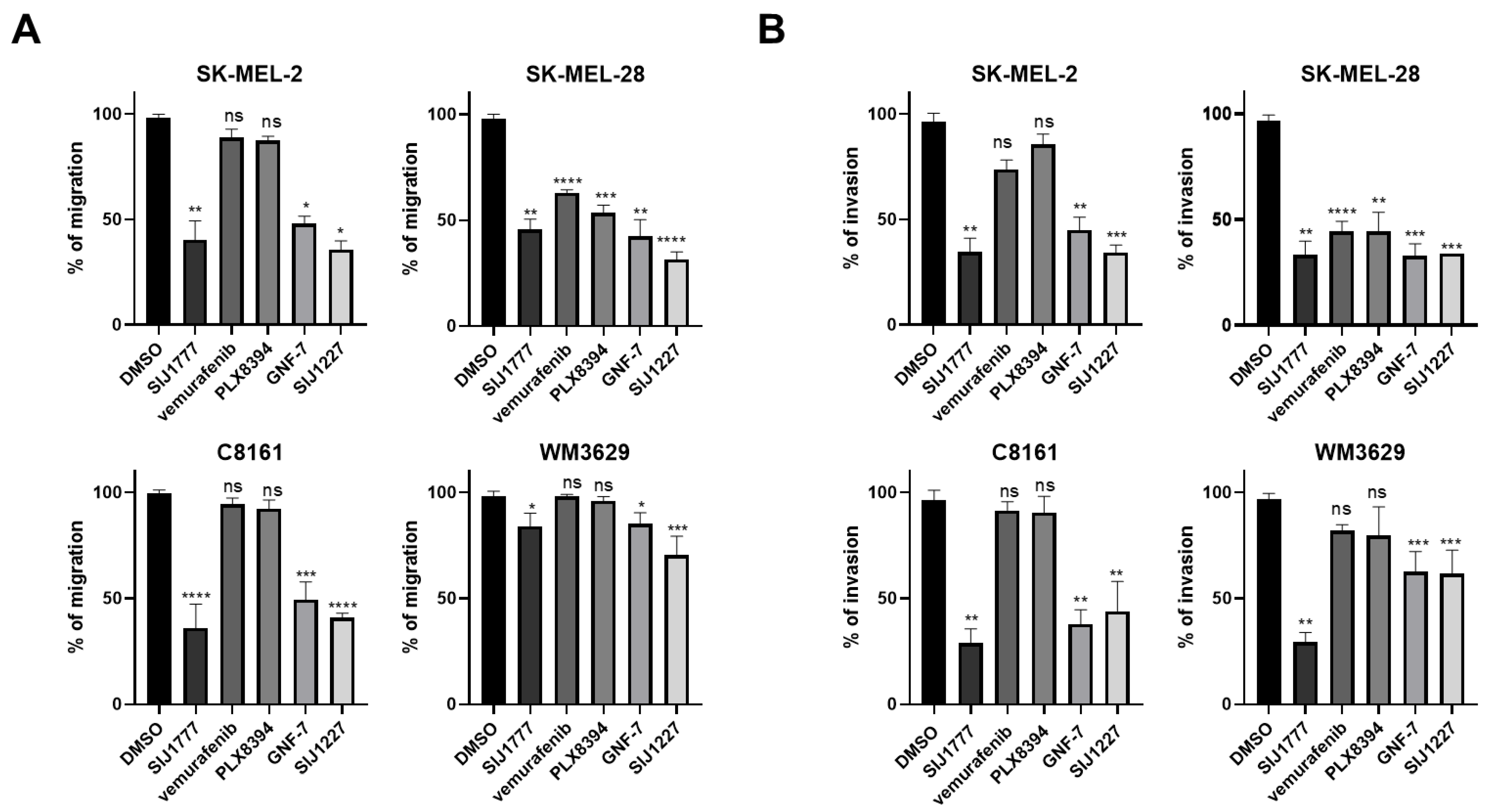
2.5. Effects of SIJ1777 on Cellular Migration and Invasion Abilities in Melanoma Cell Lines
2.6. Colony Formation Inhibitory Activities of SIJ1777
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
N-(3-(7-((1-(1-Ethylpiperidin-4-yl)-1H-pyrazol-4-yl)amino)-1-methyl-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (SIJ1227)
N-(3-(7-((1,3-Dimethyl-1H-pyrazol-5-yl)amino)-1-methyl-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (SIJ1278)
N-(4-Methyl-3-(1-methyl-7-((1-methyl-1H-pyrazol-4-yl)amino)-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)phenyl)-3-(trifluoromethyl)benzamide (SIJ1281)
N-(3-(7-((1-(3-(Dimethylamino)propyl)-1H-pyrazol-4-yl)amino)-1-methyl-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (SIJ1744)
N-(4-Methyl-3-(1-methyl-2-oxo-7-((1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazol-4-yl)amino)-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)phenyl)-3-(trifluoromethyl)benzamide (SIJ1748)
N-(4-Methyl-3-(1-methyl-7-((1-methyl-1H-pyrazol-3-yl)amino)-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)phenyl)-3-(trifluoromethyl)benzamide (SIJ1777)
N-(3-(7-((1-(1-Acetylpiperidin-4-yl)-1H-pyrazol-4-yl)amino)-1-methyl-2-oxo-1,4-dihydropyrimido[4,5-d]pyrimidin-3(2H)-yl)-4-methylphenyl)-3-(trifluoromethyl)benzamide (SIJ1787)
3.1.2. Molecular Docking Study
3.2. Biology
3.2.1. Cell Culture
3.2.2. Anti-Proliferation Assay
3.2.3. Western Blot
3.2.4. Flow Cytometry Analysis
3.2.5. Migration and Invasion Assay
3.2.6. Colony Formation Assay
3.2.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| MAPK | Mitogen-activated protein kinase |
| NSCLC | Non-small cell lung cancer |
| TNBC | Triple negative breast cancer |
| PARP | Poly(ADP-ribose) polymerase |
| Pd2(dba)3 | Tris(dibenzylideneacetone)-dipalladium(0) |
| PI | Propidium iodide |
| Xphos | 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
| DCM | Dichloromethane |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLO-BOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. Published electronically Feb 4. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The growing burden of invasive melanoma: Projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, F.; Lortet-Tieulent, J.; Schüz, J.; Zeeb, H.; Greinert, R.; Breitbart, E.W.; Bray, F. International trends in the incidence of malignant melanoma 1953-2008-are recent generations at higher or lower risk? Int. J. Cancer 2013, 132, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Linos, E.; Swetter, S.M.; Cockburn, M.G.; Colditz, G.A.; Clarke, C.A. Increasing burden of melanoma in the United States. J. Investig. Dermatol. 2009, 129, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Cassano, R.; Cuconato, M.; Calviello, G.; Serini, S.; Trombino, S. Recent advances in nanotechnology for the treatment of melanoma. Molecules 2021, 26, 785. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Castañeda, L.D.; Nova, J.A.; Tovar-Parra, J.D. Frequency of mutations in BRAF, NRAS, and KIT in different populations and histological subtypes of melanoma: A systemic review. Melanoma Res. 2020, 30, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Gao, Y.; Su, W.; Yaeger, R.; Tao, J.; Na, N.; Zhang, Y.; Zhang, C.; Rymar, A.; Tao, A.; et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 2019, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Lajoie, M.; Moldoveanu, D.; Nguyen, T.-T.; Savage, P.; Rajkumar, S.; Huang, X.; Lvova, M.; Protopopov, A.; Vuzman, D.; et al. Dual MAPK inhibition is an effective therapeutic strategy for a subset of class II BRAF mutant melanomas. Clin. Cancer Res. 2018, 24, 6483–6494. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.G.; Ren, P.; Adrian, F.; Sun, F.; Lee, H.S.; Wang, X.; Ding, Q.; Zhang, G.; Xie, Y.; Zhang, J.; et al. A type-II kinase inhibitor capable of inhibiting the T315I “gate-keeper” mutant of Bcr-Abl. J. Med. Chem. 2010, 53, 5439–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Shin, I.; Ju, E.; Choi, S.; Hur, W.; Kim, H.; Hong, E.; Kim, N.D.; Choi, H.G.; Gray, N.S.; et al. First SAR study for overriding NRAS mutant driven acute myeloid leukemia. J. Med. Chem. 2018, 61, 8353–8373. [Google Scholar] [CrossRef] [PubMed]
- Nonami, A.; Sattler, M.; Weisberg, E.; Liu, Q.; Zhang, J.; Patricelli, M.P.; Christie, A.L.; Saur, A.M.; Kohl, N.E.; Kung, A.L.; et al. Identification of novel therapeutic targets in acute leukemias with NRAS mutations using a pharmacologic approach. Blood 2015, 125, 3133–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Shin, I.; Kim, N.; Nam, Y.; Sim, T. The first small molecules capable of strongly suppressing proliferation of cancer cells harboring BRAF class I/II/III mutations. Biochem. Biophys. Res. Commun. 2020, 532, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Sala, E.; Mologni, L.; Truffa, S.; Gaetano, C.; Bollag, G.E.; Gambacorti-Passerini, C. BRAF Silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol. Cancer Res. 2008, 6, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Lassen, A.; Atefi, M.; Robert, L.; Wong, D.J.; Cerniglia, M.; Comin-Anduix, B.; Ribas, A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 2014, 13, 83. [Google Scholar] [CrossRef] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Liu, X.-H.; Wang, K.-M.; Nie, F.-Q.; Kong, R.; Yang, J.-S.; Xia, R.; Xu, T.-P.; Jin, F.-Y.; Liu, Z.-J.; et al. Downregulation of BRAF activated non-coding RNA is associated with poor prognosis for non-small cell lung cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Mol. Cancer 2014, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Mccarty, S.K.; Saji, M.; Zhang, X.; Knippler, C.M.; Kirschner, L.S.; Fernandez, S.; Ringel, M.D. BRAF activates and physically interacts with PAK to regulate cell motility. Endocr. Relat. Cancer 2014, 21, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Liu, S.; Zhang, G.; Kwong, L.N.; Zhu, Y.; Miller, J.P.; Hu, Y.; Zhong, W.; Zeng, J.; Wu, L.; et al. Oncogenic BRAF-mediated melanoma cell invasion. Cell Rep. 2016, 15, 2012–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | GI50 (μM) a | |||||
|---|---|---|---|---|---|---|
| - | Class I | Class I | Class II | Class III | Class III | |
| BRAF wt | BRAF V600E | BRAF V600E | BRAF G464E | BRAF G469E | BRAF D594G | |
| SK-MEL-2 | SK-MEL-28 | A375 | C8161 | WM3670 | WM3629 | |
| vemurafenib | 3.84 ± 0.04 | 0.49 ± 0.01 | 0.18 ± 0.02 | 5.81 ± 0.24 | 13.65 ± 0.89 | 33.10 ± 3.61 |
| PLX8394 | 19.30 ± 2.02 ** | 0.53 ± 0.01 | 0.10 ± 0.04 | 27.30 ± 1.34 ** | 17.86 ± 0.31 | 27.95 ± 2.52 |
| GNF-7 | 0.23 ± 0.08 *** | 0.15 ± 0.02 * | 0.06 ± 0.00 | 0.02 ± 0.00 **** | 0.13 ± 0.01 **** | 0.21 ± 0.00 *** |
| SIJ1227 | 0.05 ± 0.01 ** | 0.05 ± 0.01 ** | 0.04 ± 0.01 | 0.05 ± 0.01 **** | 0.04 ± 0.00 *** | 0.08 ± 0.00 ** |
| SIJ1281 | 0.12 ± 0.01 *** | 0.03 ± 0.01 *** | 0.02 ± 0.00 * | 0.02 ± 0.01 **** | 0.03 ± 0.01 *** | 0.03 ± 0.00 *** |
| SIJ1278 | 0.24 ± 0.03 *** | 0.14 ± 0.02 * | 0.09 ± 0.02 | 0.08 ± 0.01 *** | 0.15 ± 0.00 **** | 0.12 ± 0.00 *** |
| SIJ1777 | 0.02 ± 0.00 *** | 0.04 ± 0.01 ** | 0.03 ± 0.00 * | 0.03 ± 0.01 **** | 0.04 ± 0.00 **** | 0.04 ± 0.00 *** |
| SIJ1744 | 0.12 ± 0.01 *** | 0.08 ± 0.01 ** | 0.02 ± 0.01 * | 0.13 ± 0.06 ** | 0.13 ± 0.00 **** | 0.08 ± 0.02 *** |
| SIJ1748 | 0.06 ± 0.00 ** | 0.02 ± 0.00 ** | 0.02 ± 0.01 * | 0.03 ± 0.01 *** | 0.12 ± 0.01 ** | 0.11 ± 0.02 *** |
| SIJ1787 | 0.03 ± 0.00 ** | 0.15 ± 0.00 * | 0.03 ± 0.01 * | 0.04 ± 0.02 **** | 0.13 ± 0.01 **** | 0.07 ± 0.01 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Shin, I.; Lee, J.; Jeon, E.; Kim, Y.; Ryu, S.; Ju, E.; Cho, W.; Sim, T. Novel and Potent Small Molecules against Melanoma Harboring BRAF Class I/II/III Mutants for Overcoming Drug Resistance. Int. J. Mol. Sci. 2021, 22, 3783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073783
Kim N, Shin I, Lee J, Jeon E, Kim Y, Ryu S, Ju E, Cho W, Sim T. Novel and Potent Small Molecules against Melanoma Harboring BRAF Class I/II/III Mutants for Overcoming Drug Resistance. International Journal of Molecular Sciences. 2021; 22(7):3783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073783
Chicago/Turabian StyleKim, Namkyoung, Injae Shin, Jiwon Lee, Eunhye Jeon, Younghoon Kim, Seongshick Ryu, Eunhye Ju, Wonjeong Cho, and Taebo Sim. 2021. "Novel and Potent Small Molecules against Melanoma Harboring BRAF Class I/II/III Mutants for Overcoming Drug Resistance" International Journal of Molecular Sciences 22, no. 7: 3783. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073783