
Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Metabolism of Fatty Acids
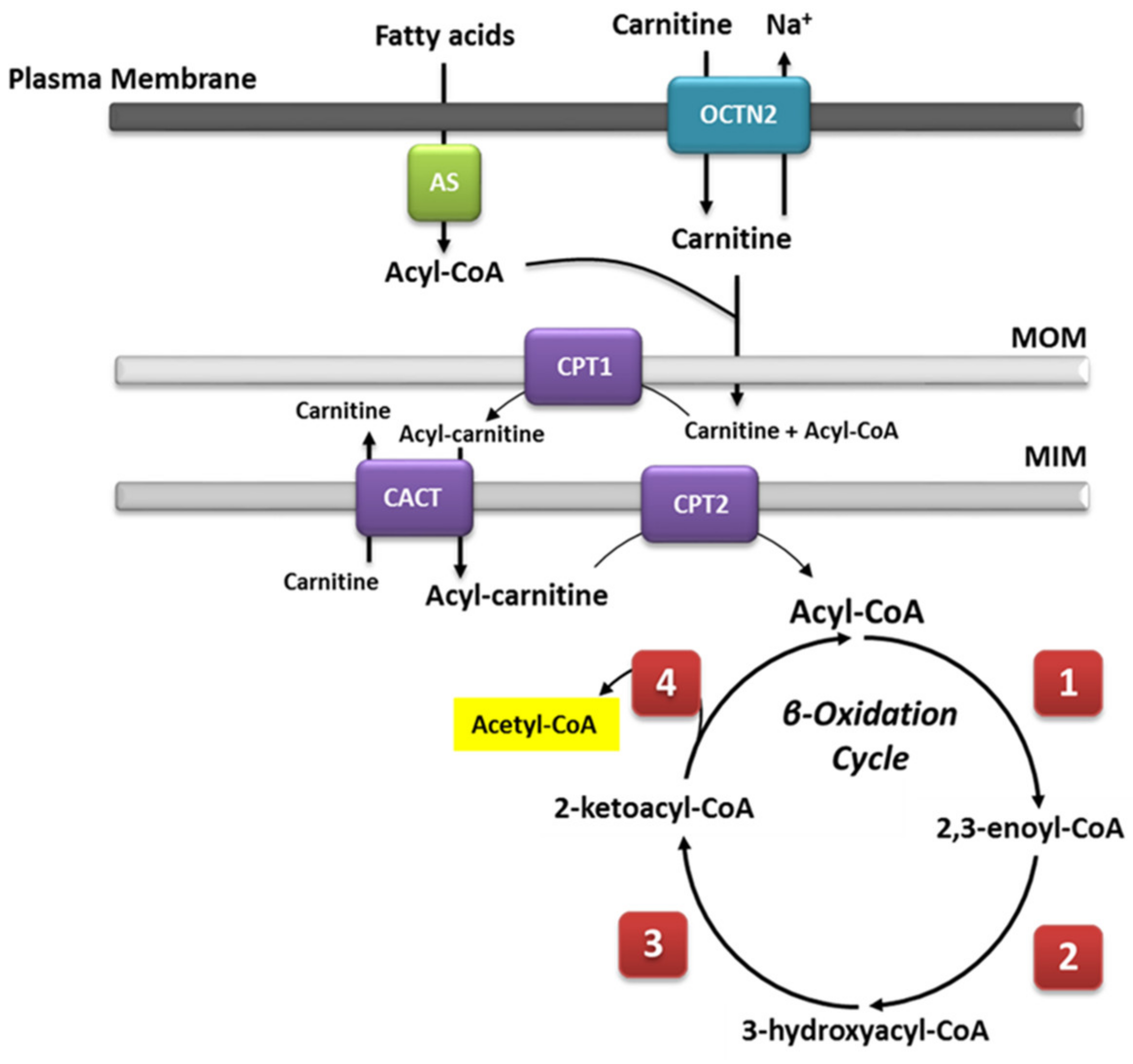
2.1. Mitochondrial β-Oxidation
2.2. Very Long-Chain Acyl-CoA Dehydrogenase Deficiency (VLCADD)
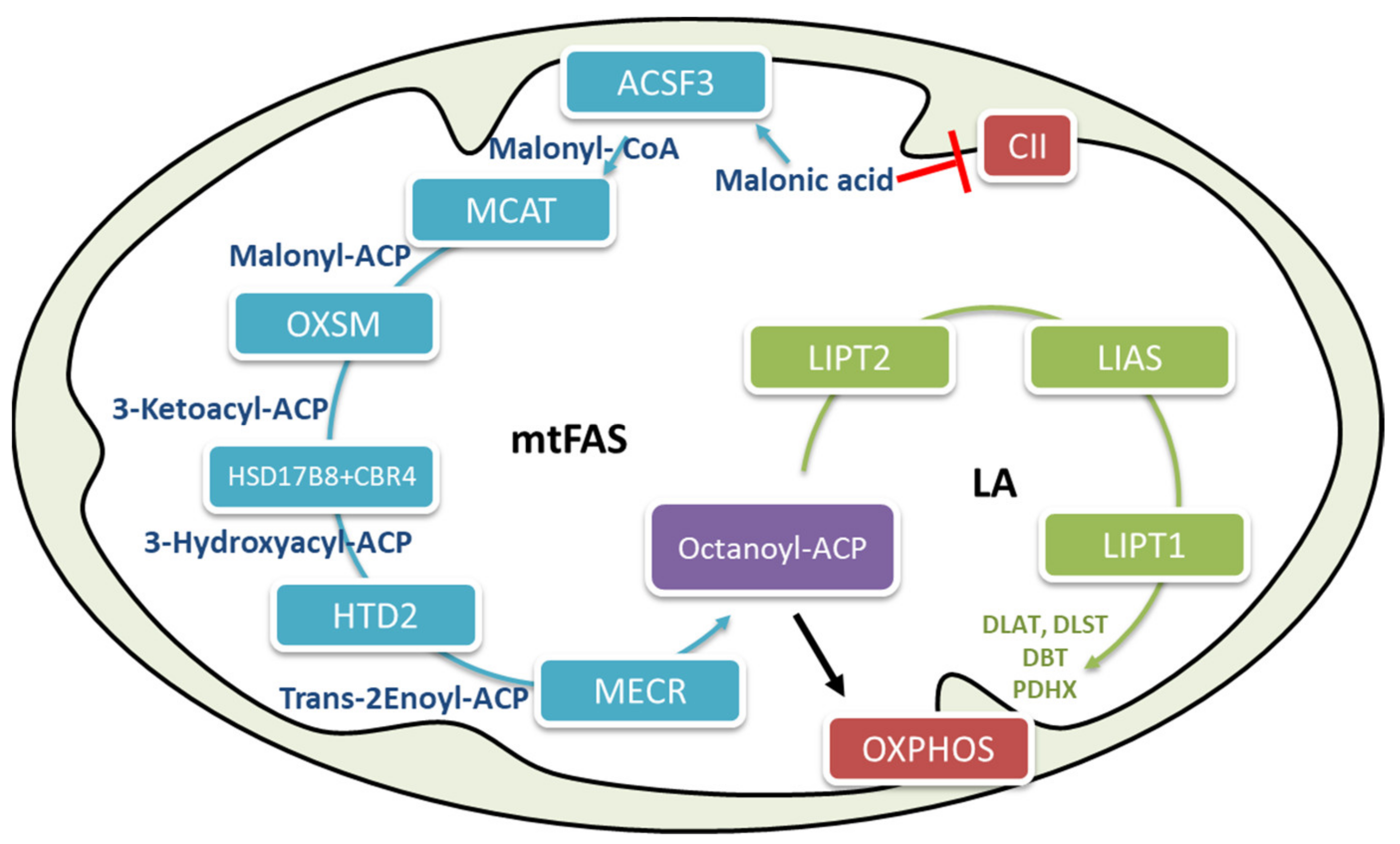
2.3. Mitochondrial Fatty Acid Biosynthesis
2.4. Metabolic Flexibility and Interconnection between Mitochondrial β-Oxidation and Fatty Acid Biosynthesis
2.5. The Effect of Diet and Sex on Metabolic Flexibility in VLCAD Deficiency
2.6. The Biological Role of Mitochondrial Fatty Acid Biosynthesis (mtFAS) in Energy Regulation
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, J.A.; Hargreaves, M.; Joyner, M.J.; Zierath, J.R. Integrative Biology of Exercise. Cell 2014, 159, 738–749. [Google Scholar] [CrossRef] [Green Version]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac Metabolism and its Interactions with Contraction, Growth, and Survival of Cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Pearson, T.; Wattis, J.A.; King, J.R.; Macdonald, I.A.; Mazzatti, D.J. A Mathematical Model of the Human Metabolic System and Metabolic Flexibility. Bull. Math. Biol. 2014, 76, 2091–2121. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.P.; Foletta, V.C.; Snow, R.J.; Wadley, G.D. Skeletal muscle mitochondria: A major player in exercise, health and disease. Biochim. Biophys. Acta 2014, 1840, 1276–1284. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [Green Version]
- Diekman, E.F.; Visser, G.; Schmitz, J.P.J.; Nievelstein, R.A.J.; de Sain van der Velden, M.; Wardrop, M.; Van Der Pol, W.L.; Houten, S.M.; Van Riel, N.A.W.; Takken, T.; et al. Altered Energetics of Exercise Explain Risk of Rhabdomyolysis in Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. PLoS ONE 2016, 11, 7818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, S.; Flögel, U.; Hermann, S.; Sturm, M.; Schäfers, M.; Spiekerkoetter, U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice. Biochim. Biophys. Acta (BBA) 2014, 1842, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, F.V.; Ruiter, J.P.N.; Ijlst, L.; De Almeida, I.T.; Wanders, R.J.A. Lactic acidosis in long-chain fatty acid β-oxidation disorders. J. Inherit. Metab. Dis. 1998, 21, 645–654. [Google Scholar] [CrossRef]
- Wakabayashi, M.; Kamijo, Y.; Nakajima, T.; Tanaka, N.; Sugiyama, E.; Yangyang, T.; Kimura, T.; Aoyama, T. Fatty Acid Accumulation and Resulting PPARαActivation in Fibroblasts due to Trifunctional Protein Deficiency. PPAR Res. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Palmfeldt, J.; Mohsen, A.-W.; Gregersen, N.; Vockley, J. Fasting induces prominent proteomic changes in liver in very long chain Acyl-CoA dehydrogenase deficient mice. Biochem. Biophys. Rep. 2016, 8, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Christianson, J.; Liu, Z.-X.; Tian, L.; Choi, C.S.; Neschen, S.; Dong, J.; Wood, P.A.; Shulman, G.I. Resistance to High-Fat Diet-Induced Obesity and Insulin Resistance in Mice with Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Cell Metab. 2010, 11, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Holloway, G.P.; Lally, J.; Nickerson, J.G.; Alkhateeb, H.; Snook, L.A.; Heigenhauser, G.J.F.; Calles-Escandon, J.; Glatz, J.F.C.; Luiken, J.J.F.P.; Spriet, L.L.; et al. Fatty acid binding protein facilitates sarcolemmal fatty acid transport but not mitochondrial oxidation in rat and human skeletal muscle. J. Physiol. 2007, 582, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Berk, P.; Stump, D. Mechanisms of cellular uptake of long chain free fatty acids. Mol. Cell. Biochem. 1999, 192, 17–31. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Shoukry, K.; Schulz, H. Significance of the Reductase-dependent Pathway for the β-Oxidation of Unsaturated Fatty Acids with Odd-numbered Double Bonds. J. Biol. Chem. 1998, 273, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.; Vreken, P.; den Boer, M.E.; Wijburg, F.A.; van Gennip, A.H.; Ijist, L. Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J. Inherit. Metab. Dis. 1999, 22, 442–487. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, U. Mitochondrial fatty acid oxidation disorders: Clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J. Inherit. Metab. Dis. 2010, 33, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, M.B.; Heitner, S.B.; Martin, J.; Rose, S.; Goldstein, A.; El-Gharbawy, A.H.; DeWard, S.; Lasarev, M.R.; Pollaro, J.; Delany, J.P.; et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 2017, 40, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.K.; Scott, A.I.; Sullivan, S.; Chang, I.J.; Lam, C.; Sun, A.; Hahn, S.; Thies, J.M.; Gunnarson, M.; McKean, K.N.; et al. Tutorial: Triheptanoin and Nutrition Management for Treatment of Long-Chain Fatty Acid Oxidation Disorders. J. Parenter. Enter. Nutr. 2020, 45, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Chapman, K.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; et al. Effects of triheptanoin (UX007) in patients with long-chain fatty acid oxidation disorders: Results from an open-label, long-term extension study. J. Inherit. Metab. Dis. 2020, 44, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.B.; Hamm, D.A.; Millington, D.S.; Matern, D.; Vockley, J.; Rinaldo, P.; Pinkert, C.A.; Rhead, W.J.; Lindsey, J.R.; Wood, P.A. Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet. 2001, 10, 2069–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exil, V.J.; Roberts, R.L.; Sims, H.; McLaughlin, J.E.; Malkin, R.A.; Gardner, C.D.; Ni, G.; Rottman, J.N.; Strauss, A.W. Very-Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency in Mice. Circ. Res. 2003, 93, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatibi, K.I.; Wehbe, Z.; Spiekerkoetter, U.; Tucci, S. Sex-specific perturbation of complex lipids in response to medium-chain fatty acids in very long-chain acyl-CoA dehydrogenase deficiency. FEBS J. 2020, 287, 3511–3525. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S. Very long-chain acyl-CoA dehydrogenase (VLCAD-) deficiency–studies on treatment effects and long-term outcomes in mouse models. J. Inherit. Metab. Dis. 2017, 40, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S.; Behringer, S.; Spiekerkoetter, U. De novo fatty acid biosynthesis and elongation in very long-chain acyl-CoA dehydrogenase- (VLCAD) deficient mice supplemented with odd or even medium-chain fatty acids. FEBS J. 2015, 282, 4242–4253. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Floegel, U.; Beermann, F.; Behringer, S.; Spiekerkoetter, U. Triheptanoin: Long-term effects in the very long-chain acyl-CoA dehydrogenase (VLCAD−/−)-deficient mouse. J. Lipid Res. 2017, 58, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, S.; Flögel, U.; Spiekerkoetter, U. Sexual dimorphism of lipid metabolism in very long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice in response to medium-chain triglycerides (MCT). Biochim. Biophys. Acta 2015, 1852, 1442–1450. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Flögel, U.; Sturm, M.; Borsch, E.; Spiekerkoetter, U. Disrupted fat distribution and composition due to medium-chain triglycerides in mice with a β-oxidation defect. Am. J. Clin. Nutr. 2011, 94, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Herebian, D.; Sturm, M.; Seibt, A.; Spiekerkoetter, U. Tissue-Specific Strategies of the Very-Long Chain Acyl-CoA Dehydrogenase-Deficient (VLCAD−/−) Mouse to Compensate a Defective Fatty Acid β-Oxidation. PLoS ONE 2012, 7, e45429. [Google Scholar] [CrossRef]
- Tucci, S.; Mingirulli, N.; Wehbe, Z.; Dumit, V.I.; Kirschner, J.; Spiekerkoetter, U. Mitochondrial fatty acid biosynthesis and muscle fiber plasticity in very long-chain acyl-CoA dehydrogenase-deficient mice. FEBS Lett. 2018, 592, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Pearson, S.; Herebian, D.; Spiekerkoetter, U. Long-term dietary effects on substrate selection and muscle fiber type in very-long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice. Biochim. Biophys. Acta 2013, 1832, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Primassin, S.; Spiekerkoetter, U. Fasting-induced oxidative stress in very long chain acyl-CoA dehydrogenase-deficient mice. FEBS J. 2010, 277, 4699–4708. [Google Scholar] [CrossRef]
- Tucci, S.; Primassin, S.; Ter Veld, F.; Spiekerkoetter, U. Medium-chain triglycerides impair lipid metabolism and induce hepatic steatosis in very long-chain acyl-CoA dehydrogenase (VLCAD)-deficient mice. Mol. Genet. Metab. 2010, 101, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, Z.; Alatibi, K.; Jellusova, J.; Spiekerkoetter, U.; Tucci, S. The fate of medium-chain fatty acids in very long-chain acyl-CoA dehydrogenase deficiency (VLCADD): A matter of sex? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Kastaniotis, A.J.; Autio, K.J.; Kerätär, J.M.; Monteuuis, G.; Mäkelä, A.M.; Nair, R.R.; Pietikäinen, L.P.; Shvetsova, A.; Chen, Z.; Hiltunen, J.K. Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Airenne, T.T.; Torkko, J.M.; Van den Plas, S.; Sormunen, R.T.; Kastaniotis, A.J.; Wierenga, R.K.; Hiltunen, J.K. Structure–function Analysis of Enoyl Thioester Reductase Involved in Mitochondrial Maintenance. J. Mol. Biol. 2003, 327, 47–59. [Google Scholar] [CrossRef]
- Autio, K.J.; Kastaniotis, A.J.; Pospiech, H.; Miinalainen, I.J.; Schonauer, M.S.; Dieckmann, C.L.; Hiltunen, J.K. An ancient genetic link between vertebrate mitochondrial fatty acid synthesis and RNA processing. FASEB J. 2007, 22, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Leskinen, H.; Liimatta, E.; Sormunen, R.T.; Miinalainen, I.J.; Hassinen, I.E.; Hiltunen, J.K. Myocardial Overexpression of Mecr, a Gene of Mitochondrial FAS II Leads to Cardiac Dysfunction in Mouse. PLoS ONE 2009, 4, 5589. [Google Scholar] [CrossRef] [Green Version]
- Runswick, M.J.; Fearnley, I.M.; Skehel, J.; Walker, J.E. Presence of an acyl carrier protein in NADH:ubiquinone oxidoreductase from bovine heart mitochondria. FEBS Lett. 1991, 286, 121–124. [Google Scholar] [CrossRef]
- Sackmann, U.; Zensen, R.; Rohlen, D.; Jahnke, U.; Weiss, H. The acyl-carrier protein in Neurospora crassa mitochondria is a subunit of NADH: Ubiquinone reductase (complex I). Eur. J. Biochem. 1991, 200, 463–469. [Google Scholar] [CrossRef]
- Cronan, J.E. Progress in the Enzymology of the Mitochondrial Diseases of Lipoic Acid Requiring Enzymes. Front. Genet. 2020, 11, 510. [Google Scholar] [CrossRef]
- Mayr, J.A.; Feichtinger, R.G.; Tort, F.; Ribes, A.; Sperl, W. Lipoic acid biosynthesis defects. J. Inherit. Metab. Dis. 2014, 37, 553–563. [Google Scholar] [CrossRef]
- Sloan, J.L.; Johnston, J.J.; Manoli, I.; Chandler, R.J.; Krause, C.; Carrillo-Carrasco, N.; Chandrasekaran, S.D.; Sysol, J.R.; O’Brien, K.; Nauser, N.S.; et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat. Genet. 2011, 43, 883–886. [Google Scholar] [CrossRef] [Green Version]
- Alfares, A.; Nunez, L.D.; Al-Thihli, K.; Mitchell, J.; Melançon, S.; Anastasio, N.; Ha, K.C.H.; Majewski, J.; Rosenblatt, D.S.; Braverman, N. Combined malonic and methylmalonic aciduria: Exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. J. Med. Genet. 2011, 48, 602–605. [Google Scholar] [CrossRef] [Green Version]
- Levtova, A.; Waters, P.J.; Buhas, D.; Lévesque, S.; Auray-Blais, C.; Clarke, J.T.; LaFramboise, R.; Maranda, B.; Mitchell, G.A.; Brunel-Guitton, C.; et al. Combined malonic and methylmalonic aciduria due to ACSF3 mutations: Benign clinical course in an unselected cohort. J. Inherit. Metab. Dis. 2019, 42, 107–116. [Google Scholar] [CrossRef]
- Monteuuis, G.; Suomi, F.; Kerätär, J.M.; Masud, A.J.; Kastaniotis, A.J. A conserved mammalian mitochondrial isoform of acetyl-CoA carboxylase ACC1 provides the malonyl-CoA essential for mitochondrial biogenesis in tandem with ACSF3. Biochem. J. 2017, 474, 3783–3797. [Google Scholar] [CrossRef] [Green Version]
- Reinders, J.; Rozemuller, E.H.; Van Der Weide, P.; Oka, A.; Slootweg, P.J.; Inoko, H.; Tilanus, M.G. Genes in the HLA region indicative for head and neck squamous cell carcinoma. Mol. Immunol. 2007, 44, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, S.; Minegishi, Y.; Suga, A.; Yoshitake, K.; Sheng, X.; Ye, J.; Smith, S.; Bunkoczi, G.; Yamamoto, M.; et al. Novel mutations in malonyl-CoA-acyl carrier protein transacylase provoke autosomal recessive optic neuropathy. Hum. Mol. Genet. 2020, 29, 444–458. [Google Scholar] [CrossRef]
- Habarou, F.; Hamel, Y.; Haack, T.B.; Feichtinger, R.G.; Lebigot, E.; Marquardt, I.; Busiah, K.; Laroche, C.; Madrange, M.; Grisel, C.; et al. Biallelic Mutations in LIPT2 Cause a Mitochondrial Lipoylation Defect Associated with Severe Neonatal Encephalopathy. Am. J. Hum. Genet. 2017, 101, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.D.; Kraemer, R.R.; Nelson, A.G. Metabolic Dysfunction in Diabetic Offspring: Deviations in Metabolic Flexibility. Med. Sci. Sports Exerc. 2013, 45, 8–15. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.-A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. Metab. 1999, 277, e1130–e1141. [Google Scholar] [CrossRef]
- Tsugane, S.; Inoue, M. Insulin resistance and cancer: Epidemiological evidence. Cancer Sci. 2010, 101, 1073–1079. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Scicluna, B.P.; Arts, R.J.W.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.L.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M. Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallberg-Henriksson, H.; Zierath, J.R. Exercise remodels subcutaneous fat tissue and improves metabolism. Nat. Rev. Endocrinol. 2015, 11, 198–200. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK—Sensing Energy while Talking to Other Signaling Pathways. Cell Metab. 2014, 20, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Dubé, J.J.; Coen, P.M.; DiStefano, G.; Chacon, A.C.; Helbling, N.L.; Desimone, M.E.; Stafanovic-Racic, M.; Hames, K.C.; Despines, A.A.; Toledo, F.G.S.; et al. Effects of acute lipid overload on skeletal muscle insulin resistance, metabolic flexibility, and mitochondrial performance. Am. J. Physiol. Metab. 2014, 307, e1117–e1124. [Google Scholar] [CrossRef] [Green Version]
- Poussin, C.; Ibberson, M.; Hall, D.; Ding, J.; Soto, J.; Abel, E.D.; Thorens, B. Oxidative Phosphorylation Flexibility in the Liver of Mice Resistant to High-Fat Diet-Induced Hepatic Steatosis. Diabetes 2011, 60, 2216–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehat, I.; Molkentin, J.D. Molecular Pathways Underlying Cardiac Remodeling During Pathophysiological Stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, H.M. Response of the fetal heart to changes in load: From hyperplasia to heart failure. Heart 2005, 91, 871–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, Z.; Behringer, S.; Alatibi, K.; Watkins, D.; Rosenblatt, D.; Spiekerkoetter, U.; Tucci, S. The emerging role of the mitochondrial fatty-acid synthase (mtFASII) in the regulation of energy metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1629–1643. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Nowinski, S.M.; Clowers, K.J.; Jeong, M.-Y.; Ouyang, Y.; Berg, J.A.; Gygi, J.P.; Gygi, S.P.; Winge, D.R.; Rutter, J. ACP Acylation Is an Acetyl-CoA-Dependent Modification Required for Electron Transport Chain Assembly. Mol. Cell 2018, 71, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, S. Brain metabolism and neurological symptoms in combined malonic and methylmalonic aciduria. Orphanet J. Rare Dis. 2020, 15, 1–3. [Google Scholar] [CrossRef]
- Heimer, G.; Kerätär, J.M.; Riley, L.G.; Balasubramaniam, S.; Eyal, E.; Pietikäinen, L.P.; Hiltunen, J.K.; Marek-Yagel, D.; Hamada, J.; Gregory, A.; et al. MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder. Am. J. Hum. Genet. 2016, 99, 1229–1244. [Google Scholar] [CrossRef] [Green Version]
- Bowman, C.E.; Wolfgang, M.J. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv. Biol. Regul. 2019, 71, 34–40. [Google Scholar] [CrossRef]
- Kursu, V.A.S.; Pietikäinen, L.P.; Fontanesi, F.; Aaltonen, M.J.; Suomi, F.; Nair, R.R.; Schonauer, M.S.; Dieckmann, C.L.; Barrientos, A.; Hiltunen, J.K.; et al. Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis inSaccharomyces cerevisiae. Mol. Microbiol. 2013, 90, 824–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, L.; Cadenas, E. Lipoic acid: Energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 2010, 48, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrad, D.; Somwar, R.; Sweeney, G.; Yaworsky, K.; Hayashi, M.; Ramlal, T.; Klip, A. The antihy-perglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: Potential role of p38 mitogen-activated protein kinase in GLUT4 activation. Diabetes 2001, 50, 1464–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular Mechanisms and Clinical Applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, G.M.; Zheng, D.; Hickner, R.C.; Houmard, J.A. Effect of exercise training on metabolic flexibility in response to a high-fat diet in obese individuals. Am. J. Physiol. Metab. 2012, 303, e1440–e1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pr. 2011, 93 (Suppl. S1), S52–S59. [Google Scholar] [CrossRef]
- Lemarié, F.; Beauchamp, E.; Legrand, P.; Rioux, V. Revisiting the metabolism and physiological functions of caprylic acid (C8:0) with special focus on ghrelin octanoylation. Biochimie 2016, 120, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Guevara, R.; Santandreu, F.M.; Valle, A.; Gianotti, M.; Oliver, J.; Roca, P. Sex-dependent differences in aged rat brain mitochondrial function and oxidative stress. Free Radic. Biol. Med. 2009, 46, 169–175. [Google Scholar] [CrossRef]
- Sanz, A.; Hiona, A.; Kujoth, G.C.; Seo, A.Y.; Hofer, T.; Kouwenhoven, E.; Kalani, R.; Prolla, T.A.; Barja, G.; Leeuwenburgh, C. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp. Gerontol. 2007, 42, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Sbert-Roig, M.; Bauzá-Thorbrügge, M.; Galmés-Pascual, B.M.; Capllonch-Amer, G.; García-Palmer, F.J.; Lladó, I.; Proenza, A.M.; Gianotti, M. GPER mediates the effects of 17β-estradiol in cardiac mitochondrial biogenesis and function. Mol. Cell. Endocrinol. 2016, 420, 116–124. [Google Scholar] [CrossRef] [PubMed]
- McNab, B.K. On the Utility of Uniformity in the Definition of Basal Rate of Metabolism. Physiol. Zool. 1997, 70, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Sastre, J.; García-Sala, D.; Lloret, A.; Pallardó, F.V.; Viña, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic. Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef]
- Lamming, D.W.; Sabatini, D.M. A Central Role for mTOR in Lipid Homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberato, M.V.; Nascimento, A.S.; Ayers, S.D.; Lin, J.Z.; Cvoro, A.; Silveira, R.L.; Martínez, L.; Souza, P.C.T.; Saidemberg, D.; Deng, T.; et al. Medium Chain Fatty Acids Are Selective Peroxisome Proliferator Activated Receptor (PPAR) γ Activators and Pan-PPAR Partial Agonists. PLoS ONE 2012, 7, 6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, W.F.; Garg, S.; Zimorski, V. Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140330. [Google Scholar] [CrossRef] [Green Version]
- Hiltunen, J.K.; Autio, K.J.; Schonauer, M.S.; Kursu, V.S.; Dieckmann, C.L.; Kastaniotis, A.J. Mitochondrial fatty acid synthesis and respiration. Biochim. Biophys. Acta 2010, 1797, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Bowman, C.E.; Rodriguez, S.; Alpergin, E.S.S.; Acoba, M.G.; Zhao, L.; Hartung, T.; Claypool, S.M.; Watkins, P.A.; Wolfgang, M.J. The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem. Biol. 2017, 24, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Parodi-Rullán, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Baker, P.R., II; Friederich, M.W.; Swanson, M.A.; Shaikh, T.; Bhattacharya, K.; Scharer, G.H.; Aicher, J.; Creadon-Swindell, G.; Geiger, E.; MacLean, K.N.; et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 2014, 137, 366–379. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Zimmermann, F.A.; Fauth, C.; Bergheim, C.; Meierhofer, D.; Radmayr, D.; Zschocke, J.; Koch, J.; Sperl, W. Lipoic Acid Synthetase Deficiency Causes Neonatal-Onset Epilepsy, Defective Mitochondrial Energy Metabolism, and Glycine Elevation. Am. J. Hum. Genet. 2011, 89, 792–797. [Google Scholar] [CrossRef] [Green Version]
- Tort, F.; Ferrer-Cortès, X.; Thió, M.; Navarro-Sastre, A.; Matalonga, L.; Quintana, E.; Bujan, N.; Arias, A.; García-Villoria, J.; Acquaviva, C.; et al. Mutations in the lipoyltransferase LIPT1 gene cause a fatal disease associated with a specific lipoylation defect of the 2-ketoacid dehydrogenase complexes. Hum. Mol. Genet. 2014, 23, 1907–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.-Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 2017, 52, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.L.N.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucci, S.; Alatibi, K.I.; Wehbe, Z. Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 3799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073799
Tucci S, Alatibi KI, Wehbe Z. Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism. International Journal of Molecular Sciences. 2021; 22(7):3799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073799
Chicago/Turabian StyleTucci, Sara, Khaled Ibrahim Alatibi, and Zeinab Wehbe. 2021. "Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism" International Journal of Molecular Sciences 22, no. 7: 3799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073799