
BH3 Mimetic Sensitivity of Colorectal Cancer Cell Lines in Correlation with Molecular Features Identifies Predictors of Response

Abstract
:1. Introduction
2. Results
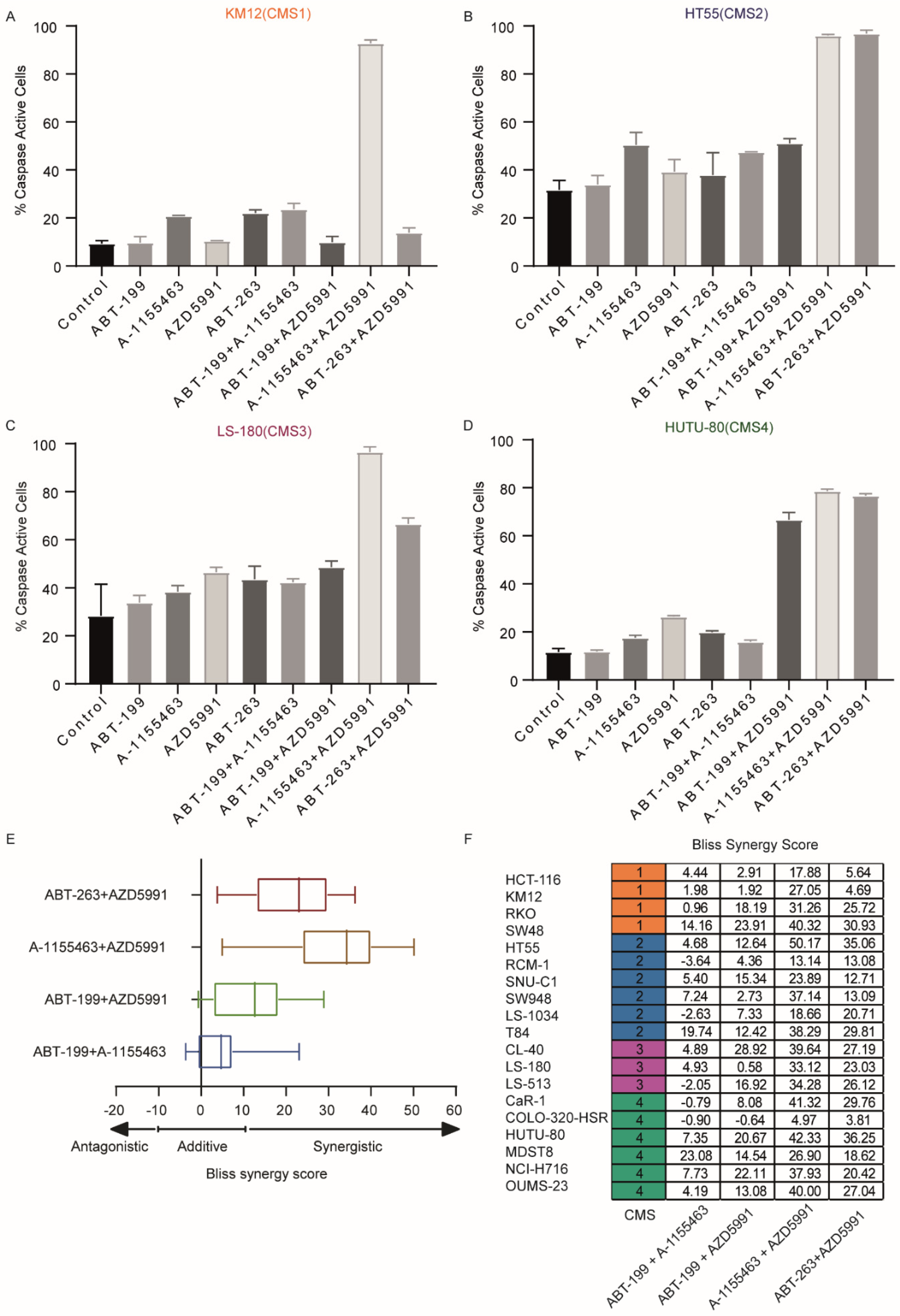
2.1. CMSs Exhibit Differential Sensitivity to BH3 Mimetics
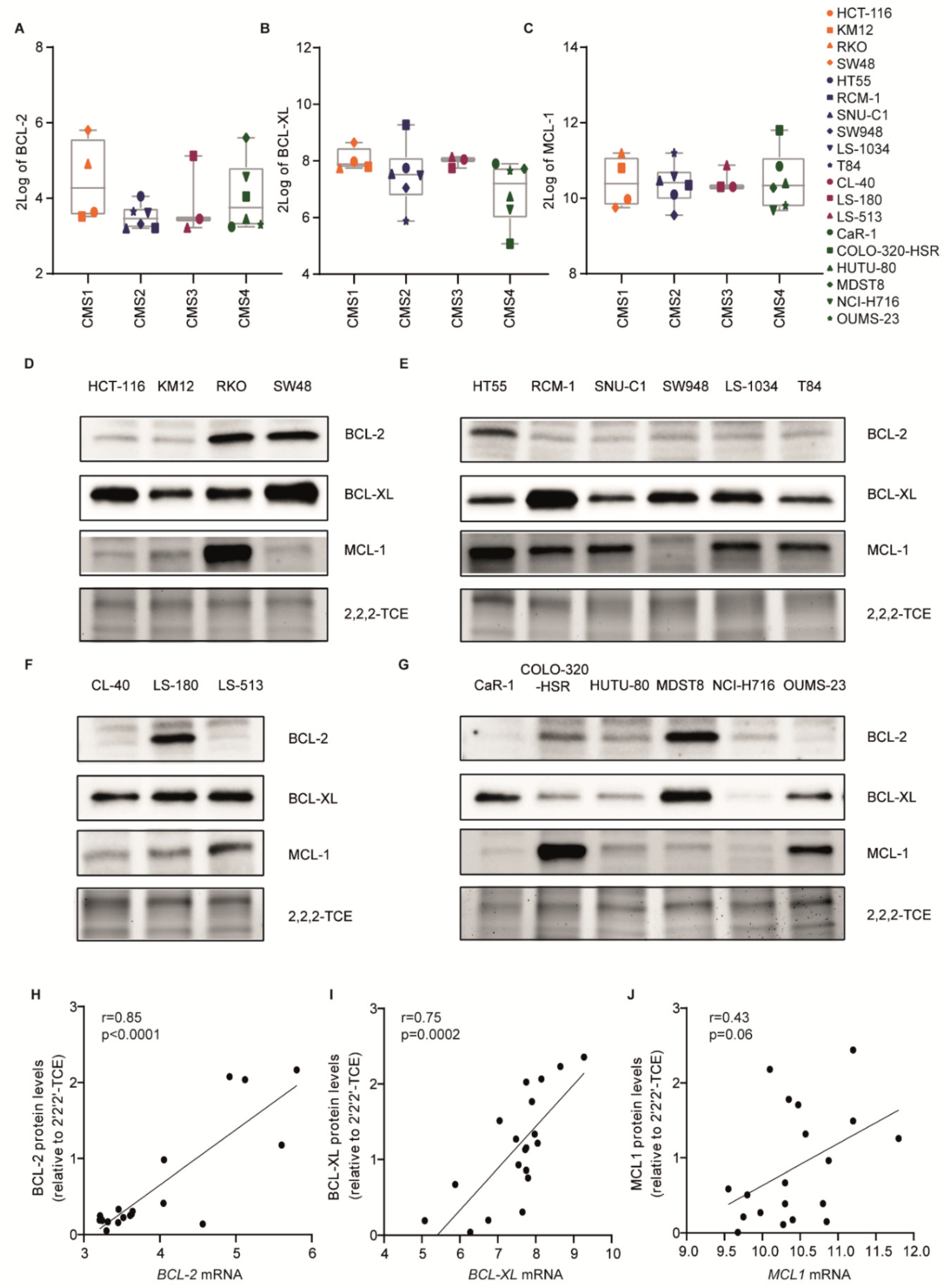
2.2. Sensitivity to BH3 Mimetics Is Not Determined by Expression of the Individual Anti-Apoptotic BCL-2 Family Members
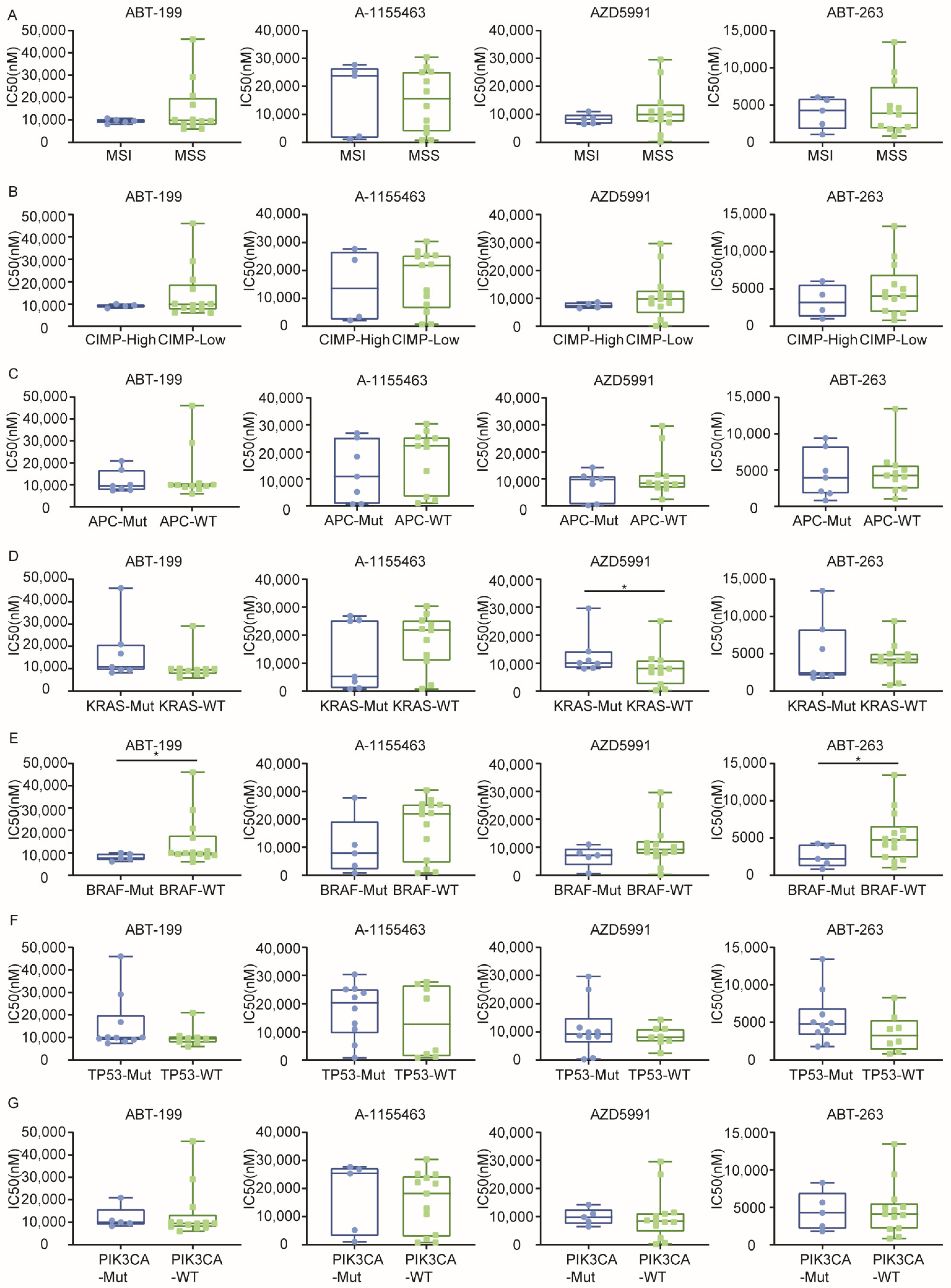
2.3. Sensitivity to BH3 Mimetics Correlates with the KRAS/BRAF Mutation Status
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Profiling
4.2. Compounds
4.3. Cell Viability Assay
4.4. Flow Cytometry and CaspaTag Staining
4.5. Bliss Synergy Scoring
4.6. The mRNA Expression Analysis
4.7. Western Blotting
4.8. Correlation Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.C.; Strasser, A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000, 103, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Maji, S.; Panda, S.; Samal, S.K.; Shriwas, O.; Rath, R.; Pellecchia, M.; Emdad, L.; Das, S.K.; Fisher, P.B.; Dash, R. Bcl-2 Antiapoptotic Family Proteins and Chemoresistance in Cancer. Adv. Cancer Res. 2018, 137, 37–75. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- De Jong, Y.; Monderer, D.; Brandinelli, E.; Monchanin, M.; van den Akker, B.E.; van Oosterwijk, J.G.; Blay, J.Y.; Dutour, A.; Bovée, J. Bcl-xl as the most promising Bcl-2 family member in targeted treatment of chondrosarcoma. Oncogenesis 2018, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; Hooff, S.R.V.; Prasetyanti, P.R.; Kandimalla, R.; Buikhuisen, J.Y.; Fessler, E.; Ramesh, P.; Lee, K.; Bochove, G.G.W.; de Jong, J.H.; et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018, 25, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colak, S.; Zimberlin, C.D.; Fessler, E.; Hogdal, L.; Prasetyanti, P.R.; Grandela, C.M.; Letai, A.; Medema, J.P. Decreased mitochondrial priming determines chemoresistance of colon cancer stem cells. Cell Death Differ. 2014, 21, 1170–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Kawano, Y.; Arihara, Y.; Kubo, T.; Takada, K.; Murase, K.; Miyanishi, K.; Kobune, M.; Kato, J. BH3 profiling discriminates the anti‑apoptotic status of 5‑fluorouracil‑resistant colon cancer cells. Oncol. Rep. 2019, 42, 2416–2425. [Google Scholar] [CrossRef]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way--non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.J.; Palmieri, M.; Riffkin, C.D.; Sakthianandeswaren, A.; Djajawi, T.M.; Hirokawa, Y.; Shuttleworth, V.; Segal, D.H.; White, C.A.; Nhu, D.; et al. Defining the susceptibility of colorectal cancers to BH3-mimetic compounds. Cell Death Dis. 2020, 11, 735. [Google Scholar] [CrossRef]
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, D.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-X(L). Cell Death Differ. 2019, 26, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rahman, S.F.; Muniandy, K.; Soo, Y.K.; Tiew, E.Y.H.; Tan, K.X.; Bates, T.E.; Mohana-Kumaran, N. Co-inhibition of BCL-XL and MCL-1 with selective BCL-2 family inhibitors enhances cytotoxicity of cervical cancer cell lines. Biochem. Biophys. Rep. 2020, 22, 100756. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.F.; Harris, T.J.; Tran, S.; Evangelista, M.; Arulananda, S.; John, T.; Ramnac, C.; Hobbs, C.; Zhu, H.; Gunasingh, G.; et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.M.; Dietz, A.; Henz, K.; Bruecher, D.; Jackson, R.; Kowald, L.; van Wijk, S.J.L.; Jayne, S.; Macip, S.; Fulda, S.; et al. Specific interactions of BCL-2 family proteins mediate sensitivity to BH3-mimetics in diffuse large B-cell lymphoma. Haematologica 2020, 105, 2150–2163. [Google Scholar] [CrossRef]
- Touzeau, C.; Ryan, J.; Guerriero, J.; Moreau, P.; Chonghaile, T.N.; Le Gouill, S.; Richardson, P.; Anderson, K.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Soderquist, R.S.; Crawford, L.; Liu, E.; Lu, M.; Agarwal, A.; Anderson, G.R.; Lin, K.H.; Winter, P.S.; Cakir, M.; Wood, K.C. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nat. Commun. 2018, 9, 3513. [Google Scholar] [CrossRef] [Green Version]
- Okumura, K.; Huang, S.; Sinicrope, F.A. Induction of Noxa sensitizes human colorectal cancer cells expressing Mcl-1 to the small-molecule Bcl-2/Bcl-xL inhibitor, ABT-737. Clin. Cancer Res. 2008, 14, 8132–8142. [Google Scholar] [CrossRef] [Green Version]
- Lindner, A.U.; Salvucci, M.; Morgan, C.; Monsefi, N.; Resler, A.J.; Cremona, M.; Curry, S.; Toomey, S.; O’Byrne, R.; Bacon, O.; et al. BCL-2 system analysis identifies high-risk colorectal cancer patients. Gut 2017, 66, 2141–2148. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Zaanan, A.; Kawakami, H.; Huang, S.; Sinicrope, F.A. Reversal of Mutant KRAS-Mediated Apoptosis Resistance by Concurrent Noxa/Bik Induction and Bcl-2/Bcl-xL Antagonism in Colon Cancer Cells. Mol. Cancer Res. 2015, 13, 659–669. [Google Scholar] [CrossRef] [Green Version]
- McKee, C.S.; Hill, D.S.; Redfern, C.P.; Armstrong, J.L.; Lovat, P.E. Oncogenic BRAF signalling increases Mcl-1 expression in cutaneous metastatic melanoma. Exp. Dermatol. 2013, 22, 767–769. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, T.M.; Oren, M. p53 and apoptosis. Semin. Cancer Biol. 1998, 8, 359–368. [Google Scholar] [CrossRef]
- Bliss, C. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Ladner, C.L.; Yang, J.; Turner, R.J.; Edwards, R.A. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors and Combinations | BCL-2 | BCL-XL | MCL-1 | BCL-W |
|---|---|---|---|---|
| ABT-199 | ++ | − | − | − |
| A-1155463 | − | ++ | − | − |
| AZD5991 | − | − | ++ | − |
| ABT-263 | + | + | − | + |
| ABT-199 + A-1155463 | ++ | ++ | − | − |
| ABT-199 + AZD5991 | ++ | − | ++ | − |
| A-1155463 + AZD5991 | − | ++ | ++ | − |
| ABT-263 + AZD5991 | + | + | ++ | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Ramesh, P.; Steinmetz, M.; Medema, J.P. BH3 Mimetic Sensitivity of Colorectal Cancer Cell Lines in Correlation with Molecular Features Identifies Predictors of Response. Int. J. Mol. Sci. 2021, 22, 3811. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083811
Zhang L, Ramesh P, Steinmetz M, Medema JP. BH3 Mimetic Sensitivity of Colorectal Cancer Cell Lines in Correlation with Molecular Features Identifies Predictors of Response. International Journal of Molecular Sciences. 2021; 22(8):3811. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083811
Chicago/Turabian StyleZhang, Le, Prashanthi Ramesh, Maxime Steinmetz, and Jan Paul Medema. 2021. "BH3 Mimetic Sensitivity of Colorectal Cancer Cell Lines in Correlation with Molecular Features Identifies Predictors of Response" International Journal of Molecular Sciences 22, no. 8: 3811. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083811