
Active Transport of Hepatotoxic Pyrrolizidine Alkaloids in HepaRG Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
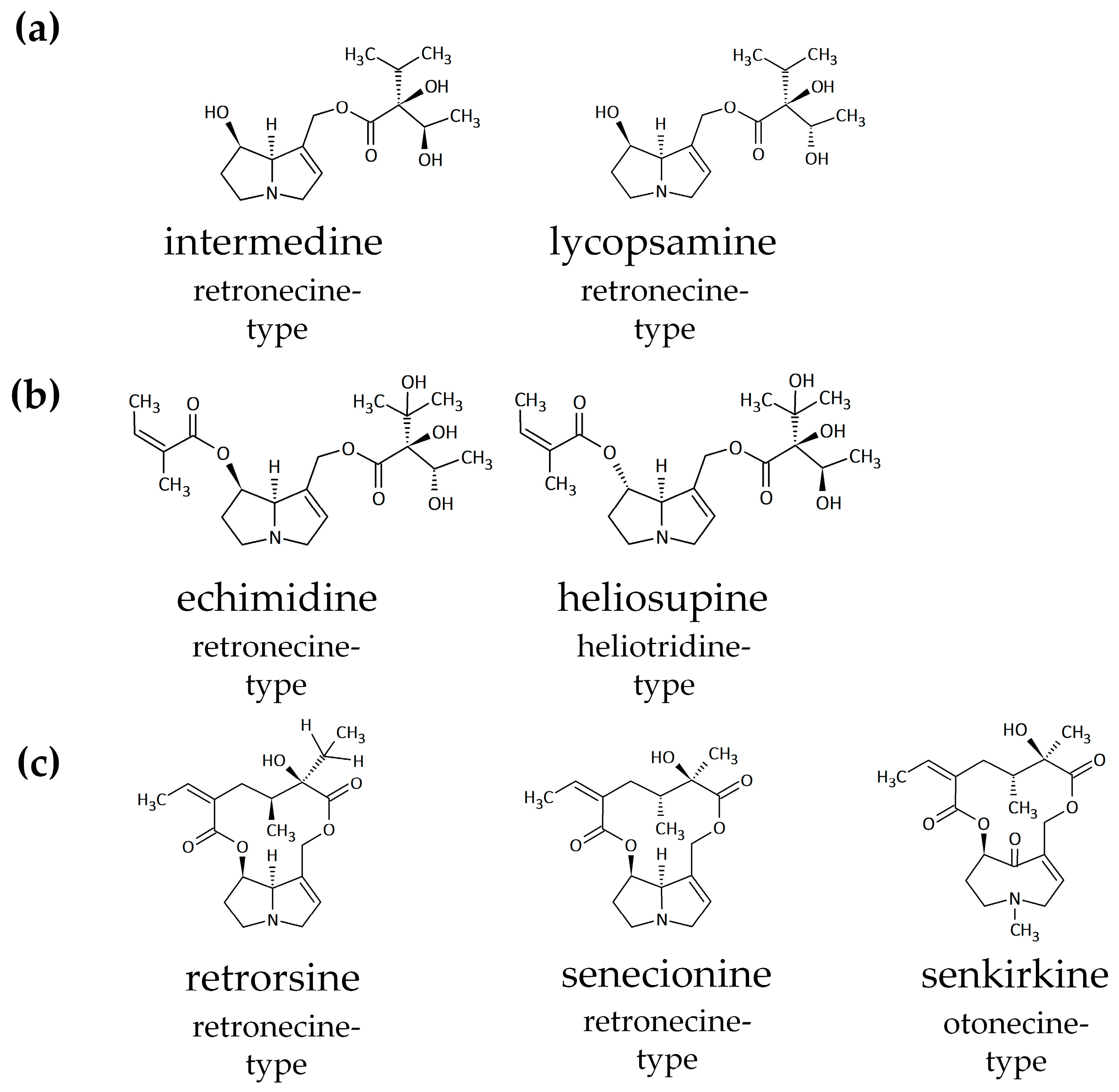
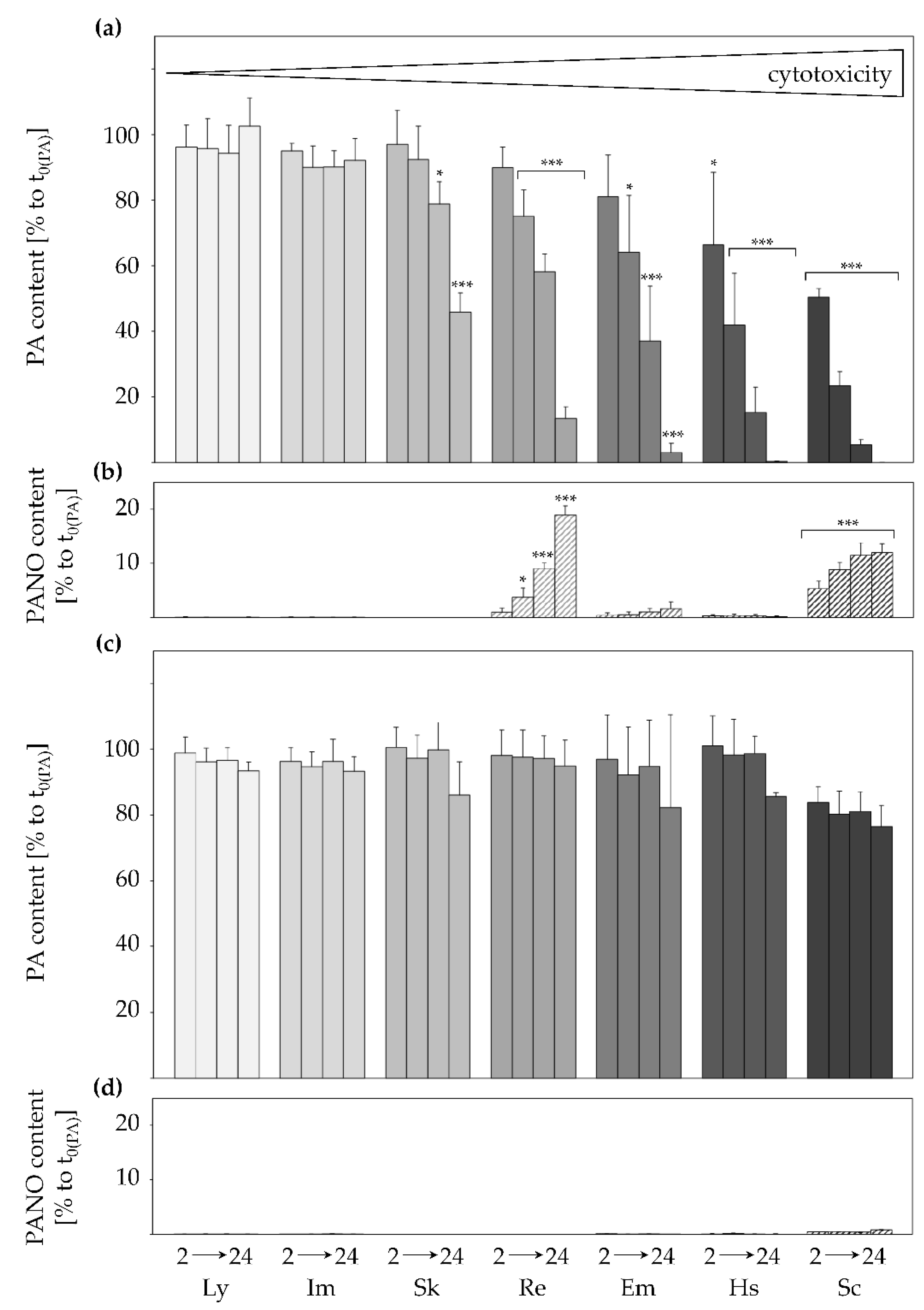
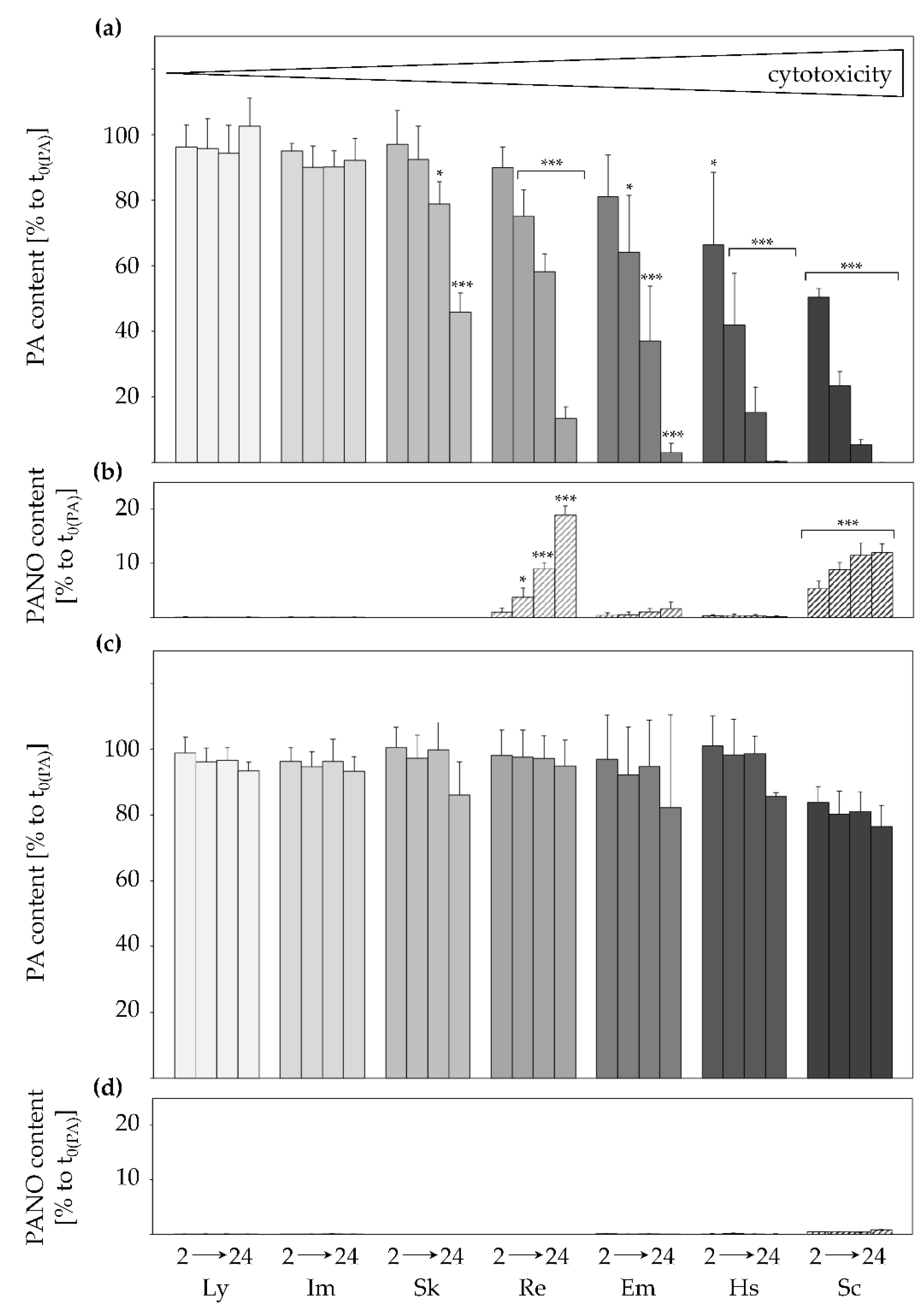
2.1. Active and Structure-Dependent Uptake of PAs in HepaRG Cells
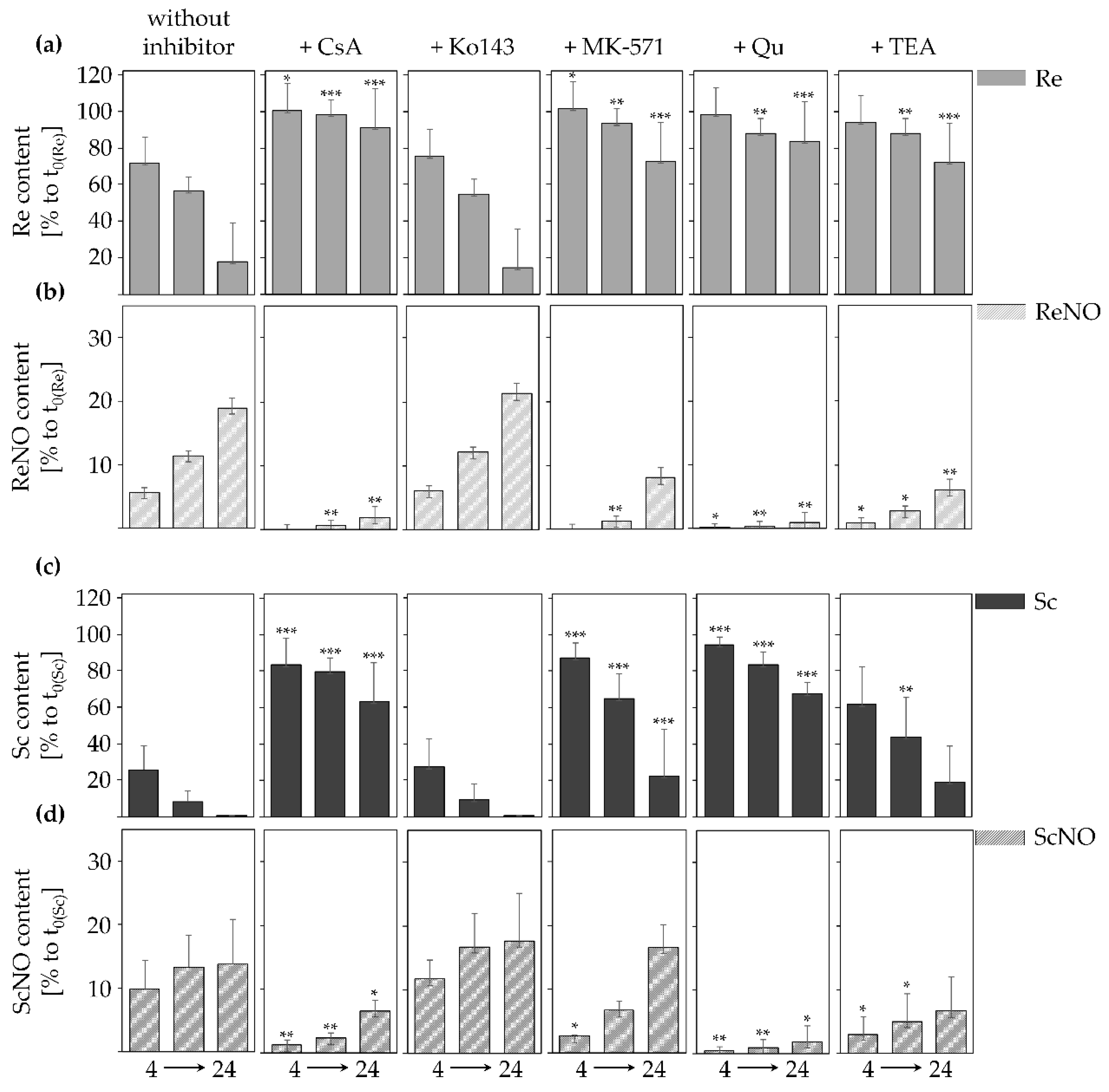
2.2. Inhibition of Transporters Decelerates PA Uptake
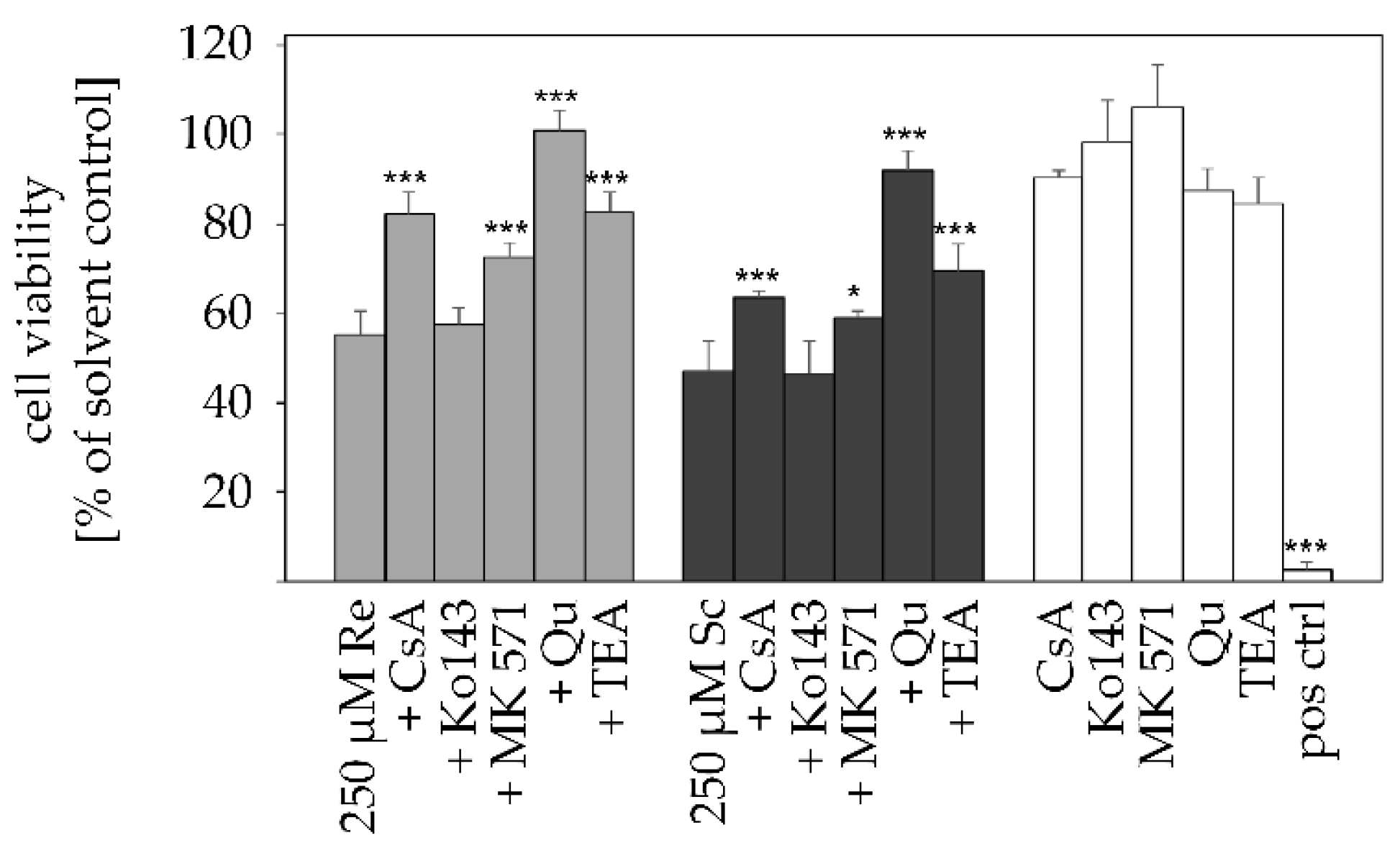
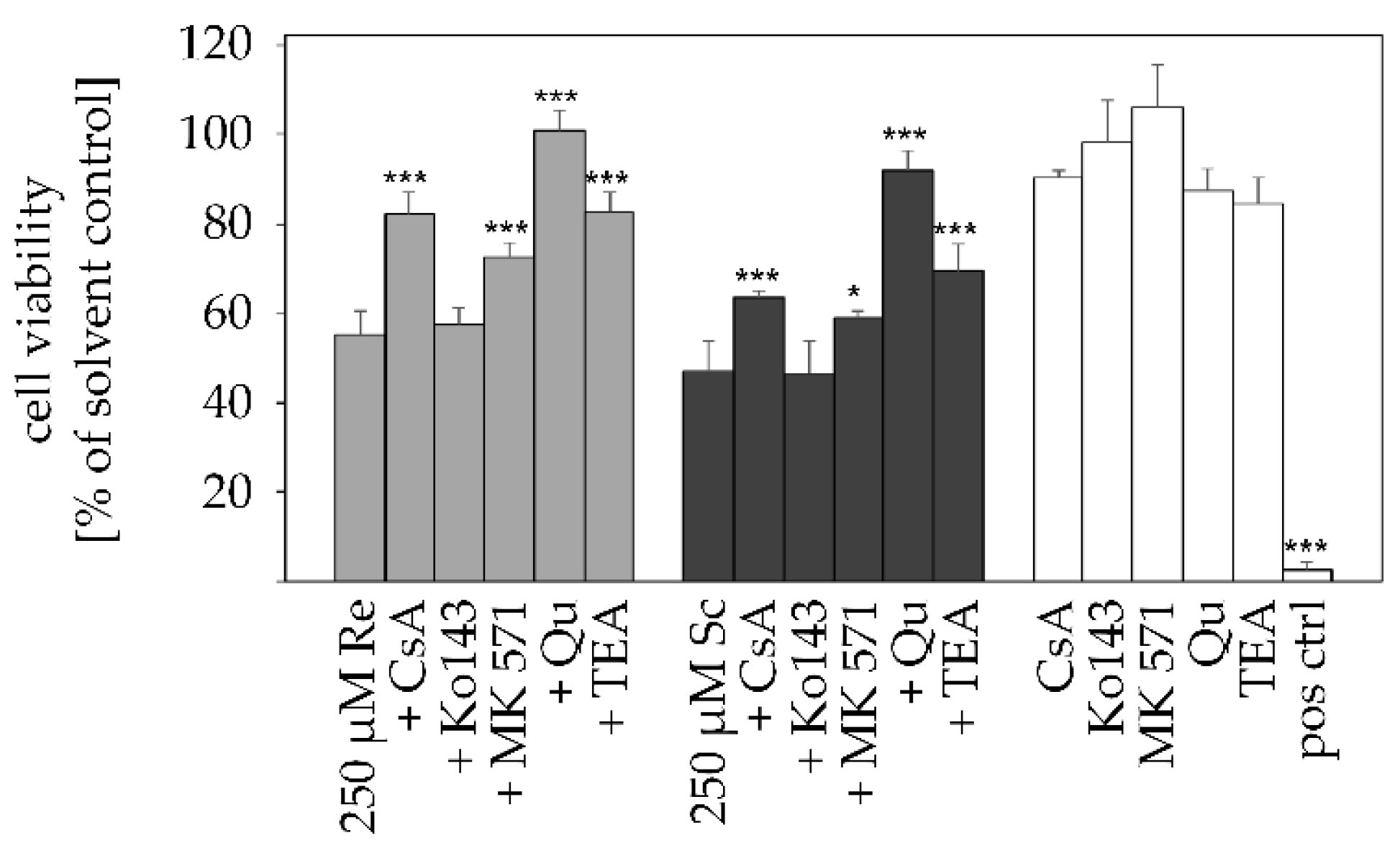
2.3. Transporter Inhibition Attenuates PA-Mediated Cytotoxicity
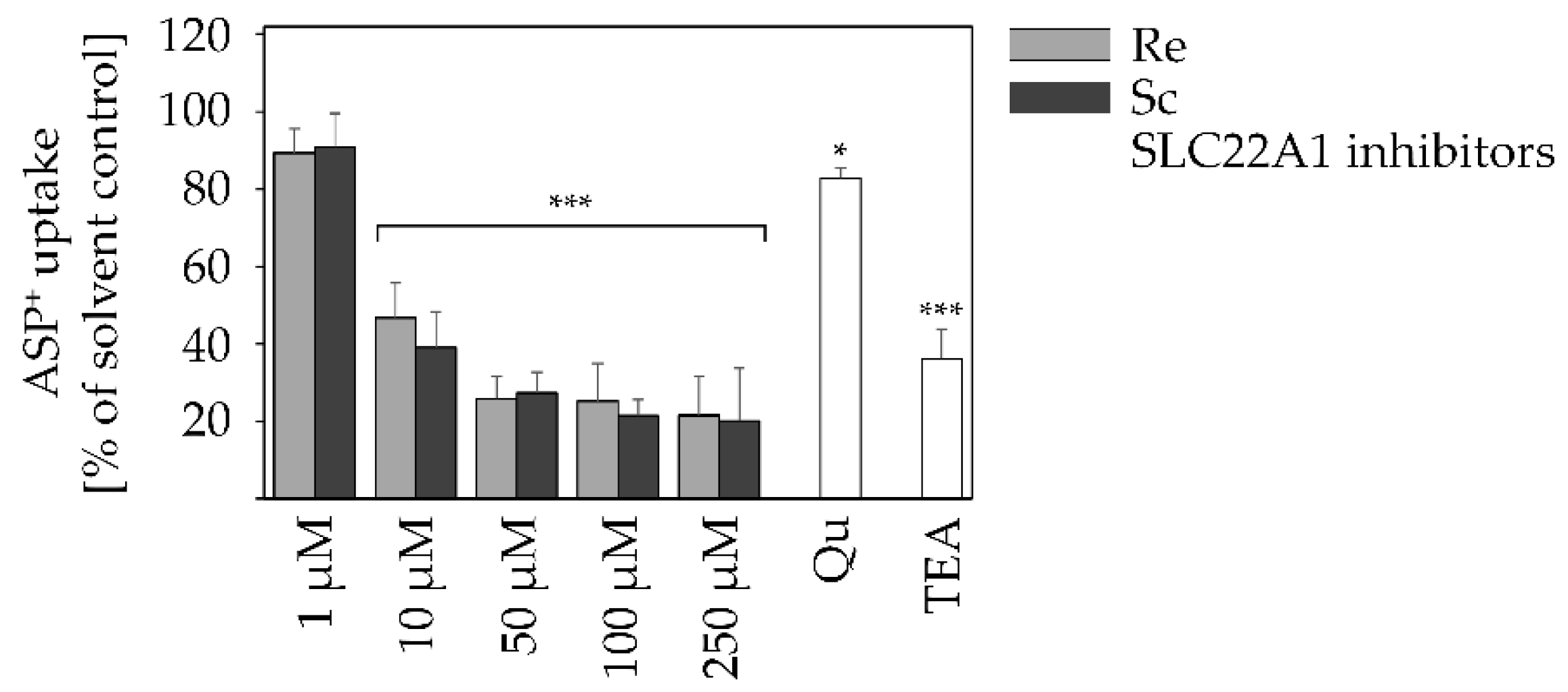
2.4. PAs Reduce SLC22A1 Substrate Uptake in a Concentration-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Inhibition Assay with Pharmacological Inhibitors
4.4. Cell Viability Assay
4.5. Transport Analysis of PAs in HepaRG Cells
4.6. Analysis of PA and PANO Content in Cell Culture Supernatant Using LC-MS/MS
4.7. ASP+ Uptake Measurements
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette transporter |
| ABCB1 | multidrug-resistance protein 1 (MDR1/P-Gp) |
| ABCB11 | bile salt export pump (BSEP) |
| ABCC | multidrug resistance-associated protein (MRP) |
| ABCG2 | breast cancer resistance protein (BCRP) |
| ACN | acetonitrile |
| ASP+ | 4-(4-dimethylaminostyryl)-N-methylpyridinium iodide |
| CsA | cyclosporine A |
| CYP | cytochrome P450-monooxygenases |
| DHP | (±)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizidine |
| DMSO | dimethyl sulfoxide |
| Em | echimidine |
| FBS | fetal bovine serum |
| GSH | glutathione |
| Hs | heliosupine |
| Im | intermedine |
| Ly | lycopsamine |
| Mc | monocrotaline |
| MDCK | Madin-Darby canine kidney cells |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid |
| PA | 1,2-unsaturated pyrrolizidine alkaloid |
| PANO | 1,2-unsaturated pyrrolizidine alkaloid N-oxide |
| PAPS | 3′-phosphoadenosine-5′-phosphosulfate |
| Re | retrorsine |
| Sc | senecionine |
| SDS | sodium dodecyl sulfate |
| SLC | solute carrier transporter |
| SLC10A1 | Na+/taurocholate co-transporting polypeptide (NTCP) |
| SLC21/SLCO | organic anion transporting polypetides (OATP) |
| SLC22 | organic anion/cation/zwitterion transporter family (OAT and OCT) |
| SLC22A1 | organic cation transporter 1 (OCT1) |
| Sk | senkirkine |
| TEA | tetraethylammonium |
| Qu | quinidine |
References
- Mattocks, A. Chemistry and Toxicology of Pyrrolizidine Alkaloids; Academic Press: London, UK, 1986. [Google Scholar]
- Culvenor, C.C.; Edgar, J.A.; Smith, L.W. Pyrrolizidine alkaloids in honey from Echium plantagineum L. J. Agric. Food Chem. 1981, 29, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.P.J.; López, P.; Castellari, M.; Bodi, D.; Ronczka, S.; Preiss-Weigert, A.; These, A. Occurrence of pyrrolizidine alkaloids in animal- and plant-derived food: Results of a survey across Europe. Food Addit. Contam. Part A 2018, 35, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Bodi, D.; Ronczka, S.; Gottschalk, C.; Behr, N.; Skibba, A.; Wagner, M.; Lahrssen-Wiederholt, M.; Preiss-Weigert, A.; These, A. Determination of pyrrolizidine alkaloids in tea, herbal drugs and honey. Food Addit. Contam. Part A 2014, 31, 1886–1895. [Google Scholar] [CrossRef]
- Wiedenfeld, H. Plants containing pyrrolizidine alkaloids: Toxicity and problems. Food Addit. Contam. Part A 2011, 28, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Kakar, F.; Akbarian, Z.; Leslie, T.; Mustafa, M.L.; Watson, J.; van Egmond, H.P.; Omar, M.F.; Mofleh, J. An outbreak of hepatic veno-occlusive disease in Western Afghanistan associated with exposure to wheat flour contaminated with pyrrolizidine alkaloids. J. Toxicol. 2010, 2010, 313280. [Google Scholar] [CrossRef]
- Stegelmeier, B.L.; Edgar, J.A.; Colegate, S.M.; Gardner, D.R.; Schoch, T.K.; Coulombe, R.A.; Molyneux, R.J. Pyrrolizidine alkaloid plants, metabolism and toxicity. J. Nat. Toxins 1999, 8, 95–116. [Google Scholar]
- Steenkamp, V.; Stewart, M.J.; Zuckerman, M. Clinical and analytical aspects of pyrrolizidine poisoning caused by South African traditional medicines. Drug Monit. 2000, 22, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Bah, M.; Bye, R.; Pereda-Miranda, R. Hepatotoxic pyrrolizidine alkaloids in the Mexican medicinal plant Packera candidissima (Asteraceae: Senecioneae). J. Ethnopharmacol. 1994, 43, 19–30. [Google Scholar] [CrossRef]
- Edgar, J.A.; Lin, H.J.; Kumana, C.R.; Ng, M.M. Pyrrolizidine alkaloid composition of three Chinese medicinal herbs, Eupatorium cannabinum, E. japonicum and Crotalaria assamica. Am. J. Chin. Med. 1992, 20, 281–288. [Google Scholar] [CrossRef]
- Huxtable, R.J.; Lüthy, J.; Zweifel, U. Toxicity of comfrey-pepsin preparations. N. Engl. J. Med. 1986, 315, 1095. [Google Scholar]
- Arseculeratne, S.N.; Gunatilaka, A.A.; Panabokke, R.G. Studies on medicinal plants of Sri Lanka: Occurrence of pyrrolizidine alkaloids and hepatotoxic properties in some traditional medicinal herbs. J. Ethnopharmacol. 1981, 4, 159–177. [Google Scholar] [CrossRef]
- Arseculeratne, S.N.; Gunatilaka, A.A.L.; Panabokke, R.G. Studies on medicinal plants of Sri Lanka. Part 14: Toxicity of some traditional medicinal herbs. J. Ethnopharmacol. 1985, 13, 323–335. [Google Scholar] [CrossRef]
- Mattocks, A.R. Toxicity of Pyrrolizidine Alkaloids. Nature 1968, 217, 723–728. [Google Scholar] [CrossRef] [PubMed]
- White, I.N.H.; Mattocks, A.R.; Butler, W.H. The conversion of the pyrrolizidine alkaloid retrorsine to pyrrolic derivatives in vivo and in vitro and its acute toxicity to various animal species. Chem. Biol. Interact. 1973, 6, 207–218. [Google Scholar] [CrossRef]
- Miranda, C.L.; Reed, R.L.; Guengerich, F.P.; Buhler, D.R. Role of cytochrome P450IIIA4 in the metabolism of the pyrrolizidine alkaloid senecionine in human liver. Carcinogenesis 1991, 12, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.P.; Xia, Q.; Lin, G.; Chou, M.W. Pyrrolizidine alkaloids--genotoxicity, metabolism enzymes, metabolic activation, and mechanisms. Drug Metab. Rev. 2004, 36, 1–55. [Google Scholar] [CrossRef]
- Edgar, J.A.; Molyneux, R.J.; Colegate, S.M. Pyrrolizidine Alkaloids: Potential Role in the Etiology of Cancers, Pulmonary Hypertension, Congenital Anomalies, and Liver Disease. Chem. Res. Toxicol. 2015, 28, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Yang, M.; Fu, P.; Ye, Y.; Lin, G. Metabolic activation of pyrrolizidine alkaloids: Insights into the structural and enzymatic basis. Chem. Res. Toxicol. 2014, 27, 1030–1039. [Google Scholar] [CrossRef]
- Chen, T.; Mei, N.; Fu, P.P. Genotoxicity of pyrrolizidine alkaloids. J. Appl. Toxicol. 2010, 30, 183–196. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Merz, K.H.; Schrenk, D. Interim relative potency factors for the toxicological risk assessment of pyrrolizidine alkaloids in food and herbal medicines. Toxicol. Lett. 2016, 263, 44–57. [Google Scholar] [CrossRef]
- Clerbaux, L.A.; Paini, A.; Lumen, A.; Osman-Ponchet, H.; Worth, A.P.; Fardel, O. Membrane transporter data to support kinetically-informed chemical risk assessment using non-animal methods: Scientific and regulatory perspectives. Environ. Int. 2019, 126, 659–671. [Google Scholar] [CrossRef]
- Bai, X.; Moraes, T.F.; Reithmeier, R.A.F. Structural biology of solute carrier (SLC) membrane transport proteins. Mol. Membr. Biol. 2017, 34, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, P.J.; Stieger, B. Bile salt transporters. Annu. Rev. Physiol. 2002, 64, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Boyer, J.L. Bile salt transporters: Molecular characterization, function, and regulation. Physiol. Rev. 2003, 83, 633–671. [Google Scholar] [CrossRef] [Green Version]
- Döring, B.; Petzinger, E. Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Scherrmann, J.-M. Chapter 26—Drug Transport Mechanisms and their Impact on the Disposition and Effects of Drugs. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 615–629. [Google Scholar] [CrossRef]
- Gerloff, T.; Stieger, B.; Hagenbuch, B.; Madon, J.; Landmann, L.; Roth, J.; Hofmann, A.F.; Meier, P.J. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J. Biol. Chem. 1998, 273, 10046–10050. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, A.; Cusato, J.; D’Avolio, A.; Bonora, S. Genetic Polymorphisms Affecting the Pharmacokinetics of Antiretroviral Drugs. Clin. Pharm. 2017, 56, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Hira, D.; Terada, T. BCRP/ABCG2 and high-alert medications: Biochemical, pharmacokinetic, pharmacogenetic, and clinical implications. Biochem. Pharm. 2018, 147, 201–210. [Google Scholar] [CrossRef]
- Tu, M.; Li, L.; Lei, H.; Ma, Z.; Chen, Z.; Sun, S.; Xu, S.; Zhou, H.; Zeng, S.; Jiang, H. Involvement of organic cation transporter 1 and CYP3A4 in retrorsine-induced toxicity. Toxicology 2014, 322, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Sun, S.; Wang, K.; Peng, X.; Wang, R.; Li, L.; Zeng, S.; Zhou, H.; Jiang, H. Organic cation transporter 1 mediates the uptake of monocrotaline and plays an important role in its hepatotoxicity. Toxicology 2013, 311, 225–230. [Google Scholar] [CrossRef]
- Hessel, S.; Gottschalk, C.; Schumann, D.; These, A.; Preiss-Weigert, A.; Lampen, A. Structure–activity relationship in the passage of different pyrrolizidine alkaloids through the gastrointestinal barrier: ABCB1 excretes heliotrine and echimidine. Mol. Nutr. Food Res. 2014, 58, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, H.; Kusuhara, H.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J. Pharm. Exp. 2010, 333, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Polli, J.W.; Olson, K.L.; Chism, J.P.; John-Williams, L.S.; Yeager, R.L.; Woodard, S.M.; Otto, V.; Castellino, S.; Demby, V.E. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab. Dispos. 2009, 37, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef] [Green Version]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wetering, K.; Feddema, W.; Helms, J.B.; Brouwers, J.F.; Borst, P. Targeted metabolomics identifies glucuronides of dietary phytoestrogens as a major class of MRP3 substrates in vivo. Gastroenterology 2009, 137, 1725–1735. [Google Scholar] [CrossRef]
- Funk, C. The role of hepatic transporters in drug elimination. Expert Opin. Drug Metab. Toxicol. 2008, 4, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Glück, J.; Waizenegger, J.; Braeuning, A.; Hessel-Pras, S. Pyrrolizidine Alkaloids Induce Cell Death in Human HepaRG Cells in a Structure-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 202. [Google Scholar] [CrossRef] [PubMed]
- Geburek, I.; Preiss-Weigert, A.; Lahrssen-Wiederholt, M.; Schrenk, D.; These, A. In vitro metabolism of pyrrolizidine alkaloids—Metabolic degradation and GSH conjugate formation of different structure types. Food Chem. Toxicol. 2020, 135, 110868. [Google Scholar] [CrossRef]
- Kaltner, F.; Rychlik, M.; Gareis, M.; Gottschalk, C. Influence of Storage on the Stability of Toxic Pyrrolizidine Alkaloids and Their N-Oxides in Peppermint Tea, Hay, and Honey. J. Agric. Food Chem. 2018, 66, 5221–5228. [Google Scholar] [CrossRef]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer 2002, 1, 417–425. [Google Scholar]
- Watashi, K.; Sluder, A.; Daito, T.; Matsunaga, S.; Ryo, A.; Nagamori, S.; Iwamoto, M.; Nakajima, S.; Tsukuda, S.; Borroto-Esoda, K.; et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 2014, 59, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Sharanek, A.; Burban, A.; Humbert, L.; Bachour-El Azzi, P.; Felix-Gomes, N.; Rainteau, D.; Guillouzo, A. Cellular Accumulation and Toxic Effects of Bile Acids in Cyclosporine A-Treated HepaRG Hepatocytes. Toxicol. Sci. 2015, 147, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, U.; Abou El-Ela, A.; Guo, L.J.; Glavinas, H.; Krajcsi, P.; Baron, J.M.; Tillmann, C.; Hiemke, C.; Langguth, P.; Härtter, S. Cyclosporine A (CsA) affects the pharmacodynamics and pharmacokinetics of the atypical antipsychotic amisulpride probably via inhibition of P-glycoprotein (P-gp). J. Neural Transm. 2006, 113, 787–801. [Google Scholar] [CrossRef]
- Bodo, A.; Bakos, E.; Szeri, F.; Varadi, A.; Sarkadi, B. Differential modulation of the human liver conjugate transporters MRP2 and MRP3 by bile acids and organic anions. J. Biol. Chem. 2003, 278, 23529–23537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.; Theile, D.; Ketabi-Kiyanvash, N.; Lindenmaier, H.; Haefeli, W.E. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non-nucleoside reverse transcriptase inhibitors. Drug Metab. Dispos. 2007, 35, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic cation transporters in intestine, kidney, liver, and brain. Annu. Rev. Physiol. 1998, 60, 243–266. [Google Scholar] [CrossRef]
- Ahlin, G.; Karlsson, J.; Pedersen, J.M.; Gustavsson, L.; Larsson, R.; Matsson, P.; Norinder, U.; Bergström, C.A.; Artursson, P. Structural requirements for drug inhibition of the liver specific human organic cation transport protein 1. J. Med. Chem. 2008, 51, 5932–5942. [Google Scholar] [CrossRef]
- Hendrickx, R.; Johansson, J.G.; Lohmann, C.; Jenvert, R.M.; Blomgren, A.; Börjesson, L.; Gustavsson, L. Identification of novel substrates and structure-activity relationship of cellular uptake mediated by human organic cation transporters 1 and 2. J. Med. Chem. 2013, 56, 7232–7242. [Google Scholar] [CrossRef]
- Bui, K.; She, F.; Zhou, D.; Butler, K.; Al-Huniti, N.; Sostek, M. The effect of quinidine, a strong P-glycoprotein inhibitor, on the pharmacokinetics and central nervous system distribution of naloxegol. J. Clin. Pharm. 2016, 56, 497–505. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Struwe, K.; Arndt, P.; Gorboulev, V.; Koepsell, H.; Schlatter, E.; Hirsch, J.R. Regulation of the human organic cation transporter hOCT1. J. Cell Physiol. 2004, 201, 420–428. [Google Scholar] [CrossRef]
- Magrassi, L.; Purves, D.; Lichtman, J.W. Fluorescent probes that stain living nerve terminals. J. Neurosci. 1987, 7, 1207–1214. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; et al. Risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 2017, 15, e04908. [Google Scholar] [CrossRef] [Green Version]
- BFR. Pyrrolizidine Alkaloids in Herbal Teas and Teas. German Federal Institute for Risk Assessment. BFR Opin. 2013, 18, 1–31. [Google Scholar]
- Allemang, A.; Mahony, C.; Lester, C.; Pfuhler, S. Relative potency of fifteen pyrrolizidine alkaloids to induce DNA damage as measured by micronucleus induction in HepaRG human liver cells. Food Chem. Toxicol. 2018, 121, 72–81. [Google Scholar] [CrossRef]
- Louisse, J.; Rijkers, D.; Stoopen, G.; Holleboom, W.J.; Delagrange, M.; Molthof, E.; Mulder, P.P.J.; Hoogenboom, R.L.A.P.; Audebert, M.; Peijnenburg, A.A.C.M. Determination of genotoxic potencies of pyrrolizidine alkaloids in HepaRG cells using the γH2AX assay. Food Chem. Toxicol. 2019, 131, 110532. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Rutz, L.; Schrenk, D. Structure-dependent hepato-cytotoxic potencies of selected pyrrolizidine alkaloids in primary rat hepatocyte culture. Food Chem. Toxicol. 2020, 135, 110923. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG cell line: A unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef]
- Le Vee, M.; Jigorel, E.; Glaise, D.; Gripon, P.; Guguen-Guillouzo, C.; Fardel, O. Functional expression of sinusoidal and canalicular hepatic drug transporters in the differentiated human hepatoma HepaRG cell line. Eur. J. Pharm. Sci. 2006, 28, 109–117. [Google Scholar] [CrossRef]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Chen, L.; Mulder, P.P.J.; Louisse, J.; Peijnenburg, A.; Wesseling, S.; Rietjens, I.M.C.M. Risk assessment for pyrrolizidine alkaloids detected in (herbal) teas and plant food supplements. Regul. Toxicol. Pharmacol. 2017, 86, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ma, J.; Ruan, J.; Zhang, C.; Ye, Y.; Pi-Cheng Fu, P.; Lin, G. Absorption difference between hepatotoxic pyrrolizidine alkaloids and their N-oxides—Mechanism and its potential toxic impact. J. Ethnopharmacol. 2020, 249, 112421. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.S. Intestinal absorption and metabolism of xenobiotics. Environ. Health Perspect. 1979, 33, 61–69. [Google Scholar] [CrossRef]
- Lüthy, J.; Heim, T.; Schlatter, C. Transfer of [3H]pyrrolizidine alkaloids from Senecio vulgaris L. and metabolites into rat milk and tissues. Toxicol. Lett. 1983, 17, 283–288. [Google Scholar] [CrossRef]
- Bruce Alberts, A.J.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Frézard, F.; Garnier-Suillerot, A. Permeability of lipid bilayer to anthracycline derivatives. Role of the bilayer composition and of the temperature. Biochim. Biophys. Acta 1998, 1389, 13–22. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2001, 46, 27–43. [Google Scholar] [CrossRef]
- Ebmeyer, J.; Braeuning, A.; Glatt, H.; These, A.; Hessel-Pras, S.; Lampen, A. Human CYP3A4-mediated toxification of the pyrrolizidine alkaloid lasiocarpine. Food Chem. Toxicol. 2019, 130, 79–88. [Google Scholar] [CrossRef]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouët, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Isom, H.C.; Secott, T.; Georgoff, I.; Woodworth, C.; Mummaw, J. Maintenance of differentiated rat hepatocytes in primary culture. Proc. Natl. Acad. Sci. USA 1985, 82, 3252–3256. [Google Scholar] [CrossRef] [Green Version]
- Anthérieu, S.; Chesné, C.; Li, R.; Camus, S.; Lahoz, A.; Picazo, L.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Guguen-Guillouzo, C.; et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab. Dispos. 2010, 38, 516–525. [Google Scholar] [CrossRef]
- Jossé, R.; Aninat, C.; Glaise, D.; Dumont, J.; Fessard, V.; Morel, F.; Poul, J.M.; Guguen-Guillouzo, C.; Guillouzo, A. Long-term functional stability of human HepaRG hepatocytes and use for chronic toxicity and genotoxicity studies. Drug Metab. Dispos. 2008, 36, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Braeuning, A. Regulation of cytochrome P450 expression by Ras- and beta-catenin-dependent signaling. Curr. Drug Metab. 2009, 10, 138–158. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kaltner, F.; Stiglbauer, B.; Rychlik, M.; Gareis, M.; Gottschalk, C. Development of a sensitive analytical method for determining 44 pyrrolizidine alkaloids in teas and herbal teas via LC-ESI-MS/MS. Anal. Bioanal. Chem. 2019, 411, 7233–7249. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enge, A.-M.; Kaltner, F.; Gottschalk, C.; Braeuning, A.; Hessel-Pras, S. Active Transport of Hepatotoxic Pyrrolizidine Alkaloids in HepaRG Cells. Int. J. Mol. Sci. 2021, 22, 3821. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083821
Enge A-M, Kaltner F, Gottschalk C, Braeuning A, Hessel-Pras S. Active Transport of Hepatotoxic Pyrrolizidine Alkaloids in HepaRG Cells. International Journal of Molecular Sciences. 2021; 22(8):3821. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083821
Chicago/Turabian StyleEnge, Anne-Margarethe, Florian Kaltner, Christoph Gottschalk, Albert Braeuning, and Stefanie Hessel-Pras. 2021. "Active Transport of Hepatotoxic Pyrrolizidine Alkaloids in HepaRG Cells" International Journal of Molecular Sciences 22, no. 8: 3821. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083821