
PSEN1 Compound Heterozygous Mutations Associated with Cerebral Amyloid Angiopathy and Cognitive Decline Phenotype

 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
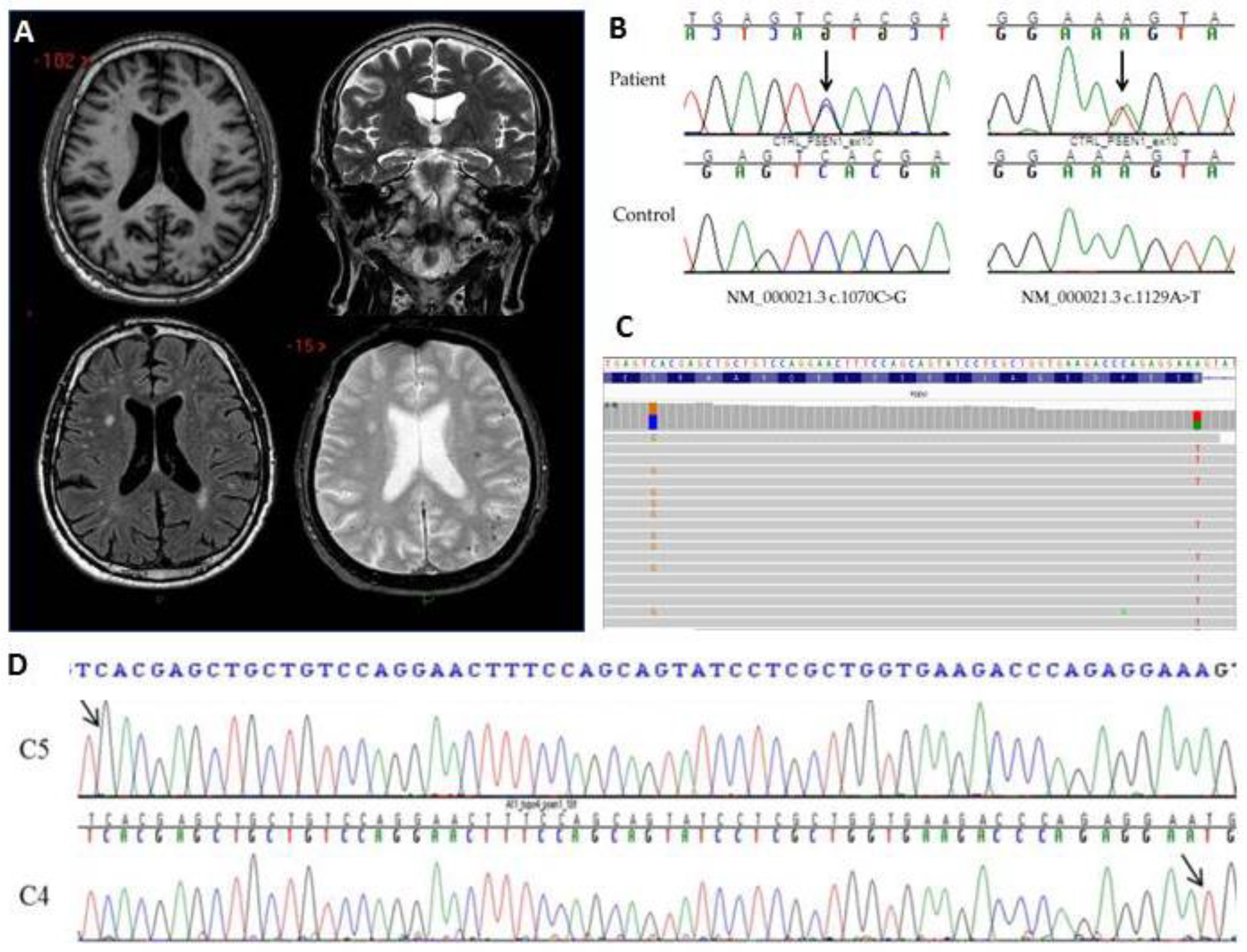
2.1. Clinical Findings
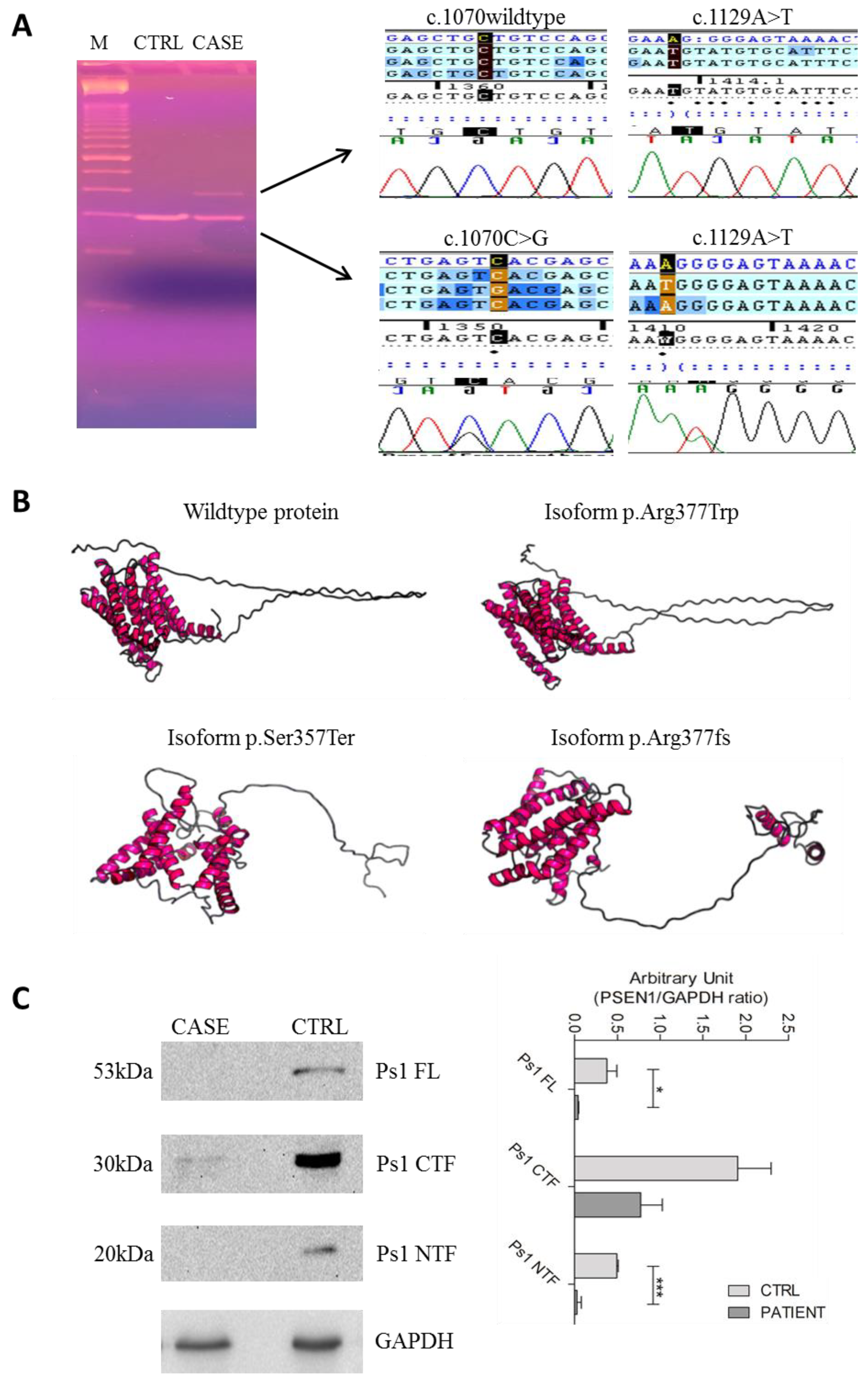
2.2. Genetic Analysis and In Silico Prediction
2.3. Variants Segregation
2.4. Alteration of Splicing Analysis
2.5. D Protein Structure
3. Discussion
4. Materials and Methods
4.1. Genetic and In Silico Analyses
4.2. Allele Sequencing
4.3. TOPO®TA Cloning
4.4. Protein Characterization
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| CAA | cerebral amyloid angiopathy |
| EEG | electroencephalogram |
| FDG-PET | fluorodeoxyglucose-positron emission tomography |
| ICH | intracerebral hemorrhages |
| MRI | magnetic resonance imaging |
| MTA | medial temporal lobe atrophy |
| NGS | next-generation sequencing |
| PBMCs | peripheral blood mononuclear cells |
| PSEN1 | presenilin 1 |
| TM | transmembrane |
References
- Razek, A.A.K.A.; Elsebaie, N.A. Imaging of vascular cognitive impairment. Clin. Imaging 2021, 74, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.; Sharman, T. Cerebral Amyloid Angiopathy. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; Van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Leys, D. Vascular Cognitive Impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A.; Pickering-Brown, S.M.; Takeuchi, A.; Iwatsubo, T.; Members of the Familial Alzheimer’s Disease Pathology Study Group. Amyloid Angiopathy and Variability in Amyloid β Deposition Is Determined by Mutation Position in Presenilin-1-Linked Alzheimer’s Disease. Am. J. Pathol. 2001, 158, 2165–2175. [Google Scholar] [CrossRef]
- Saito, S.; Ihara, M. Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatry 2016, 29, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Quillard-Muraine, M.; Guyant-Maréchal, L.; Martinaud, O.; Pariente, J.; Puel, M.; Rollin-Sillaire, A.; Pasquier, F.; et al. The French Series of Autosomal Dominant Early Onset Alzheimer’s Disease Cases: Mutation Spectrum and Cerebrospinal Fluid Biomarkers. J. Alzheimer’s Dis. 2012, 30, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, B.; Pilotto, A.; Bonvicini, C.; Archetti, S.; Alberici, A.; Lupi, A.; Gennarelli, M.; Padovani, A. Atypical presentation of a novel Presenilin 1 R377W mutation: Sporadic, late-onset Alzheimer disease with epilepsy and frontotemporal atrophy. Neurol. Sci. 2011, 33, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Scarioni, M.; Arighi, A.; Fenoglio, C.; Sorrentino, F.; Serpente, M.; Rotondo, E.; Mercurio, M.; Marotta, G.; Dijkstra, A.A.; Pijnenburg, Y.A.L.; et al. Lateonset presentation and phenotypic heterogeneity of the rare R377W PSEN1 mutation. Eur. J. Neurol. 2020, 27, 2630–2634. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Launer, L.J.; Barkhof, F.; Weinstein, H.C.; Van Gool, W.A. Visual assessment of medial temporal lobe atrophy on magnetic resonance imaging: Interobserver reliability. J. Neurol. 1995, 242, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Piccinelli, S.C.; Palmieri, I.; Necchini, N.; Valente, M.; Zanella, I.; Biasiotto, G.; Di Lorenzo, D.; Cereda, C.; Padovani, A. A Novel Mutation in the Stalk Domain of KIF5A Causes a Slowly Progressive Atypical Motor Syndrome. J. Clin. Med. 2018, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, I.; Valente, M.; Farina, L.M.; Gana, S.; Minafra, B.; Zangaglia, R.; Pansarasa, O.; Sproviero, D.; Costa, A.; Pacchetti, C.; et al. PSEN1 Compound Heterozygous Mutations Associated with Cerebral Amyloid Angiopathy and Cognitive Decline Phenotype. Int. J. Mol. Sci. 2021, 22, 3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083870
Palmieri I, Valente M, Farina LM, Gana S, Minafra B, Zangaglia R, Pansarasa O, Sproviero D, Costa A, Pacchetti C, et al. PSEN1 Compound Heterozygous Mutations Associated with Cerebral Amyloid Angiopathy and Cognitive Decline Phenotype. International Journal of Molecular Sciences. 2021; 22(8):3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083870
Chicago/Turabian StylePalmieri, Ilaria, Marialuisa Valente, Lisa Maria Farina, Simone Gana, Brigida Minafra, Roberta Zangaglia, Orietta Pansarasa, Daisy Sproviero, Alfredo Costa, Claudio Pacchetti, and et al. 2021. "PSEN1 Compound Heterozygous Mutations Associated with Cerebral Amyloid Angiopathy and Cognitive Decline Phenotype" International Journal of Molecular Sciences 22, no. 8: 3870. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083870