
FeIII, CuII and ZnII Complexes of the Rigid 9-Oxido-phenalenone Ligand—Spectroscopy, Electrochemistry, and Cytotoxic Properties

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
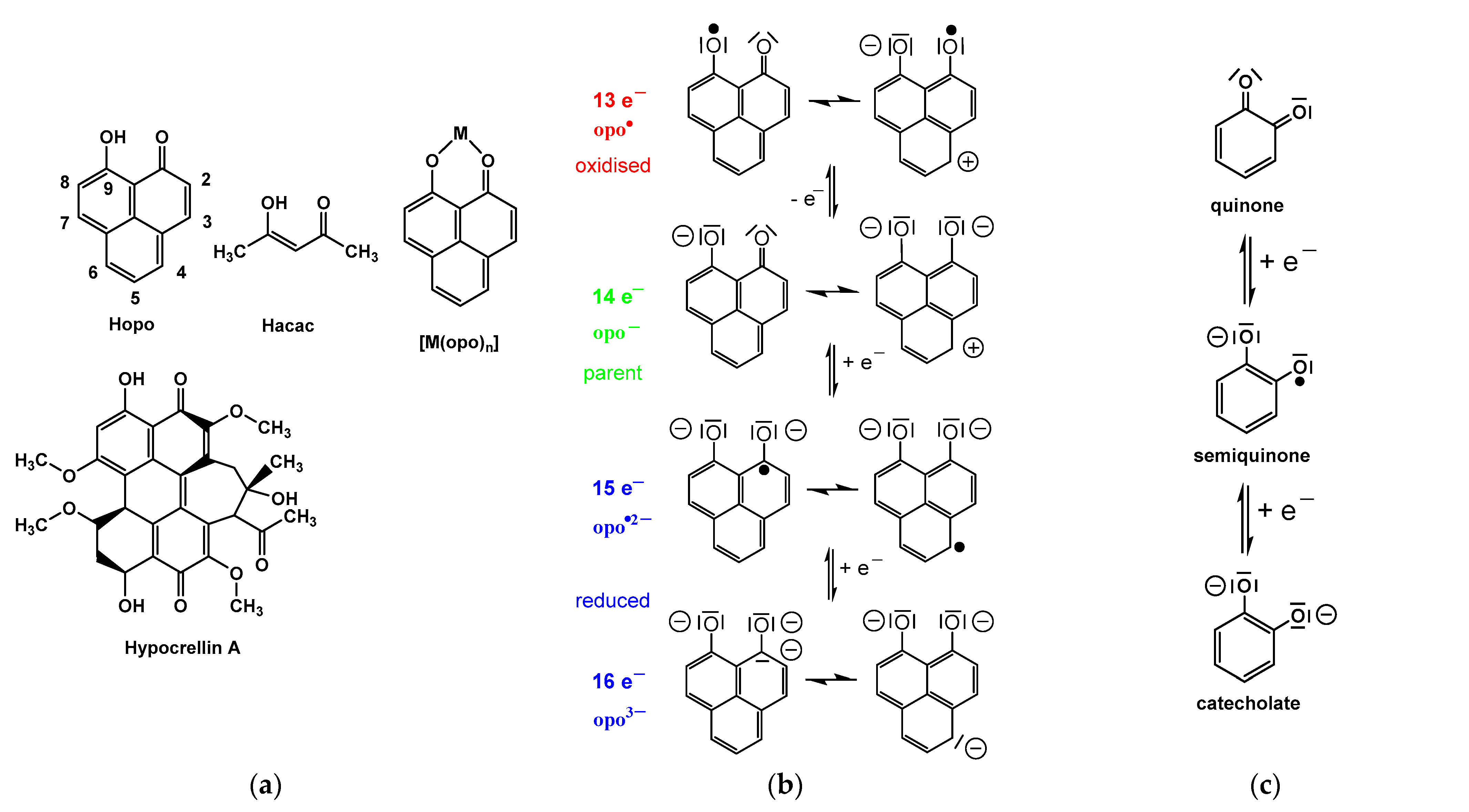
2.1. Determination of the pKa values of Hopo and Hacac
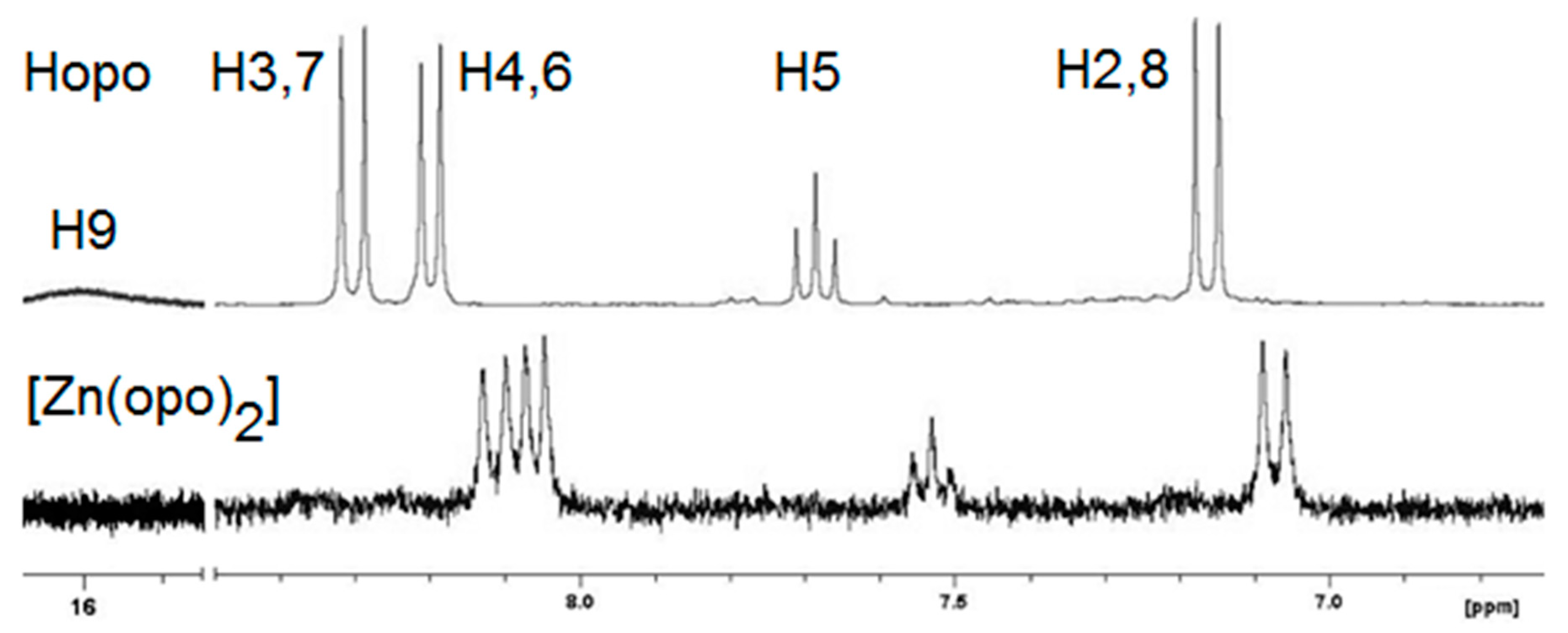
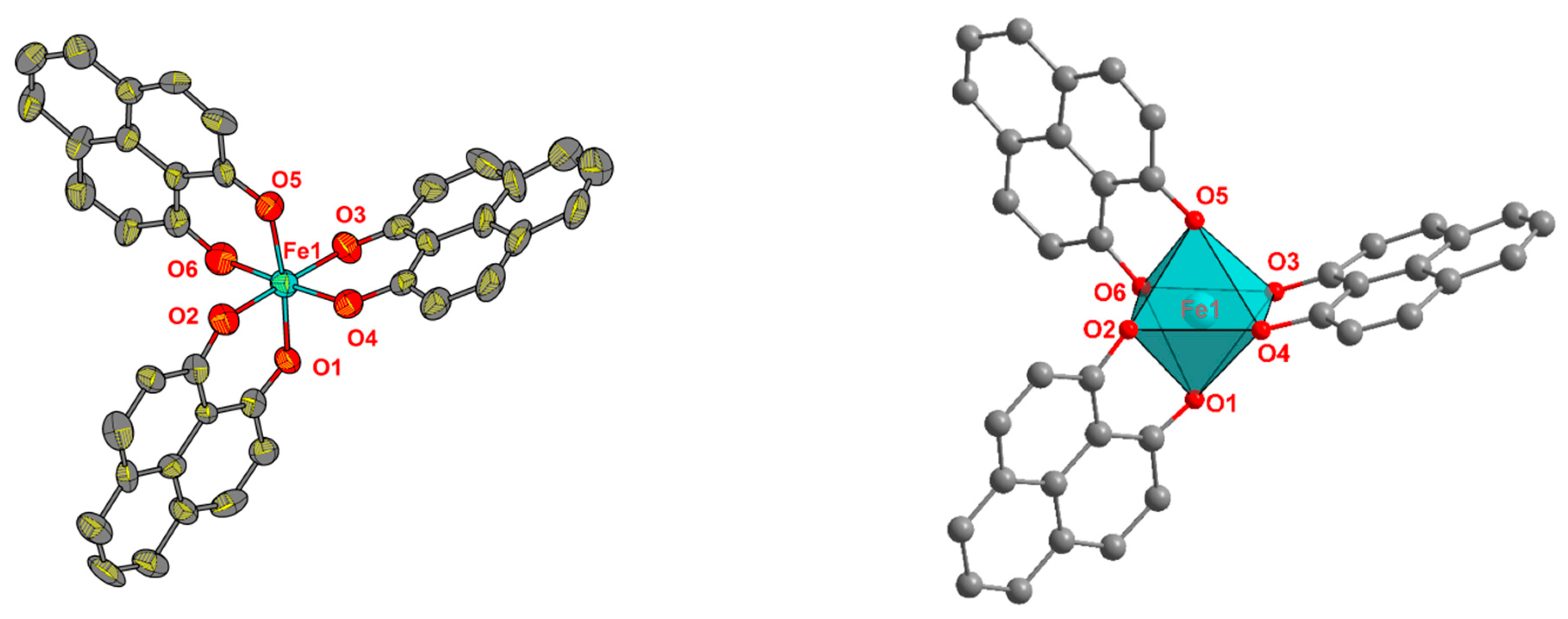
2.2. Synthesis and Structure Analysis of the Opo Complexes
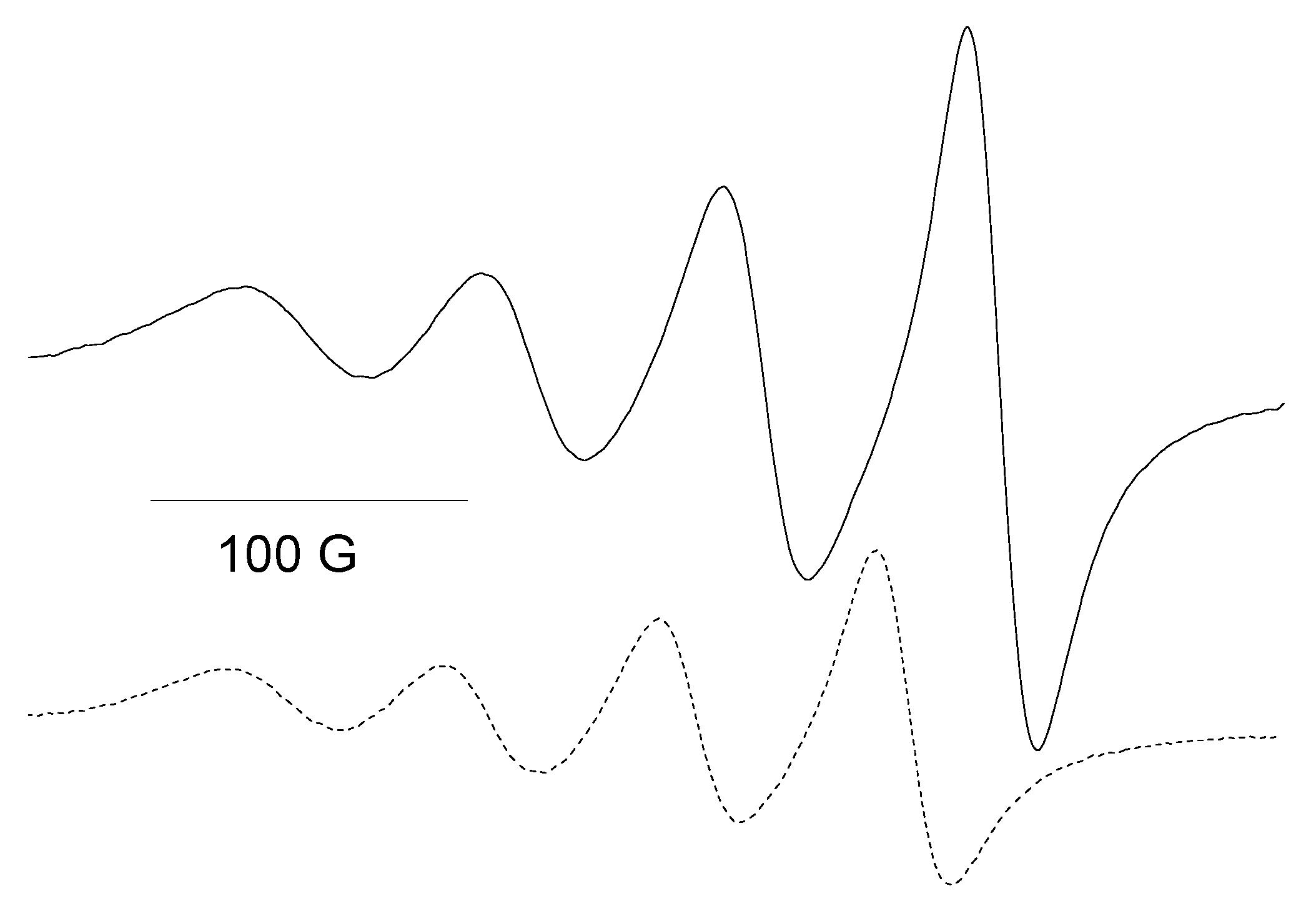
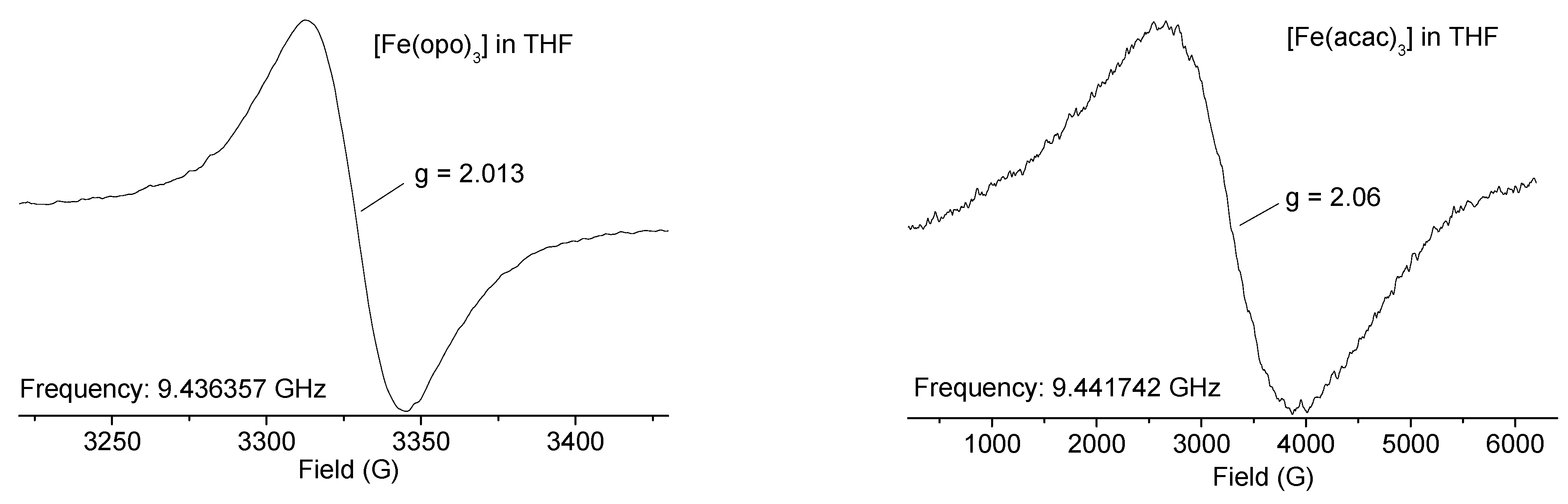
2.3. EPR Spectroscopy for [Cu(opo)2] and [Fe(opo)3]
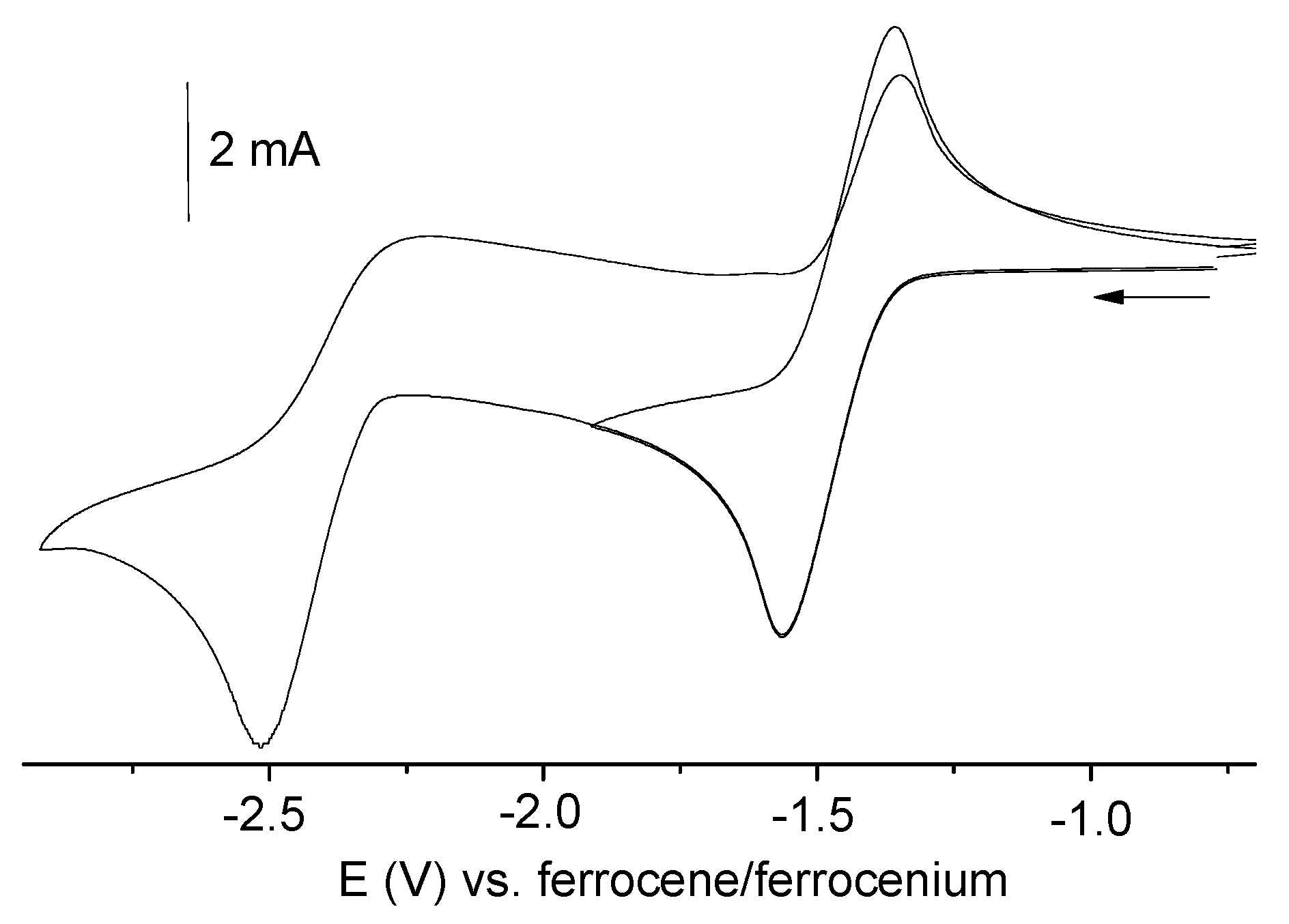
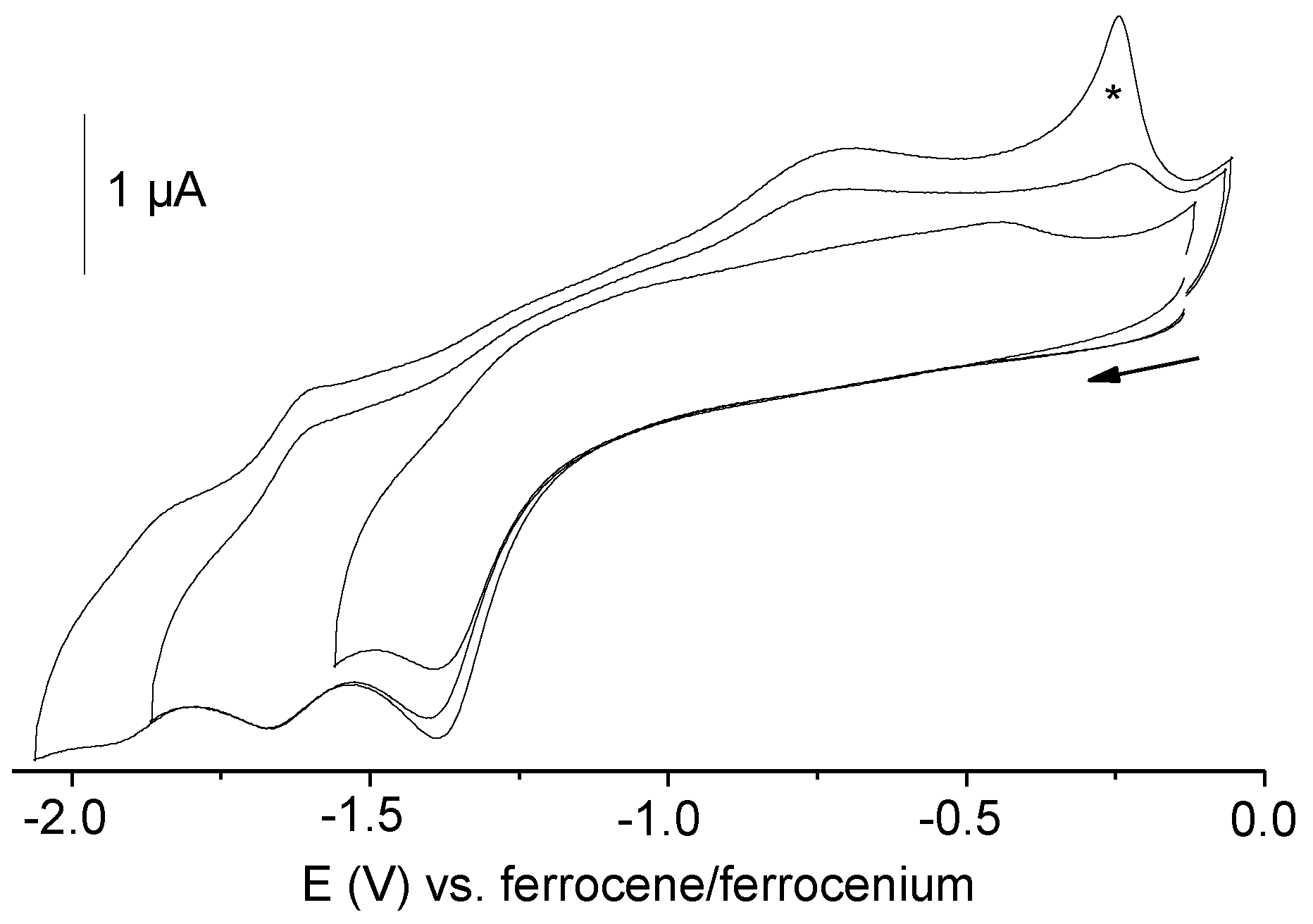
2.4. Electrochemistry
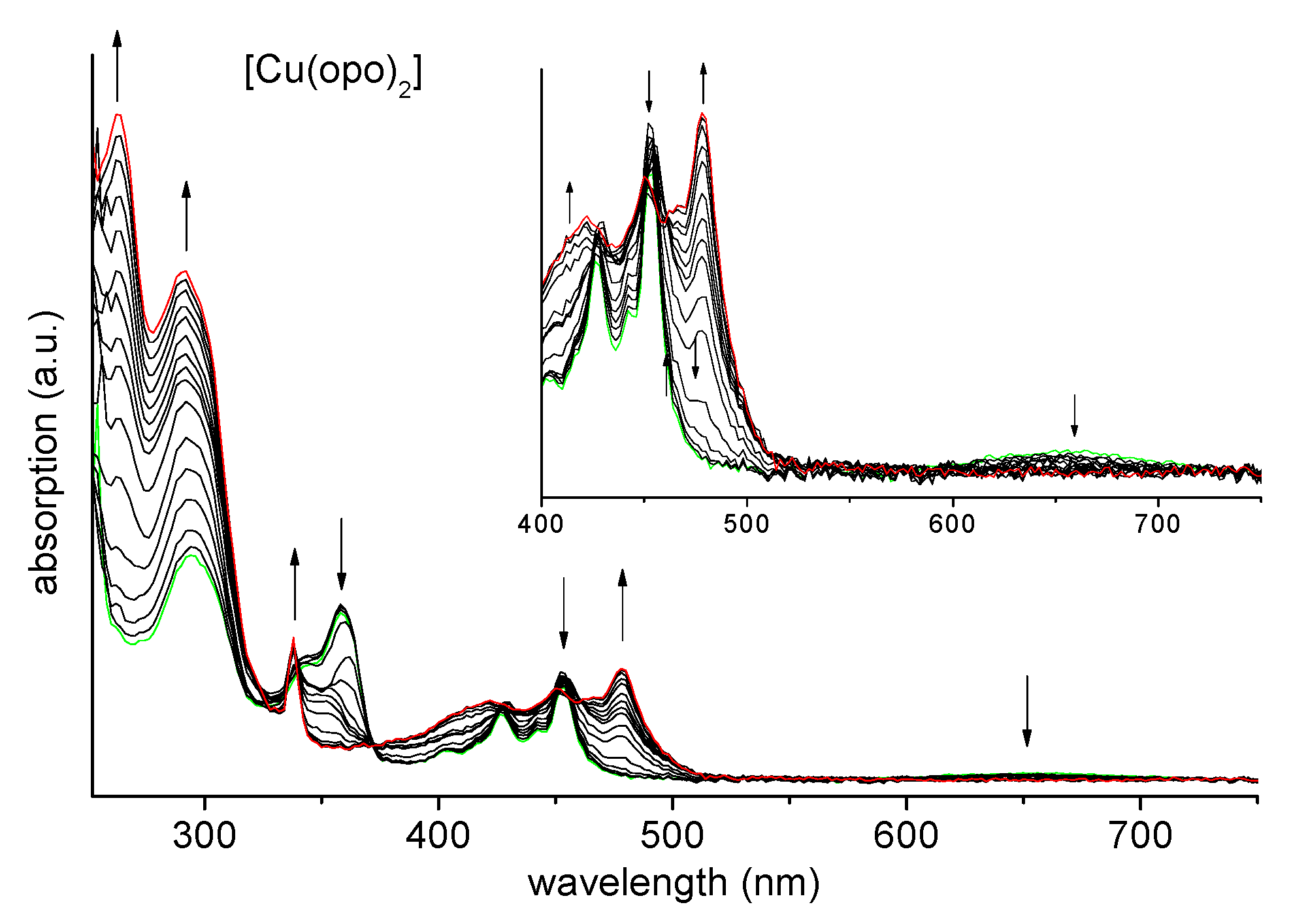
2.5. Absorption Spectroscopy and Spectroelectrochemistry
2.6. Antiproliferative Properties
3. Conclusions
4. Experimental Section
4.1. Instrumentation
4.2. Crystal Structure Determination
4.3. Antiproliferative Activities.
4.4. Materials and Syntheses
4.5. Synthesis of the Opo Complexes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koelsch, C.F.; Antes, J.A. Studies in the perinaphthalene series. IV. Some attempts to synthesize 9-phenyl-perinaphthanone-7. J. Org. Chem. 1941, 6, 558–565. [Google Scholar] [CrossRef]
- Jackman, L.M.; Trewella, J.C.; Haddon, R.C. Studies in Nuclear Magnetic Resonance Spectroscopy. 17. Deuteron Quadrupole Coupling Constants in Intramolecularly Hydrogen Bonded Systems. J. Am. Chem. Soc. 1980, 102, 2519–2525. [Google Scholar] [CrossRef]
- Rossetti, R.; Rayford, R.; Haddon, R.C.; Brus, L.E. Proton Localization in an Asymmetric Double-Minimum Potential: 2-Methyl-9-hydroxyphenalenone. J. Am. Chem. Soc. 1981, 103, 4303–4307. [Google Scholar] [CrossRef]
- Kunze, K.L.; de la Vega, J.R. Intramolecular Proton Exchange in 9-Hydroxyphenalen-1-one and Methyl-9-hydroxyphenalen-1-one. J. Am. Chem. Soc. 1984, 106, 6528–6537. [Google Scholar] [CrossRef]
- Engdahl, C.; Gogoll, A.; Edlund, U. Long-Range Deuterium Isotope Effects on 13C NMR Shifts of Intramolecularly Hydrogen-Bonded 9-Hydroxyphenalen-1-ones. Magn. Reson. Chem. 1991, 29, 54–62. [Google Scholar] [CrossRef]
- Nakai, H.; Sodeyama, K. Energy density analysis (EDA) of proton transfer reactions in malonaldehyde, tropolone, and 9-hydroxyphenalenone. J. Mol. Struct. (Theochem.) 2003, 637, 27–35. [Google Scholar] [CrossRef]
- Wang, D.-P.; Chen, S.-G.; Chen, D.-Z. Theoretical studies of conjugate and substituent effects on the intramolecular proton transfer: An HF/CIS study. J. Photochem. Photobiol. A 2004, 162, 407–414. [Google Scholar] [CrossRef]
- Kuwahara, D.; Koyano, H.; Manaka, T.; Nakamura, H.; Mochida, T.; Sugawara, T. Dynamics of 9-Hydroxyphenalenone Studied by One-Dimensional Solid-State Spin Exchange NMR. J. Phys. Chem. A 2006, 110, 13731–13735. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Schroeder, J.; Troe, J. Intramolecular Hydrogen Bonding in 1,8-Dihydroxyanthraquinone, 1-Aminoanthraquinone, and 9-Hydroxyphenalenone Studied by Picosecond Time-Resolved Fluorescence, Spectroscopy in a Supersonic Jet. J. Phys. Chem. B 2006, 110, 19820–19832. [Google Scholar] [CrossRef] [PubMed]
- Weihrich, R.; Limage, M.H.; Parker, F.; Fillaux, F. Proton tunnelling in the intramolecular hydrogen bond of 9-Hydroxyphenalenone. J. Mol. Struct. 2004, 700, 147–149. [Google Scholar] [CrossRef]
- Haddon, R.C.; Chichester, S.V.; Marshall, J.H. Electron Delocalization in 9-Oxidophenalenone Complexes of Boron and Beryllium. Tetrahedron 1986, 42, 6293–6300. [Google Scholar] [CrossRef]
- Chi, X.; Itkis, M.E.; Kirschbaum, K.; Pinkerton, A.A.; Oakley, R.T.; Cordes, A.W.; Haddon, R.C. Dimeric Phenalenyl-Based Neutral Radical Molecular Conductors. J. Am. Chem. Soc. 2001, 123, 4041–4048. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Itkis, M.E.; Tham, F.S.; Oakley, R.T.; Corders, A.W.; Haddon, R.C. Synthesis, Structure, and Physical Properties of a New Phenalenyl-Based Neutral Radical Crystal: Correlation between Structure and Transport Properties in Carbon-Based Molecular Conductors. Int. J. Quantum Chem. 2003, 95, 853–865. [Google Scholar] [CrossRef]
- Mandal, S.K.; Samanta, S.; Itkis, M.E.; Jensen, D.W.; Reed, R.W.; Oakley, R.T.; Tham, F.S.; Donnadieu, B.; Haddon, R.C. Resonating Valence Bond Ground State in Oxygen-Functionalized Phenalenyl-Based Neutral Radical Molecular Conductors. J. Am. Chem. Soc. 2006, 128, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Itkis, M.E.; Tham, F.S.; Reed, R.W.; Oakley, R.T.; Donnadieu, B.; Haddon, R.C. Phenalenyl-Based Neutral Radical Molecular Conductors: Substituent Effects on Solid-State Structures and Properties. J. Am. Chem. Soc. 2007, 129, 7163–7174. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Itkis, M.E.; Patrick, B.O.; Barclay, T.M.; Reed, R.W.; Oakley, R.T.; Cordes, A.W.; Haddon, R.C. The First Phenalenyl-Based Neutral Radical Molecular Conductor. J. Am. Chem. Soc. 1999, 121, 10395–10402. [Google Scholar] [CrossRef]
- Stary, J.; Liljenzin, J.O. Critical evaluation of equilibrium constants involving acetylacetone and its metal chelates. Pure Appl. Chem. 1982, 54, 2557–2592. [Google Scholar] [CrossRef]
- Kaim, W. Chelate rings of different sizes with non-innocent ligands. Dalton Trans. 2019, 48, 8521–8529. [Google Scholar] [CrossRef]
- Vigato, P.A.; Peruzzo, V.; Tamburini, S. The evolution of β-diketone or β-diketophenol ligands and related complexes. Coord. Chem. Rev. 2009, 253, 1099–1201. [Google Scholar] [CrossRef]
- Hynes, M.J. Reactions of of β-diketone complexes in solution. Rev. Inorg. Chem. 1990, 11, 21–78. [Google Scholar] [CrossRef]
- Demura, Y.; Kawato, T.; Kanatomi, H.; Murase, I. Metal Chelates of 9-Hydroxy-1-phenalenone. Bull. Chem. Soc. Jpn. 1975, 48, 2820–2824. [Google Scholar] [CrossRef] [Green Version]
- Neidlein, R.; Behzadi, Z. Synthesis and properties of some metal chelates of 9-hydroxyphenalen-1-one. Chemiker-Zeitung 1976, 100, 388–389. [Google Scholar]
- Hofmann, M.; Musso, H. Synthesis of metal complexes with bis(phenalenyl)imine ligands. Liebigs Ann. 1990, 11, 1119–1124. [Google Scholar] [CrossRef]
- Pal, S.K.; Tham, F.S.; Reed, R.W.; Oakley, R.T.; Haddon, R.C. Synthesis and characterization of germanium (IV) and silicon (IV) complexes derived from 9-hydroxyphenalenone: X-ray crystal and molecular structure of tris-(9-oxophenalenone)-germanium (IV) and silicon (IV) salts. Polyhedron 2005, 24, 2076–2083. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhou, J.; Zhang, Y.; Qiao, R.; Xia, S.; Chen, J.; Wang, X.; Zhang, B. Photodynamic Properties of Hypocrellin A, Complexes with Rare Earth Trivalent Ions: Role of the Excited State Energies of the Metal Ions. J. Phys. Chem. B 2007, 111, 2688–2696. [Google Scholar] [CrossRef]
- Van Deun, R.; Fias, P.; Nockemann, P.; Van Hecke, K.; Van Meervelt, L.; Binnemans, K. Visible-Light-Sensitized Near-Infrared Luminescence from Rare-Earth Complexes of the 9-Hydroxyphenalen-1-one Ligand. Inorg. Chem. 2006, 45, 10416–10418. [Google Scholar] [CrossRef]
- Van Deun, R.; Nockemann, P.; Fias, P.; Van Hecke, K.; Van Meervelt, L.; Binnemans, K. Visible light sensitisation of europium(III) luminescence in a 9-hydroxyphenal-1-one complex. Chem. Commun. 2005, 7, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Magri, A.; Fuhr, O.; Ruben, M. Phenalenyl-based mononuclear dysprosium complexes. Beilstein J. Nanotechnol. 2016, 7, 995–1009. [Google Scholar] [CrossRef] [Green Version]
- Mochida, T.; Torigoe, R.; Koinuma, T.; Asano, C.; Satou, T.; Koike, K.; Nikaido, T. Platinum-Group Chelate Complexes with 9-Hydroxyphenalenone Derivatives: Synthesis, Structures, Spectroscopic Properties and Cytotoxic Activities. Eur. J. Inorg. Chem. 2006, 2006, 558–565. [Google Scholar] [CrossRef]
- Kokoschka, M.; Galgonek, J.; Vondrasek, J.; Hobza, P. Computational methods for the description of pharmacologically relevant platinum complexes—Molecular structure and bond dissociation. Phys. Chem. Chem. Phys. 2016, 18, 4051–4062. [Google Scholar] [CrossRef]
- Dutta, P.K.; Sharma, R.; Kumari, S.; Dubey, R.D.; Sarkar, S.; Paulraj, J.; Vijaykumar, G.; Pandey, M.; Sravanti, L.; Samarla, M.; et al. A safe and efficacious Pt(II) anticancer prodrug: Design, synthesis, in vitro efficacy, the role of carrier ligands and in vivo tumour growth inhibition. Chem. Commun. 2019, 55, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- López-Arencibia, A.; Bethencourt-Estrell, C.J.; Freijo, M.B.; Reyes-Batlle, M.; Sifaoui, I.; Nicolás-Hernández, D.S.; McNaughton-Smith, G.; Lorenzo-Morales, J.; Abad-Grillo, T.; Pinero, J.E. New phenalenone analogues with improved activity against Leishmania species. Biomed. Pharmacother. 2020, 132, 110814. [Google Scholar] [CrossRef] [PubMed]
- Freijo, M.B.; Lopez-Arencibia, A.; Pinero, J.E.; McNaughton-Smith, G.; Abad-Grillo, T. Design, synthesis and evaluation of amino-substituted 1H-phenalen-1-ones as anti-leishmanial agents. Eur. J. Med. Chem. 2018, 143, 1312–1324. [Google Scholar] [CrossRef]
- O’Brien, E.M.; Morgan, B.J.; Mulrooney, C.A.; Carroll, P.J.; Kozlowski, M.C. Perylenequinone Natural Products: Total Synthesis of Hypocrellin A. J. Org. Chem. 2010, 75, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Khan, S.I.; Jacob, M.R.; Tekwani, B.L.; Li, Z.; Pasco, D.S.; Walker, L.A.; Khan, I.A. Antimicrobial and Antileishmanial Activities of Hypocrellins A and B. Antimicrob. Agents Chemother. 2004, 48, 4450–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwu, Z.; Zimmermann, J.; Meyers, T.; Lown, J.W. Design, Synthesis and Investigation of Mechanisms of Action of Novel Protein Kinase C Inhibitors: Perylenequinoid Pigments. Biochem. Pharmacol. 1994, 47, 373–385. [Google Scholar] [CrossRef]
- Niu, T.; Tian, Y.; Shi, Y.; Guo, G.; Tong, Y.; Wang, G. Antifibrotic effects of Hypocrellin A combined with LED red light irradiation on keloid fibroblasts by counteracting the TGF-β/Smad/autophagy/apoptosis signalling pathway. Photodiag. Photodyn. Ther. 2021, 34, 102202. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, W.; Wu, J.; Zheng, X.; Ge, J.; Ren, H.; Zhang, W.; Lee, C.-S.; Wang, P. Near-Infrared Hypocrellin Derivatives for Synergistic Photodynamic and Photothermal Therapy. Chem.-Asian J. 2020, 15, 3456–3468. [Google Scholar] [CrossRef]
- Tian, C.; Xu, S.; Chen, S.; Shen, J.; Zhang, M.; Shen, T. Chelation of Hypocrellin B with Zinc Ions with Electron Paramagnetic Resonance (EPR) Evidence of the Photodynamic Activity of the Resulting Chelate. Free Radic. Res. 2001, 35, 543–554. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, Y.-J.; Zhou, Q.-X.; Lei, W.-H.; Chen, J.-R.; Wang, X.-S.; Zhang, B.-W. Dinuclear Cu(II) Hypocrellin B Complexes with Enhanced Photonuclease Activity. Inorg. Chem. 2010, 49, 10108–10116. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hou, Y.-J.; Zhou, Q.-X.; Chen, J.-R.; Zhang, B.-W.; Wang, X.-S. A new Co(III)-hypocrellin B complex with enhanced photonuclease activity. J. Inorg. Biochem. 2011, 105, 978–984. [Google Scholar] [CrossRef]
- Zhou, L.; Ge, X.; Liu, J.; Zhou, J.; Wei, S.; Li, F.; Shen, J. Internal heavy atom effect of Au(III) and Pt(IV) on hypocrellin A for enhanced in vitro photodynamic therapy of cancer. Bioorg. Med. Chem. Lett. 2013, 23, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, C.Z. A molecular logic gate for the highly selective recognition of pyrophosphate with a hypocrellin A-Zn(II) complex. Analyst 2010, 135, 2853–2857. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Lahiri, G.K. The coordination potential of indigo, anthraquinone and related redox-active dyes. Coord. Chem. Rev. 2019, 393, 1–8. [Google Scholar] [CrossRef]
- Hazari, A.S.; Paretzki, A.; Fiedler, J.; Zalis, S.; Kaim, W.; Lahiri, G.K. Different manifestations of enhanced π-acceptor ligation at every redox level of [Os(9-OP)L2]n, n = 2+, +, 0, − (9-OP− = 9-oxidophenalenone and L = bpy or pap). Dalton Trans. 2016, 45, 18241–18251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwala, H.; Scherer, T.M.; Mobin, S.M.; Kaim, W.; Lahiri, G.K. Bidirectional non-innocence of the β-diketonato ligand 9-oxidophenalenone (L−) in [Ru([9]aneS3)(L)(dmso)]n, [9]aneS3 = 1,4,7-trithiacyclononane. Dalton Trans. 2014, 43, 3939–3948. [Google Scholar] [CrossRef]
- Das, A.; Scherer, T.M.; Mondal, P.; Mobin, S.M.; Kaim, W.; Lahiri, G.K. Experimental and DFT Evidence for the Fractional Non-Innocence of a β-Diketonate Ligand. Chem.-Eur. J. 2012, 18, 14434–14443. [Google Scholar] [CrossRef]
- Das, A.; Scherer, T.M.; Mobin, S.M.; Kaim, W.; Lahiri, G.K. 9-Oxidophenalenone: A Noninnocent β-Diketonate Ligand? Inorg. Chem. 2012, 51, 4390–4397. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Samuel, P.P.; Schulzke, C.; Mandal, S.K. Main group chemistry of 9-hydroxophenalenone: Syntheses and structural characterization of the alkaline earth and zinc complexes. J. Chem. Sci. 2014, 126, 1581–1588. [Google Scholar] [CrossRef]
- Lehner, P.; Staudinger, C.; Borisov, S.M.; Klimant, I. Ultra-sensitive optical oxygen sensors for characterization of nearly anoxic systems. Nat. Commun. 2014, 5, 4460. [Google Scholar] [CrossRef] [Green Version]
- Yakuschenko, I.K.; Kaplunov, M.G.; Krasnikova, S.S. Some Metal Complexes of 9-Hydroxyphenalenone as Novel Electron Transporting Materials for OLEDs. Mol. Cryst. Liq. Cryst. 2014, 590, 24–28. [Google Scholar] [CrossRef]
- Bag, P.; Itkis, M.E.; Stekovic, D.; Pal, S.K.; Tham, F.S.; Haddon, R.C. Band Structure Engineering by Substitutional Doping in Solid-State Solutions of [5-Me-PLY(O,O)]2B(1−x)Bex Radical Crystals. J. Am. Chem. Soc. 2015, 137, 10000–10008. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sau, S.C.; Mandal, S.K. Exploring Closed-Shell Cationic Phenalenyl: From Catalysis to Spin Electronics. Acc. Chem. Res. 2017, 50, 1679–1691. [Google Scholar] [CrossRef]
- Stekovic, D.; Bag, P.; Shankhari, P.; Fokwa, B.P.T.; Itkis, M.E. Effect of Substitution on the Hysteretic Phase Transition in a Bistable Phenalenyl-Based Neutral Radical Molecular Conductor. Chem. Eur. J. 2019, 25, 4166–4174. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J.; Datta, P.; Das, A.; Jomy, S.; Mandal, S.K. Switching between mono and doubly reduced odd alternant hydrocarbon: Designing a redox catalyst. Chem. Sci. 2021, 12, 3039–3049. [Google Scholar] [CrossRef]
- Dey, S.K.; Honecker, A.; Mitra, P.; Mandal, S.K.; Mukherje, A. Magnetostructural Studies on Tetranuclear Manganese [MnIII2MnII2] Complexes of 9-Hydroxyphenalenone with Weak π···π Interactions. Eur. J. Inorg. Chem. 2012, 2012, 5814–5824. [Google Scholar] [CrossRef]
- Pariyar, A.; Vijaykumar, G.; Bhunia, M.; Dey, S.K.; Singh, S.K.; Kurungot, S.; Mandal, S.K. Switching Closed-Shell to Open-Shell Phenalenyl: Toward Designing Electroactive Materials. J. Am. Chem. Soc. 2015, 137, 5955–5960. [Google Scholar] [CrossRef]
- Tan, C.-M.; Zhou, Y.-H.; Chen, C.-Y.; Yu, J.-F.; Chen, K.-Q. Spin filtering and rectifying effects in the zinc methyl phenalenyl molecule between graphene nanoribbon leads. Org. Electron. 2016, 28, 244–251. [Google Scholar] [CrossRef]
- Wahl, I.M.; Westphal, E.; da Costa Gouveia, T.L.; Sousa Santana, F.; Hughes, D.L.; Ribeiro, R.R.; Piccoli, L.H.R.; Winnischofer, H. Methyl Ester Functionalized Phenalenyl Arene- and Bipyridine−Ruthenium-Based Complexes for Electroactive Langmuir−Blodgett Films. Inorg. Chem. 2019, 58, 8408–8418. [Google Scholar] [CrossRef] [PubMed]
- Vijaykumar, G.; Bhunia, M.; Mandal, S.K. A phenalenyl-based nickel catalyst for the hydroboration of olefins under ambient conditions. Dalton Trans. 2019, 48, 5779–5784. [Google Scholar] [CrossRef]
- Vardhanapu, P.K.; Ahmed, J.; Jose, A.; Shaw, B.K.; Sen, T.K.; Mathews, A.A.; Mandal, S.K. Phenalenyl Based Aluminum Compound for Catalytic C−H Arylation of Arene and Heteroarenes at Room Temperature. J. Org. Chem. 2019, 84, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Vijaykumar, G.; Pariyar, A.; Ahmed, J.; Shaw, B.K.; Adhikari, D.; Mandal, S.K. Tuning the redox non-innocence of a phenalenyl ligand toward efficient nickel-assisted catalytic hydrosilylation. Chem. Sci. 2018, 9, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Ahmed, J.; Shaw, B.K.; Jose, A.; Mandal, S.K. An Iron-Based Long-Lived Catalyst for Direct C‒H Arylation of Arenes and Heteroarenes. Chem.-Eur. J. 2018, 24, 17651–17655. [Google Scholar] [CrossRef]
- Bellan, E.V.; Thevenin, L.; Gayet, F.; Fliedel, C.; Poli, R. Catalyzed Chain Transfer in Vinyl Acetate Polymerization Mediated by 9-Oxyphenalenone Cobalt(II) Complexes. ACS Macro Lett. 2017, 6, 959–962. [Google Scholar] [CrossRef]
- Bhunia, M.; Sahoo, S.R.; Shaw, B.K.; Vaidya, S.; Pariyar, A.; Vijaykumar, G.; Adhikari, D.; Mandal, S.K. Storing redox equivalent in the phenalenyl backbone towards catalytic multi-electron reduction. Chem. Sci. 2019, 10, 7433–7441. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.K.; Mukherjee, A. Zero-Order Catechol Oxidase Activity by a Mononuclear Manganese(III) Complex Showing High Turnover Comparable to Catechol Oxidase Enzyme. ChemCatChem. 2013, 5, 3533–3537. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, T.K.; Chowdhury, A.D.; Mobin, S.M.; Lahiri, G.K. Electronic structure and catalytic aspects of [(trpy)(Cl)Ru(L)]n incorporating potential non-innocent ligands, L−: 9-Oxidophenalenone and trpy: 2,2’:6’,2”-terpyridine. Polyhedron 2013, 52, 1130–1137. [Google Scholar] [CrossRef]
- Sen, T.K.; Mukherjee, A.; Modak, A.; Mandal, S.K.; Koley, D. Substitution effect on phenalenyl backbone in the rate of organozinc catalyzed ROP of cyclic esters. Dalton Trans. 2013, 42, 1893–1904. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sen, T.K.; Ghorai, P.K.; Samuel, P.P.; Schulzke, C.; Mandal, S.K. Phenalenyl-Based Organozinc Catalysts for Intramolecular Hydroamination Reactions: A Combined Catalytic, Kinetic, and Mechanistic Investigation of the Catalytic Cycle. Chem.-Eur. J. 2012, 18, 10530–10545. [Google Scholar] [CrossRef]
- Ahmed, J.; Sreejyothi, P.; Vijaykumar, G.; Jose, A.; Raj, M.; Mandal, S.K. A new face of phenalenyl-based radicals in the transition metal-free C–H arylation of heteroarenes at room temperature: Trapping the radical initiator via C–C σ-bond formation. Chem. Sci. 2017, 8, 7798–7806. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Chung, T.D.; Kim, H. Voltammetric determination of the pKa of various acids in polar aprotic solvents using 1,4-benzoquinone. J. Electroanal. Chem. 2001, 498, 209–215. [Google Scholar] [CrossRef]
- Small, D.; Zaitsev, V.; Jung, Y.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Intermolecular π-to-π Bonding between Stacked Aromatic Dyads. Experimental and Theoretical Binding Energies and Near-IR Optical Transitions for Phenalenyl Radical/Radical versus Radical/Cation Dimerizations. J. Am. Chem. Soc. 2004, 126, 13850–13858. [Google Scholar] [CrossRef] [PubMed]
- Caes, B.; Jensen, D., Jr. Synthesis and Characterization of 9-Hydroxyphenalenone Using 2D NMR Techniques. J. Chem. Educ. 2008, 85, 413–415. [Google Scholar] [CrossRef]
- Baker, T.M.; Howard, K.M.; Brennessel, W.W.; Neidig, M.L. Crystal structure of a third polymorph of tris(acetylacetonato-κ2-O:O’)iron(III). Acta Cryst. Sect. E Struct. Rep. Online 2015, E71, m228–m229. [Google Scholar] [CrossRef]
- Bullen, G.J.; Mason, R.; Pauling, P. The Crystal and Molecular Structure of Bis(acetylacetonato)nickel(II). Inorg. Chem. 1965, 4, 456–462. [Google Scholar] [CrossRef]
- Mehrotra, R.C.; Bohra, R.; Gaur, D.P. Metal ß-Diketones and Allied Derivatives; Academic Press: London, UK; New York, NY, USA, 1978. [Google Scholar]
- Kessler, V.G.; Spijksma, G.I.; Seisenbaeva, G.A.; Hakansson, S.; Blank, D.H.A.; Bouvmeester, H.J.M. New insight in the role of modifying ligands in the sol-gel processing of metal alkoxide precursors: A possibility to approach new classes of materials. J. Sol-Gel Sci. Technol. 2006, 40, 163–179. [Google Scholar] [CrossRef]
- Montgomery, H.; Lingafaelter, E.C. The crystal structure of diaquobisacetylacetonatonickel(II). Acta Cryst. 1964, 17, 1481–1482. [Google Scholar] [CrossRef]
- Kudrat-E-Zahan, M.; Nishida, Y.; Sakiyama, H. Identification of cis/trans isomers of bis(acetylacetonato)nickel(II) complexes in solution based on electronic spectra. Inorg. Chim. Acta 2010, 363, 168–172. [Google Scholar] [CrossRef]
- Tahir, A.A.; Hamid, M.; Zeller, M.; Mazhar, M.; Hunter, A.D. Bis(µ-acetylacetonato-κ2-O:O’)bis[(acetylacetonato-κ2-O:O’)aquanickel(II)] hemihydrate. Acta Cryst. Sect. E Struct. Rep. Online 2007, E63, m272–m274. [Google Scholar] [CrossRef]
- Darensbourg, M.; Buonomo, R.M.; Reibenspies, J.H. Crystal structure of tetrakis[(µ3-methoxo-2,4-pentanedionatomethanolnickel(II)], C28H56Ni4O. Z. Kristallogr. 1995, 210, 469–470. [Google Scholar]
- El Fallah, M.S.; Rentschler, E.; Caneschi, A.; Gatteschi, D. Synthesis, crystal structure and magnetic properties of the tetranuclear complex [Ni4(OCH3)4(dbm)4(CH3OH)4]2·(C2H5)2O. Inorg. Chim. Acta 1996, 247, 231–235. [Google Scholar] [CrossRef]
- Worthy, A.; Grosjean, A.; Pfrunder, M.C.; Xu, Y.; Yan, C.; Edwards, G.; Clegg, J.K.; McMurtrie, J.C. Atomic resolution of structural changes in elastic crystals of copper(II) acetylacetonate. Nat. Chem. 2018, 10, 65–69. [Google Scholar] [CrossRef]
- Moreno, Y.; Arrue, R.; Saavedra, R.; Pivan, J.-Y.; Pena, O.; Roisnell, T. Magnetic and Structural Study of Unsolvated [Cu(acac)2], (acac = acetylacetonate). J. Chil. Chem. Soc. 2013, 58, 2122–2124. [Google Scholar] [CrossRef] [Green Version]
- Ritterskamp, N.; Sharples, K.; Richards, E.; Folli, A.; Chiesa, M.; Platts, J.A.; Murphy, D.M. Understanding the Coordination Modes of [Cu(acac)2(imidazole)n=1,2] Adducts by EPR, ENDOR, HYSCORE, and DFT Analysis. Inorg. Chem. 2017, 56, 11862–11875. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.; Sharples, K.M.; Platts, J.A.; Murphy, D.M. Structure determination of bound nitrogen-based adducts with copper(II) acetylacetonato; an EPR, ENDOR and DFT study. Phys. Chem. Chem. Phys. 2015, 17, 11445–11454. [Google Scholar] [CrossRef] [PubMed]
- Butera, R.A.; Waldeck, D.H. An EPR Experiment for the Undergraduate Physical Chemistry Laboratory. J. Chem. Educ. 2000, 77, 1489–1491. [Google Scholar] [CrossRef]
- Yordanov, N.D.; Shopov, D. EPR Spectra of Mixed-Ligand Copper(II) Complexes in Solution. J. Inorg. Nucl. Chem. 1976, 38, 137–140. [Google Scholar] [CrossRef]
- Nunes, P.; Nagy, N.V.; Alegria, E.C.B.A.; Pombeiro, A.J.L.; Correia, I. The solvation and electrochemical behavior of copper acetylacetonate complexes in ionic liquids. J. Mol. Struct. 2014, 1060, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Collison, D.; Powell, A.K. Electron Spin Resonance Studies of “FeO6” Tris Chelate Complexes: Models for the Effects of Zero-Field Splitting in Distorted S = 5/2 Spin Systems. Inorg. Chem. 1990, 29, 4735–4746. [Google Scholar] [CrossRef]
- Hedewy, S.; Hoffmann, K. Electron Paramagnetic Resonance of Ferric Acetyl Acetonate. Phys. Status Solidi A 1986, 97, 129–133. [Google Scholar] [CrossRef]
- Mohammadnezhad, G.; Amirian, A.A.; Görls, H.; Plass, W.; Sandleben, A.; Schäfer, S.; Klein, A. Redox Instability of Copper(II) Complexes of a Triazine-Based PNP Pincer. Eur. J. Inorg. Chem. 2021, 2021, 1140–1151. [Google Scholar] [CrossRef]
- Bubrin, M.; Kvapilová, H.; Fiedler, J.; Ehret, F.; Záliš, S.; Kaim, W. Hybrid α-Diimine/Bis(chalcogenoether) Ligands for Copper(I) and Copper(II) Complexes. Z. Anorg. Allg. Chem. 2018, 644, 661–670. [Google Scholar] [CrossRef]
- Butsch, B.; Klein, A.; Nitsche, S.; Stirnat, K.; Hawkett, J.R.; McInnes, E.J.L.; Bauer, M. Generation and characterisation of the phenoxyl-radical containing Cu(II) complex [Cu(triaz)2]+ (triaz− = O,N chelating triazole-phenolate). Dalton Trans. 2012, 41, 11464–11475. [Google Scholar] [CrossRef] [PubMed]
- Nagao, H.; Komeda, N.; Mukaida, M.; Suzuki, M.; Tanaka, K. Structural and Electrochemical Comparison of Copper(II) Complexes with Tripodal Ligands. Inorg. Chem. 1996, 35, 6809–6815. [Google Scholar] [CrossRef]
- Pladzyk, A.; Ponikiewski, L.; Dołega, A.; Słowy, K.; Sokołowska, A.; Dziubinska, K.; Hnatejko, Z. Structural Variety of Cobalt(II), Nickel(II), Zinc(II), and Cadmium(II) Complexes with 4,4’-Azopyridine:Synthesis, Structure and Luminescence Properties. Chem.-Asian J. 2015, 10, 2388–2396. [Google Scholar] [CrossRef]
- Brahma, S.; Shivashankar, A. Zinc acetylacetonate hydrate adducted with nitrogen donor ligands: Synthesis, spectroscopic characterization, and thermal analysis. J. Mol. Struct. 2015, 1101, 41–49. [Google Scholar] [CrossRef]
- Carlotto, S.; Floreano, L.; Cossaro, A.; Dominguez, M.; Rancan, M.; Sambia, M.; Casarin, M. The electronic properties of three popular high spin complexes [TM(acac)3, TM = Cr, Mn, and Fe] revisited: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2017, 19, 24840–24854. [Google Scholar] [CrossRef]
- Schäfer, S.; Ott, I.; Gust, R.; Sheldrick, W.S. Influence of the Polypyridyl (pp) Ligand Size on the DNA Binding Properties, Cytotoxicity and Cellular Uptake of Organoruthenium(II) Complexes of the Type [(η6-C6Me6)Ru(L)(pp)]n+ [L = Cl, n = 1; L = (NH2)2CS, n = 2]. Eur. J. Inorg. Chem. 2007, 2007, 3034–3046. [Google Scholar] [CrossRef]
- Duling, D.R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra (PEST Winsim). J. Magn. Reson. B 1994, 104, 105–110. [Google Scholar] [CrossRef]
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [Google Scholar] [CrossRef]
- Kaim, W.; Klein, A. (Eds.) Spectroelectrochemistry; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Sheldrick, G.M. ShelXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXL-2017/1, Program for the Solution of Crystal Structures; University of Göttingen: Göttingen, Germany, 2017. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- STOE X-RED. Data Reduction Program, Version 1.31/Windows; STOE & Cie: Darmstadt, Germany, 2005.
- STOE X-SHAPE. Crystal Optimisation for Numerical Absorption Correction, Version 1.06/Windows; STOE & Cie: Darmstadt, Germany, 1999.
- Haddon, R.C.; Rayford, R.; Hirani, A.M. 2-Methyl- and 5-methyl-9-hydroxyphenalenone. J. Org. Chem. 1981, 46, 4587–4588. [Google Scholar] [CrossRef]
- Gmelins Handbuch der Anorganischen Chemie; Verlag Chemie: Weinheim, Germany, 1975; p. 152ff.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E½(Mn+1/Mn) | E½(1ligand) | E½(2ligand) |
|---|---|---|---|
| Hopo | - | −1.52 | −2.51 |
| [Fe(opo)3] b | −1.19 | −1.31 | −1.60 |
| [Fe(acac)3] | −1.13 | −1.41 | - |
| [Cu(opo)2] | −0.92 c | −1.61 | −1.91 |
| [Cu(acac)2] | −1.19 d | −1.66 | −2.51 |
| [Zn(opo)2] | - | −1.42 | −2.50 |
| [Zn(acac)2] | - | −1.51 | - |
| Compound | ε/nm (ε/Lmol−1cm−1) | Solvent |
|---|---|---|
| Hopo | 350(1230), 393(333), 413(606), 428(504), 438(717), 451(133) | CH3OH |
| [Zn(opo)2] | 351(1512), 393(328), 414(589), 429(626), 438(705), 453(377) | CH3OH |
| Hopo | 350(1231), 393(331), 413(607), 428(502), 438(719), 451(131) | DMF |
| [Cu(opo)2] | 294(8600), 358(6400), 403(1070), 426(2470), 452(3520), 651(260) | DMF |
| [Cu(acac)2] | 376sh(5480), 639(1120), 642(260) | DMF |
| Hopo | 235(950), 257(607), 265(567), 350(1230), 393(296), 414 (538), 430(397), 439(607) | THF |
| [Fe(opo)3] | 265(15690), 327(4610), 361(6310), 368(6280), 453(3470), 480(2930), 571(230) | THF |
| [Fe(acac)3] | 273(28449), 353(4154), 435(4193) | THF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butsch, K.; Haseloer, A.; Schmitz, S.; Ott, I.; Schur, J.; Klein, A. FeIII, CuII and ZnII Complexes of the Rigid 9-Oxido-phenalenone Ligand—Spectroscopy, Electrochemistry, and Cytotoxic Properties. Int. J. Mol. Sci. 2021, 22, 3976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083976
Butsch K, Haseloer A, Schmitz S, Ott I, Schur J, Klein A. FeIII, CuII and ZnII Complexes of the Rigid 9-Oxido-phenalenone Ligand—Spectroscopy, Electrochemistry, and Cytotoxic Properties. International Journal of Molecular Sciences. 2021; 22(8):3976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083976
Chicago/Turabian StyleButsch, Katharina, Alexander Haseloer, Simon Schmitz, Ingo Ott, Julia Schur, and Axel Klein. 2021. "FeIII, CuII and ZnII Complexes of the Rigid 9-Oxido-phenalenone Ligand—Spectroscopy, Electrochemistry, and Cytotoxic Properties" International Journal of Molecular Sciences 22, no. 8: 3976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083976