
TRPM7-Mediated Calcium Transport in HAT-7 Ameloblasts

 ,
,  , , and
, , and 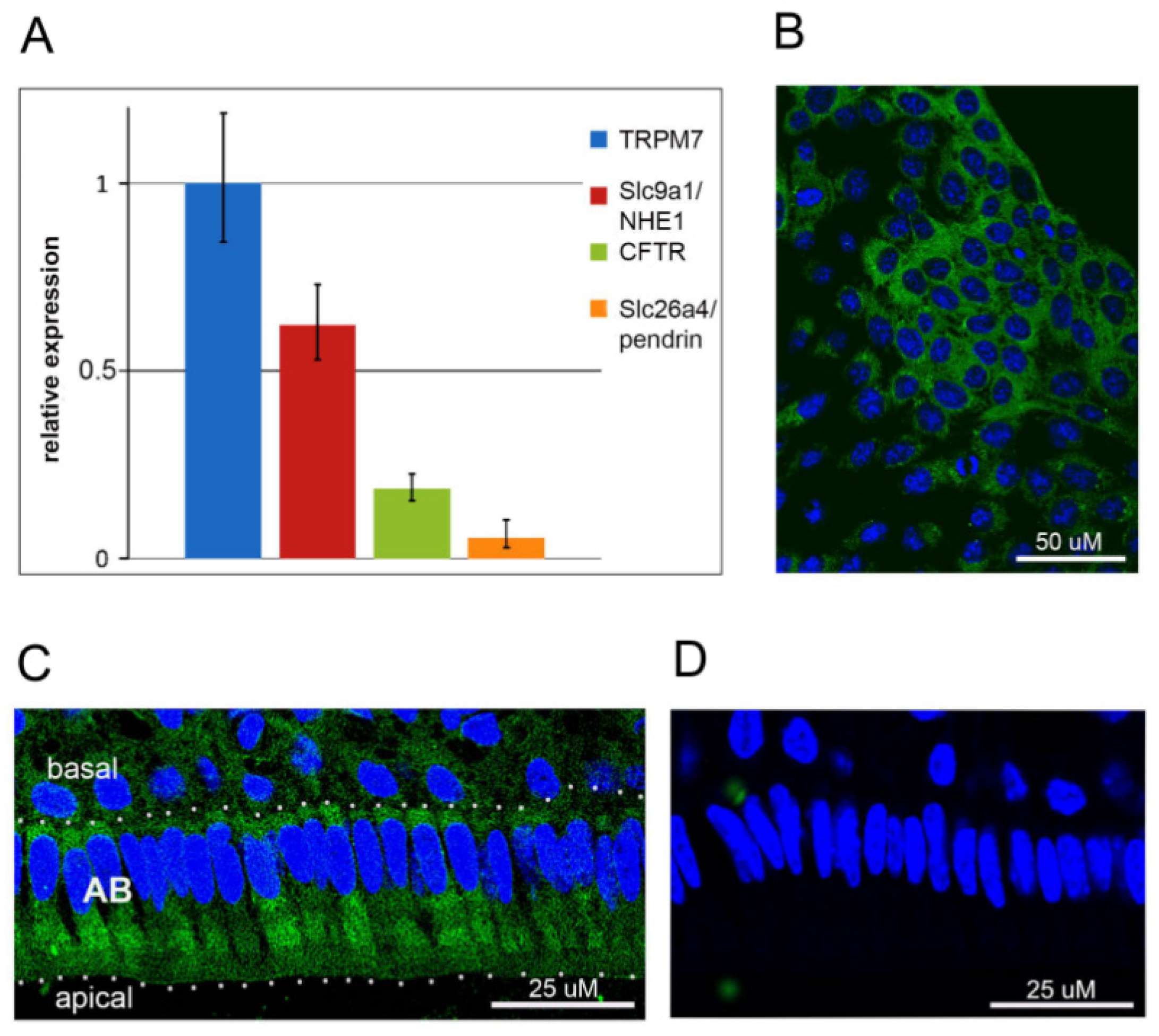{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TRPM7 Is Expressed in HAT-7 Cells and in Mouse Ameloblasts
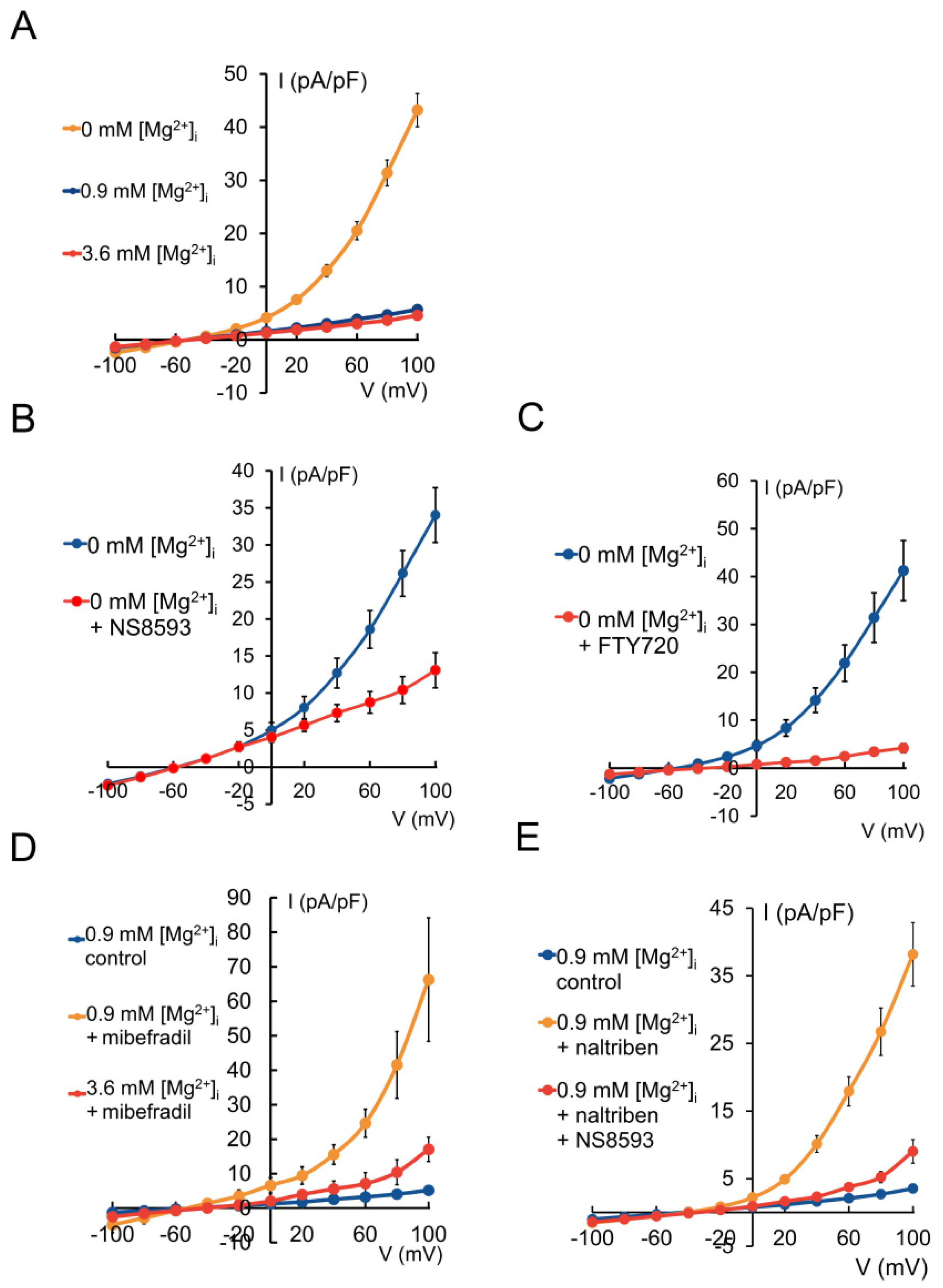
2.2. HAT-7 Cells Exhibit TRPM7-Like Ion Currents
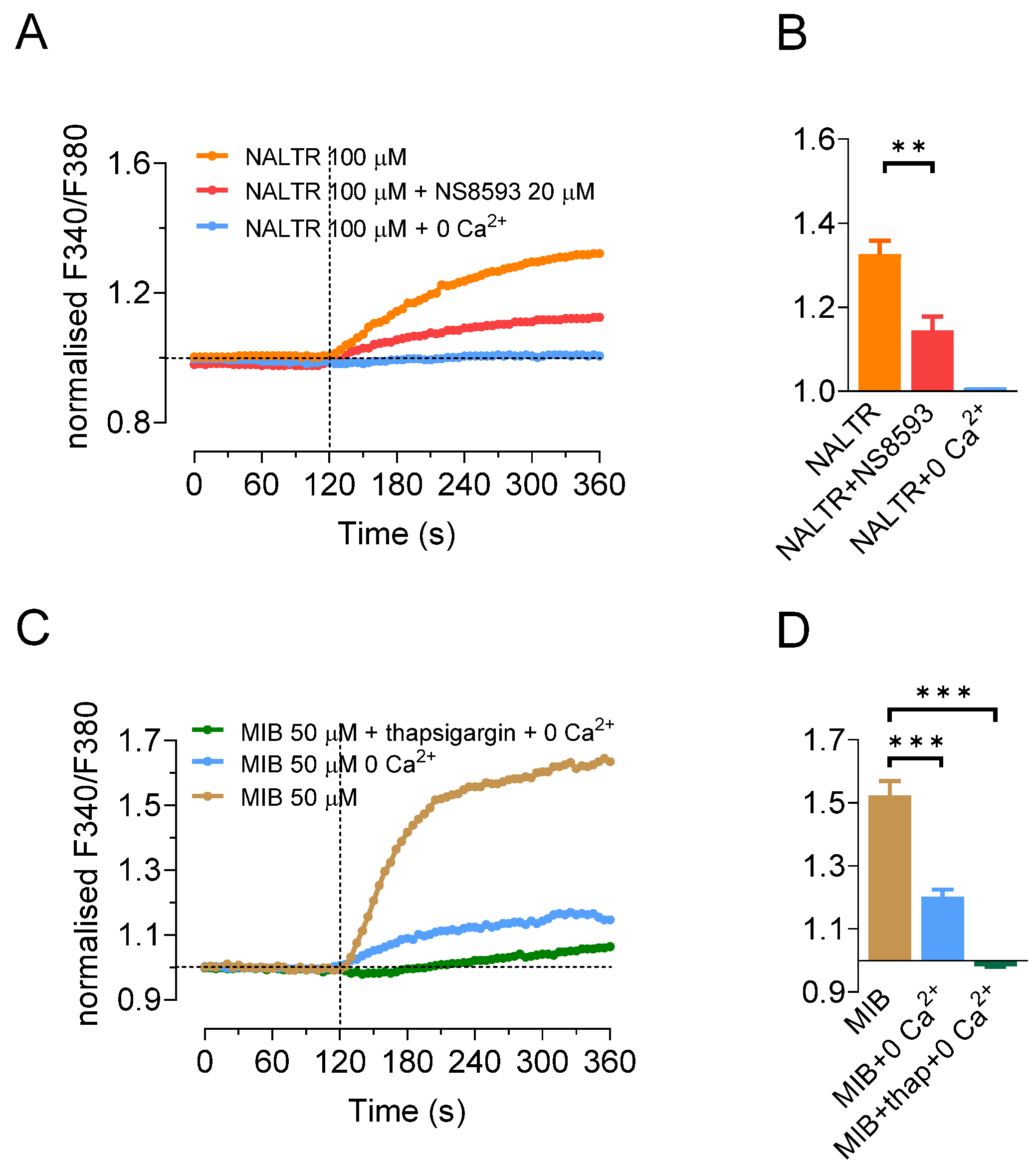
2.3. TRPM7 Activators Stimulate Ca2+ Influx in HAT-7 Cells
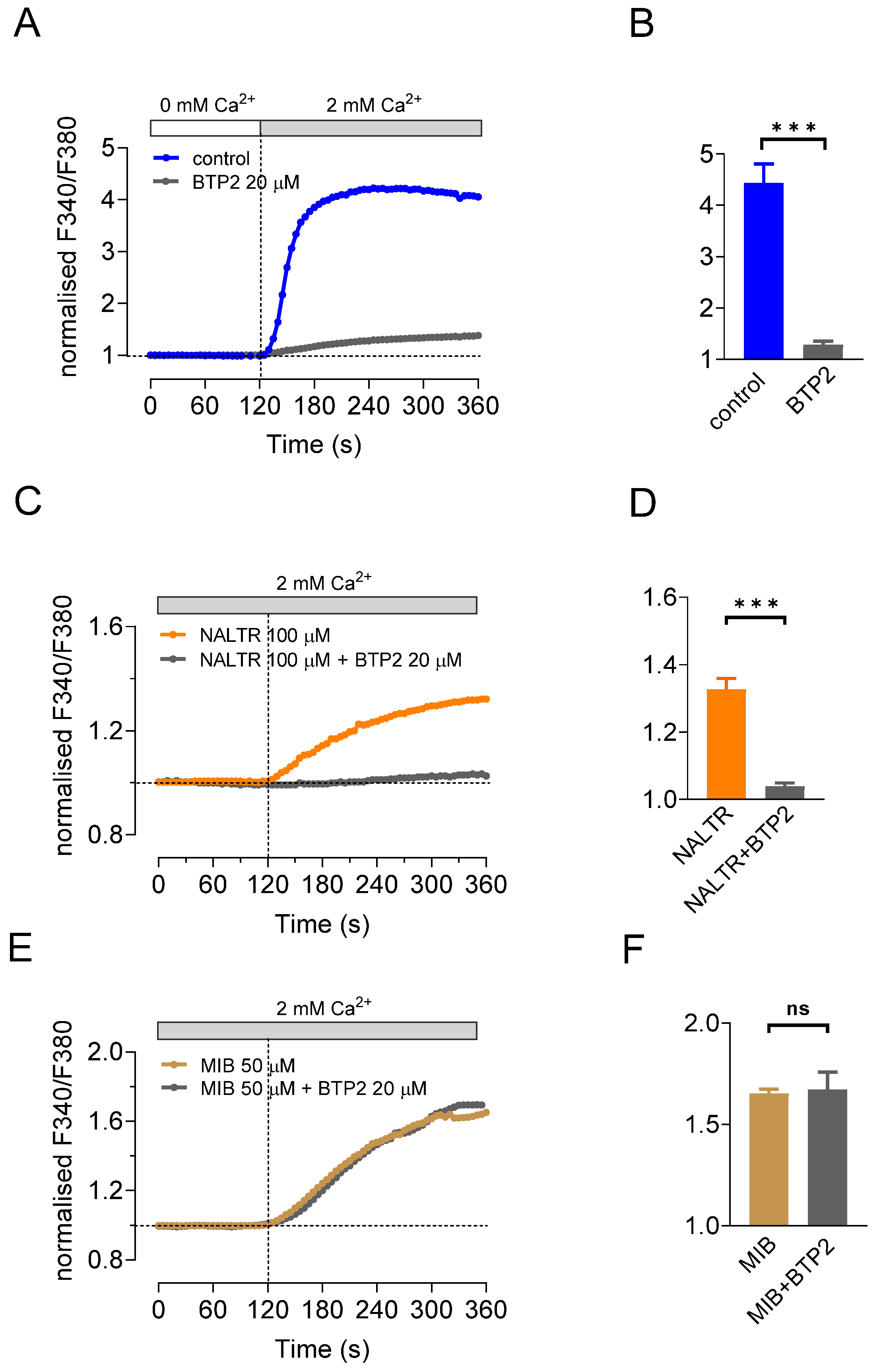
2.4. SOCE Blocker BTP2 Inhibits Ca2+ Influx Stimulated by Naltriben but Not Mibefradil
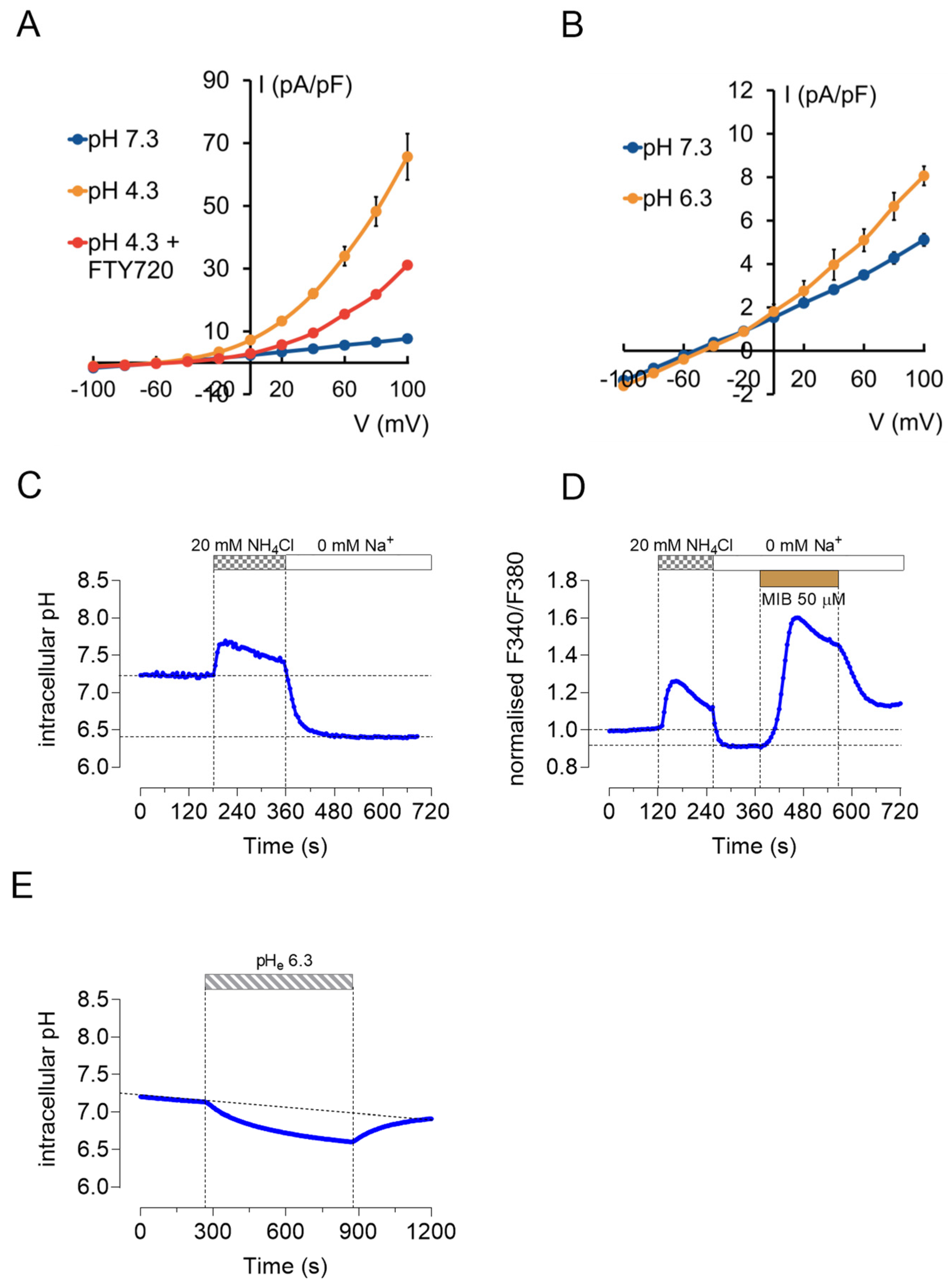
2.5. TRPM7 Currents and Ca2+ Influx Are pH Sensitive
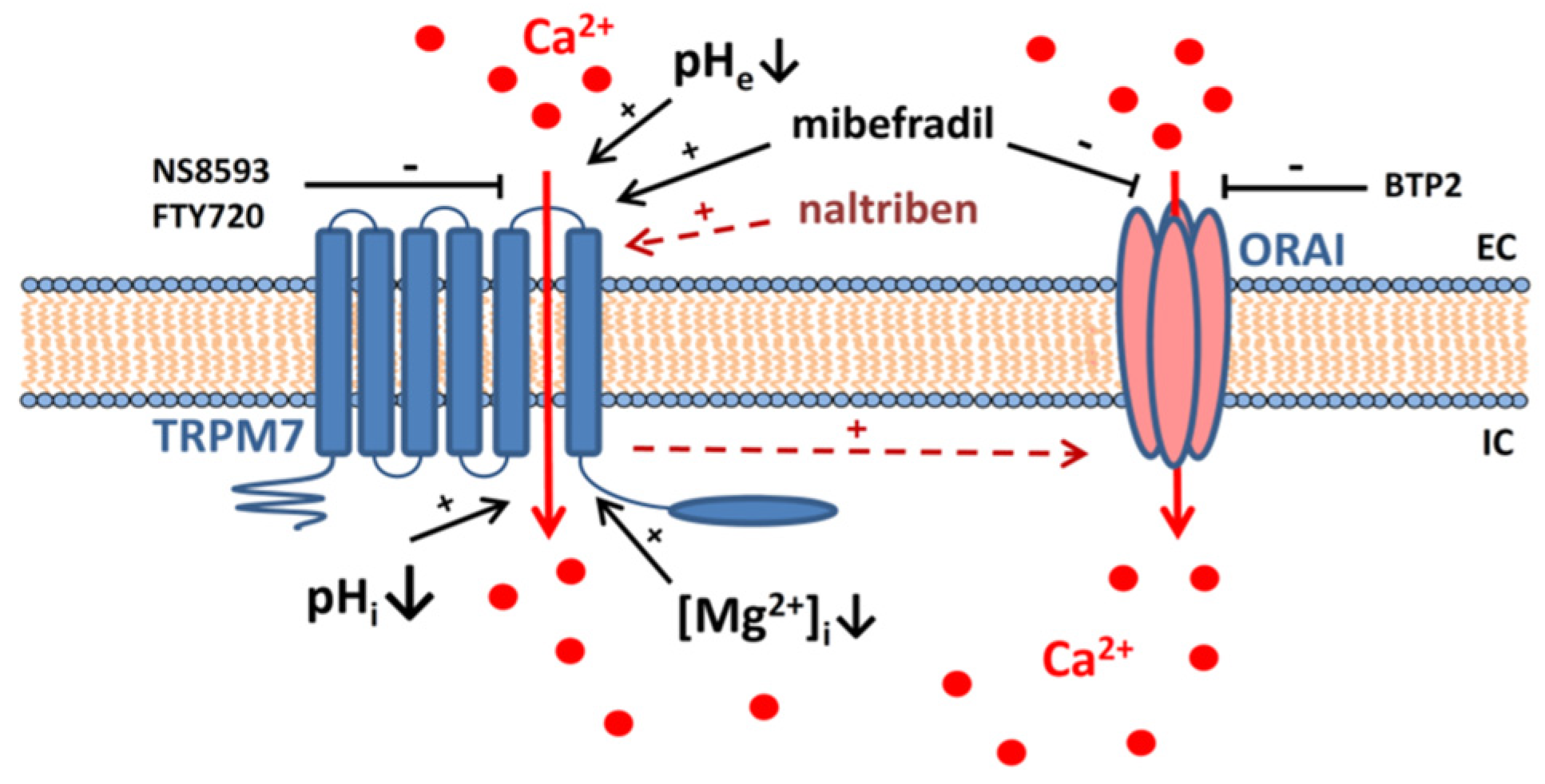
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RT-qPCR
4.3. Immunohistochemistry
4.4. Electrophysiology
4.5. Calcium Imaging
4.6. Intracellular pH Measurements
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bronckers, A.L. Ion Transport by Ameloblasts during Amelogenesis. J. Dent. Res. 2017, 96, 243–253. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Habelitz, S.; Wright, J.T.; Paine, M.L. Dental enamel formation and implications for oral health and disease. Physiol. Rev. 2017, 97, 939–993. [Google Scholar] [CrossRef]
- Varga, G.; Den Besten, P.; Racz, R.; Zsembery, A. Importance of bicarbonate transport in pH control during amelogenesis—need for functional studies. Oral Dis. 2018, 24, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepperdinger, U.; Maurer, E.; Witsch-Baumgartner, M.; Stigler, R.; Zschocke, J.; Lussi, A.; Kapferer-Seebacher, I. Expanding the phenotype of hypomaturation amelogenesis imperfecta due to a novel SLC24A4 variant. Clin. Oral Investig. 2020, 24, 3519–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, S.Y.T.; Wen, X.; Yin, K.; Chen, J.; Smith, C.E.; Paine, M.L. Multiple Calcium Export Exchangers and Pumps Are a Prominent Feature of Enamel Organ Cells. Front. Physiol. 2017, 8, 336. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, M.; Lacruz, R.S. CRAC channels in dental enamel cells. Cell Calcium 2018, 75, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Nurbaeva, M.K.; Eckstein, M.; Feske, S.; Lacruz, R.S. Ca(2+) transport and signalling in enamel cells. J. Physiol. 2017, 595, 3015–3039. [Google Scholar] [CrossRef] [Green Version]
- Nurbaeva, M.K.; Eckstein, M.; Devotta, A.; Saint-Jeannet, J.P.; Yule, D.I.; Hubbard, M.J.; Lacruz, R.S. Evidence That Calcium Entry Into Calcium-Transporting Dental Enamel Cells Is Regulated by Cholecystokinin, Acetylcholine and ATP. Front. Physiol. 2018, 9, 801. [Google Scholar] [CrossRef]
- Eckstein, M.; Vaeth, M.; Aulestia, F.J.; Costiniti, V.; Kassam, S.N.; Bromage, T.G.; Pedersen, P.; Issekutz, T.; Idaghdour, Y.; Moursi, A.M.; et al. Differential regulation of Ca(2+) influx by ORAI channels mediates enamel mineralization. Sci. Signal. 2019, 12, eaav4663. [Google Scholar] [CrossRef]
- Kim, H.E.; Hong, J.H. The overview of channels, transporters, and calcium signaling molecules during amelogenesis. Arch. Oral Biol. 2018, 93, 47–55. [Google Scholar] [CrossRef]
- Bernhardt, M.L.; Stein, P.; Carvacho, I.; Krapp, C.; Ardestani, G.; Mehregan, A.; Umbach, D.M.; Bartolomei, M.S.; Fissore, R.A.; Williams, C.J. TRPM7 and CaV3.2 channels mediate Ca(2+) influx required for egg activation at fertilization. Proc. Natl. Acad. Sci. USA 2018, 115, E10370–E10378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittermeier, L.; Demirkhanyan, L.; Stadlbauer, B.; Breit, A.; Recordati, C.; Hilgendorff, A.; Matsushita, M.; Braun, A.; Simmons, D.G.; Zakharian, E.; et al. TRPM7 is the central gatekeeper of intestinal mineral absorption essential for postnatal survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4706–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abiria, S.A.; Krapivinsky, G.; Sah, R.; Santa-Cruz, A.G.; Chaudhuri, D.; Zhang, J.; Adstamongkonkul, P.; De Caen, P.G.; Clapham, D.E. TRPM7 senses oxidative stress to release Zn(2+) from unique intracellular vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, E6079–E6088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Qu, J.; Sun, L.; Li, Q.; Ling, H.; Yang, N.; Ma, T.; Wang, Q.; Li, M.; Zhang, K.; et al. Ca2+/Mg2+ homeostasisrelated TRPM7 channel mediates chondrocyte hypertrophy via regulation of the PI3KAkt signaling pathway. Mol. Med. Rep. 2017, 16, 5699–5705. [Google Scholar] [CrossRef]
- Ogata, K.; Tsumuraya, T.; Oka, K.; Shin, M.; Okamoto, F.; Kajiya, H.; Katagiri, C.; Ozaki, M.; Matsushita, M.; Okabe, K. The crucial role of the TRPM7 kinase domain in the early stage of amelogenesis. Sci. Rep. 2017, 7, 18099. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Le, M.H.; Abduweli, D.; Ho, S.P.; Ryazanova, L.V.; Hu, Z.; Ryazanov, A.G.; Den Besten, P.K.; Zhang, Y. A Critical Role of TRPM7 As an Ion Channel Protein in Mediating the Mineralization of the Craniofacial Hard Tissues. Front. Physiol. 2016, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Yue, Z.; Sun, B.; Yang, W.; Xie, J.; Ni, E.; Feng, Y.; Mahmood, R.; Zhang, Y.; Yue, L. Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. Br. J. Pharmacol. 2013, 168, 1294–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590–595. [Google Scholar] [CrossRef]
- Jiang, J.; Li, M.; Yue, L. Potentiation of TRPM7 inward currents by protons. J. Gen. Physiol. 2005, 126, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Macianskiene, R.; Almanaityte, M.; Jekabsone, A.; Mubagwa, K. Modulation of Human Cardiac TRPM7 Current by Extracellular Acidic pH Depends upon Extracellular Concentrations of Divalent Cations. PLoS ONE 2017, 12, e0170923. [Google Scholar] [CrossRef] [Green Version]
- Kawano, S.; Morotomi, T.; Toyono, T.; Nakamura, N.; Uchida, T.; Ohishi, M.; Toyoshima, K.; Harada, H. Establishment of dental epithelial cell line (HAT-7) and the cell differentiation dependent on Notch signaling pathway. Connect. Tissue Res. 2002, 43, 409–412. [Google Scholar] [CrossRef]
- Matsumoto, A.; Harada, H.; Saito, M.; Taniguchi, A. Induction of enamel matrix protein expression in an ameloblast cell line co-cultured with a mesenchymal cell line in vitro. Cell. Dev. Biol. Anim. 2011, 47, 39–44. [Google Scholar] [CrossRef]
- Zheng, L.; Seon, Y.J.; Mourao, M.A.; Schnell, S.; Kim, D.; Harada, H.; Papagerakis, S.; Papagerakis, P. Circadian rhythms regulate amelogenesis. Bone 2013, 55, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Morita, T.; Murata, K.; Minowa, E.; Jahan, A.; Saito, M.; Tanimura, A. Effects of full-length human amelogenin on the differentiation of dental epithelial cells and osteoblastic cells. Arch. Oral Biol. 2019, 107, 104479. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Z.; Chen, G.; Li, J.; Li, H.; Yu, M.; Zhang, W.; Guo, W.; Tian, W. GSK3beta regulates ameloblast differentiation via Wnt and TGF-beta pathways. J. Cell Physiol. 2018, 233, 5322–5333. [Google Scholar] [CrossRef] [PubMed]
- Jalali, R.; Lodder, J.C.; Zandieh-Doulabi, B.; Micha, D.; Melvin, J.E.; Catalan, M.A.; Mansvelder, H.D.; Den Besten, P.; Bronckers, A. The Role of Na:K:2Cl Cotransporter 1 (NKCC1/SLC12A2) in Dental Epithelium during Enamel Formation in Mice. Front. Physiol. 2017, 8, 924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bori, E.; Guo, J.; Racz, R.; Burghardt, B.; Foldes, A.; Keremi, B.; Harada, H.; Steward, M.C.; Den, B.P.; Bronckers, A.L.; et al. Evidence for Bicarbonate Secretion by Ameloblasts in a Novel Cellular Model. J. Dent. Res. 2016, 95, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racz, R.; Foldes, A.; Bori, E.; Zsembery, A.; Harada, H.; Steward, M.C.; DenBesten, P.; Bronckers, A.L.J.J.; Gerber, G.; Varga, G. No Change in Bicarbonate Transport but Tight-Junction Formation Is Delayed by Fluoride in a Novel Ameloblast Model. Front. Physiol. 2017, 8, 940. [Google Scholar] [CrossRef] [Green Version]
- Racz, R.; Nagy, A.; Rakonczay, Z.; Dunavari, E.K.; Gerber, G.; Varga, G. Defense Mechanisms Against Acid Exposure by Dental Enamel Formation, Saliva and Pancreatic Juice Production. Curr. Pharm. Des. 2018, 24, 2012–2022. [Google Scholar] [CrossRef]
- Chokshi, R.; Matsushita, M.; Kozak, J.A. Detailed examination of Mg2+ and pH sensitivity of human TRPM7 channels. Am. J. Physiol. Cell Physiol. 2012, 302, C1004–C1011. [Google Scholar] [CrossRef] [Green Version]
- Chubanov, V.; Schnitzler, M.; Meissner, M.; Schafer, S.; Abstiens, K.; Hofmann, T.; Gudermann, T. Natural and synthetic modulators of SK (K(ca)2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 2012, 166, 1357–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.; Ferioli, S.; Hofmann, T.; Zierler, S.; Gudermann, T.; Chubanov, V. Mibefradil represents a new class of benzimidazole TRPM7 channel agonists. Pflug. Arch. 2016, 468, 623–634. [Google Scholar] [CrossRef]
- Hofmann, T.; Schafer, S.; Linseisen, M.; Sytik, L.; Gudermann, T.; Chubanov, V. Activation of TRPM7 channels by small molecules under physiological conditions. Pflug. Arch. 2014, 466, 2177–2189. [Google Scholar] [CrossRef] [PubMed]
- Souza Bomfim, G.H.; Costiniti, V.; Li, Y.; Idaghdour, Y.; Lacruz, R.S. TRPM7 activation potentiates SOCE in enamel cells but requires ORAI. Cell Calcium 2020, 87, 102187. [Google Scholar] [CrossRef] [PubMed]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels 2013, 7, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Inazu, M.; Konishi, M.; Yokoyama, U. Functional expression of TRPM7 as a Ca(2+) influx pathway in adipocytes. Physiol. Rep. 2019, 7, e14272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macianskiene, R.; Martisiene, I.; Zablockaite, D.; Gendviliene, V. Characterization of Mg(2)(+)-regulated TRPM7-like current in human atrial myocytes. J. Biomed. Sci. 2012, 19, 75. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Xu, G.; Xie, M.; Zhang, X.; Schools, G.P.; Ma, L.; Kimelberg, H.K.; Chen, H. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J. Neurosci. 2009, 29, 8551–8564. [Google Scholar] [CrossRef]
- Won, J.; Vang, H.; Kim, J.H.; Lee, P.R.; Kang, Y.; Oh, S.B. TRPM7 Mediates Mechanosensitivity in Adult Rat Odontoblasts. J. Dent. Res. 2018, 97, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Ryazanova, L.V.; Hu, Z.; Suzuki, S.; Chubanov, V.; Fleig, A.; Ryazanov, A.G. Elucidating the role of the TRPM7 alpha-kinase: TRPM7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci. Rep. 2014, 4, 7599. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Rubaiy, H.N.; Chen, G.L.; Hallett, T.; Zaibi, N.; Zeng, B.; Saurabh, R.; Xu, S.Z. Mibefradil, a T-type Ca(2+) channel blocker also blocks Orai channels by action at the extracellular surface. Br. J. Pharmacol. 2019, 176, 3845–3856. [Google Scholar] [CrossRef]
- Chubanov, V.; Gudermann, T. Mapping TRPM7 Function by NS8593. Int. J. Mol. Sci. 2020, 21, 7017. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, J.A.; Matsushita, M.; Nairn, A.C.; Cahalan, M.D. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J. Gen. Physiol. 2005, 126, 499–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Du, J.; Jiang, J.; Ratzan, W.; Su, L.T.; Runnels, L.W.; Yue, L. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 2007, 282, 25817–25830. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kádár, K.; Juhász, V.; Földes, A.; Rácz, R.; Zhang, Y.; Löchli, H.; Kató, E.; Köles, L.; Steward, M.C.; DenBesten, P.; et al. TRPM7-Mediated Calcium Transport in HAT-7 Ameloblasts. Int. J. Mol. Sci. 2021, 22, 3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083992
Kádár K, Juhász V, Földes A, Rácz R, Zhang Y, Löchli H, Kató E, Köles L, Steward MC, DenBesten P, et al. TRPM7-Mediated Calcium Transport in HAT-7 Ameloblasts. International Journal of Molecular Sciences. 2021; 22(8):3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083992
Chicago/Turabian StyleKádár, Kristóf, Viktória Juhász, Anna Földes, Róbert Rácz, Yan Zhang, Heike Löchli, Erzsébet Kató, László Köles, Martin C. Steward, Pamela DenBesten, and et al. 2021. "TRPM7-Mediated Calcium Transport in HAT-7 Ameloblasts" International Journal of Molecular Sciences 22, no. 8: 3992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22083992