
Cre mRNA Is Not Transferred by EVs from Endothelial and Adipose-Derived Stromal/Stem Cells during Vascular Network Formation

 , , and
, , and
Abstract
:1. Introduction
2. Results
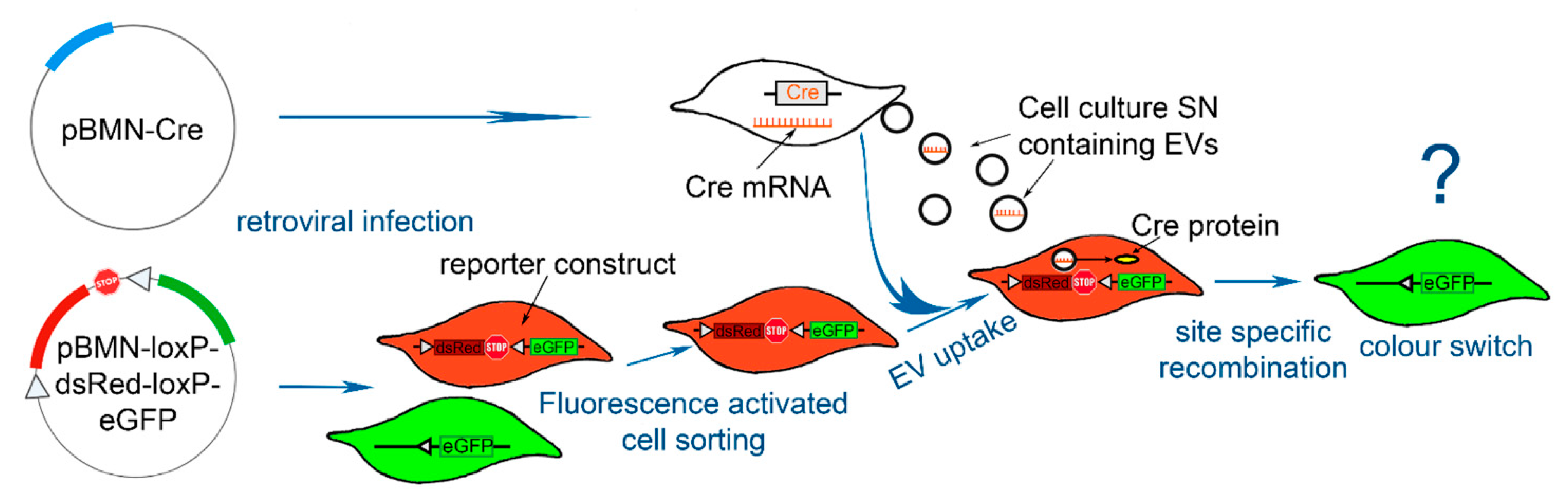
2.1. Strategy—Adaption of the Cre-loxP System to Evaluate EV Uptake
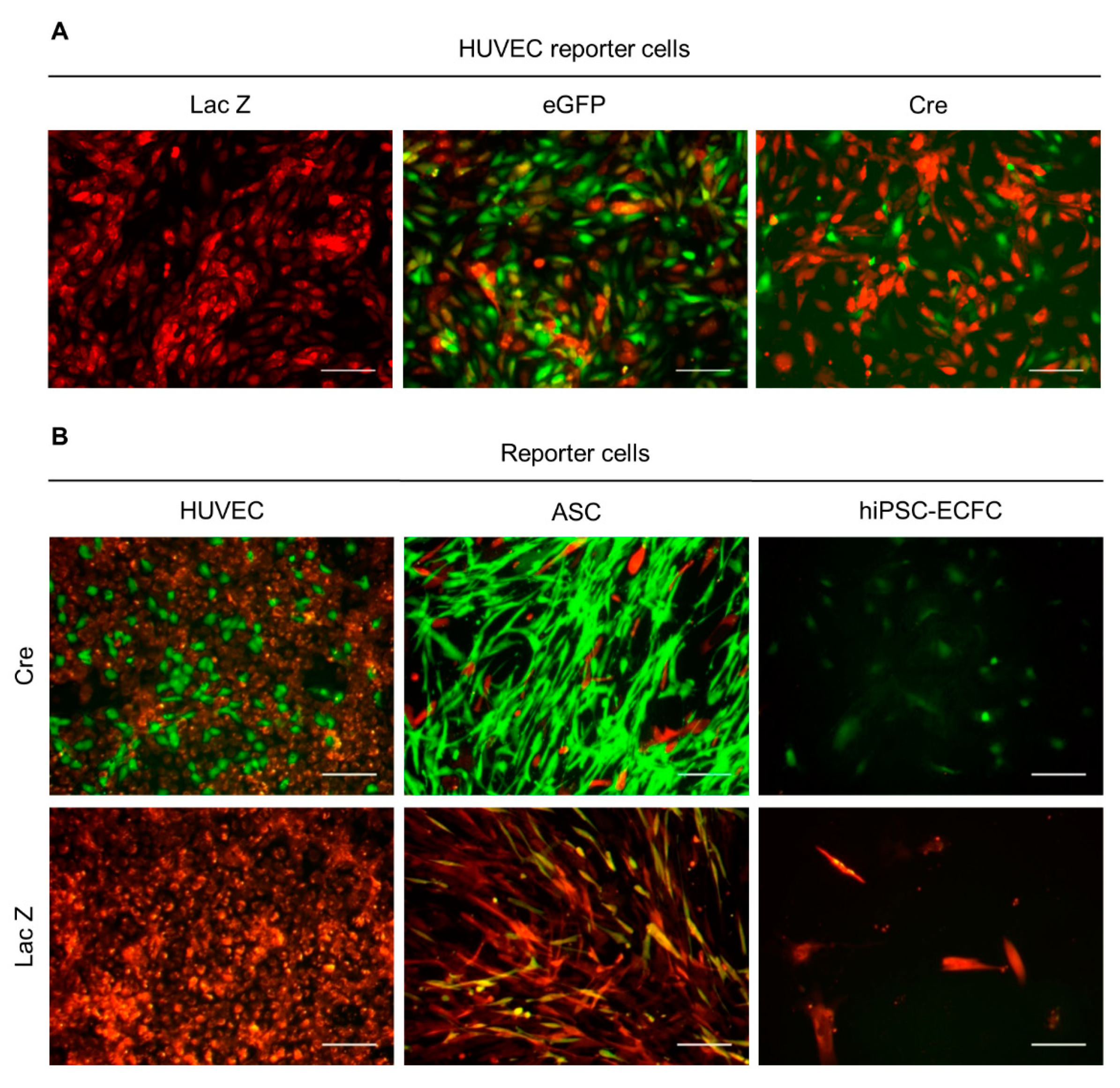
2.2. Functionality Control via Transfection and Superinfection of Reporter Cells with the Cre Construct Shows Cre Mediated Recombination
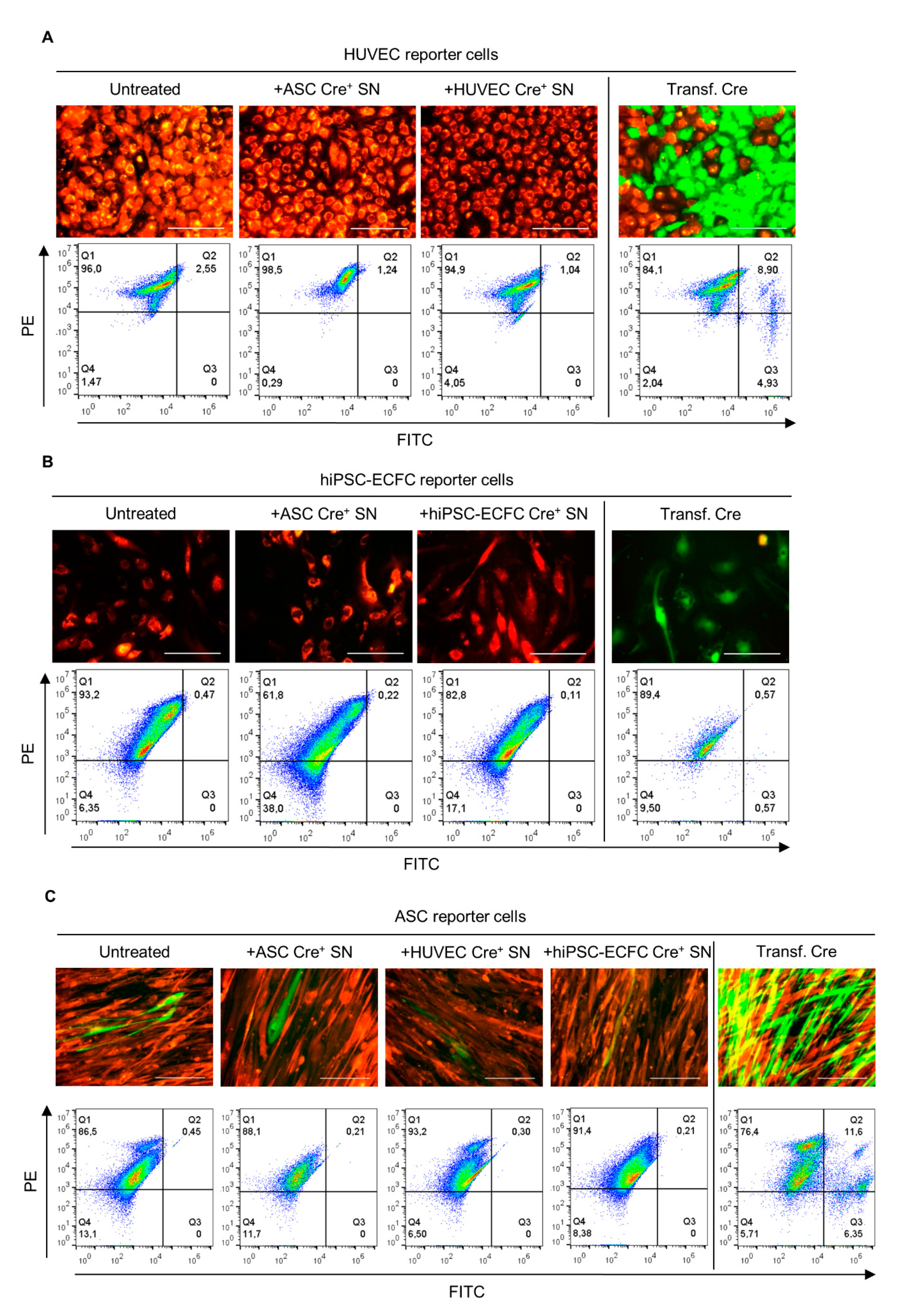
2.3. Supernatant Change from Cre-Expressing Cells to Reporter Cells—EVs in Conditioned Medium Do Not Mediate Cre Transfer
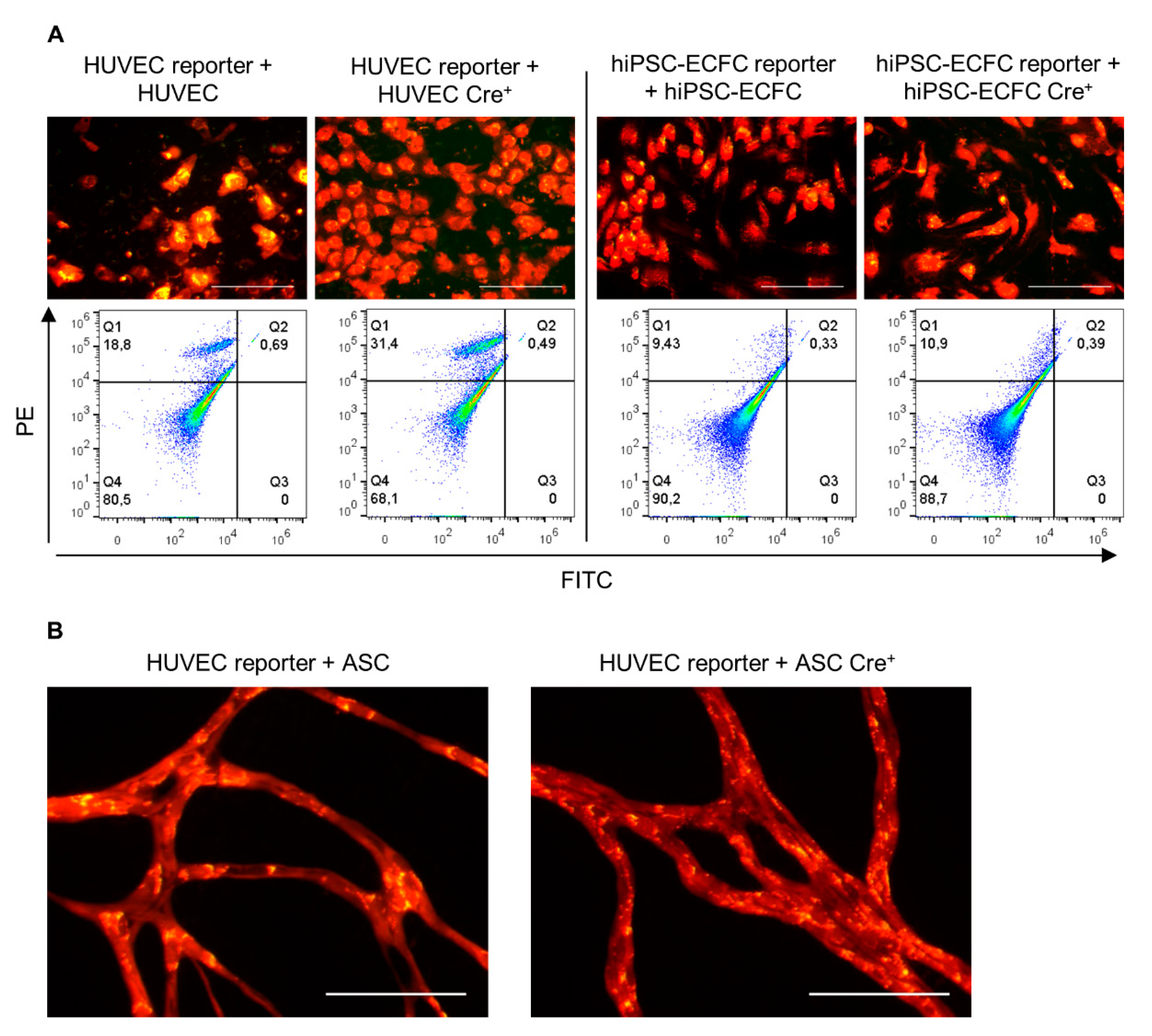
2.4. Direct Coculture of Reporter with Cre+ Cells Does Not Induce a Cre-Mediated Recombination
2.5. Supernatant Transfer from TNF-α Activated and HUVEC Cre+ to HUVEC/ASC Reporter Cells Does Not Lead to Cre Mediated Recombination
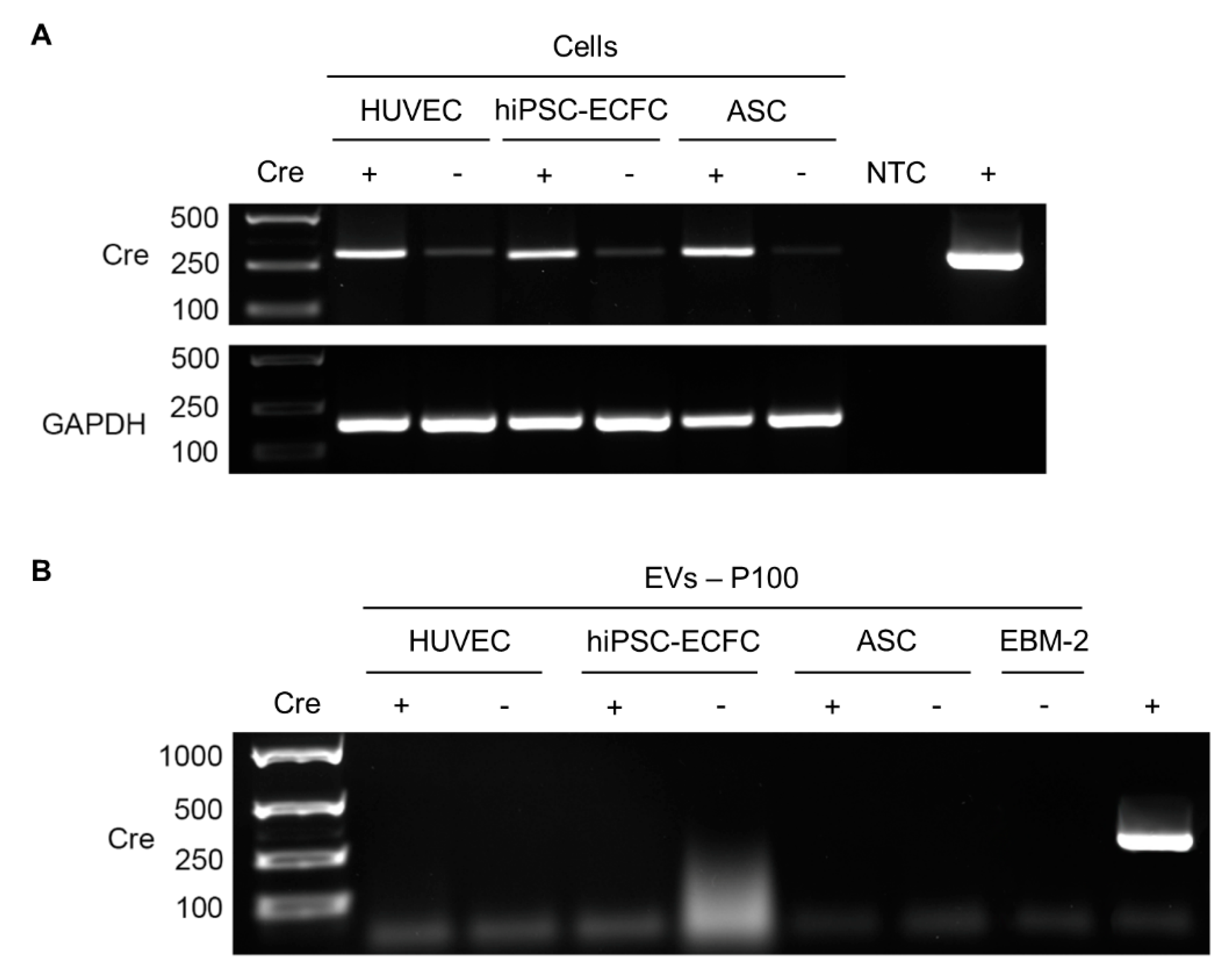
2.6. Detection of Cre in Cells and EVs
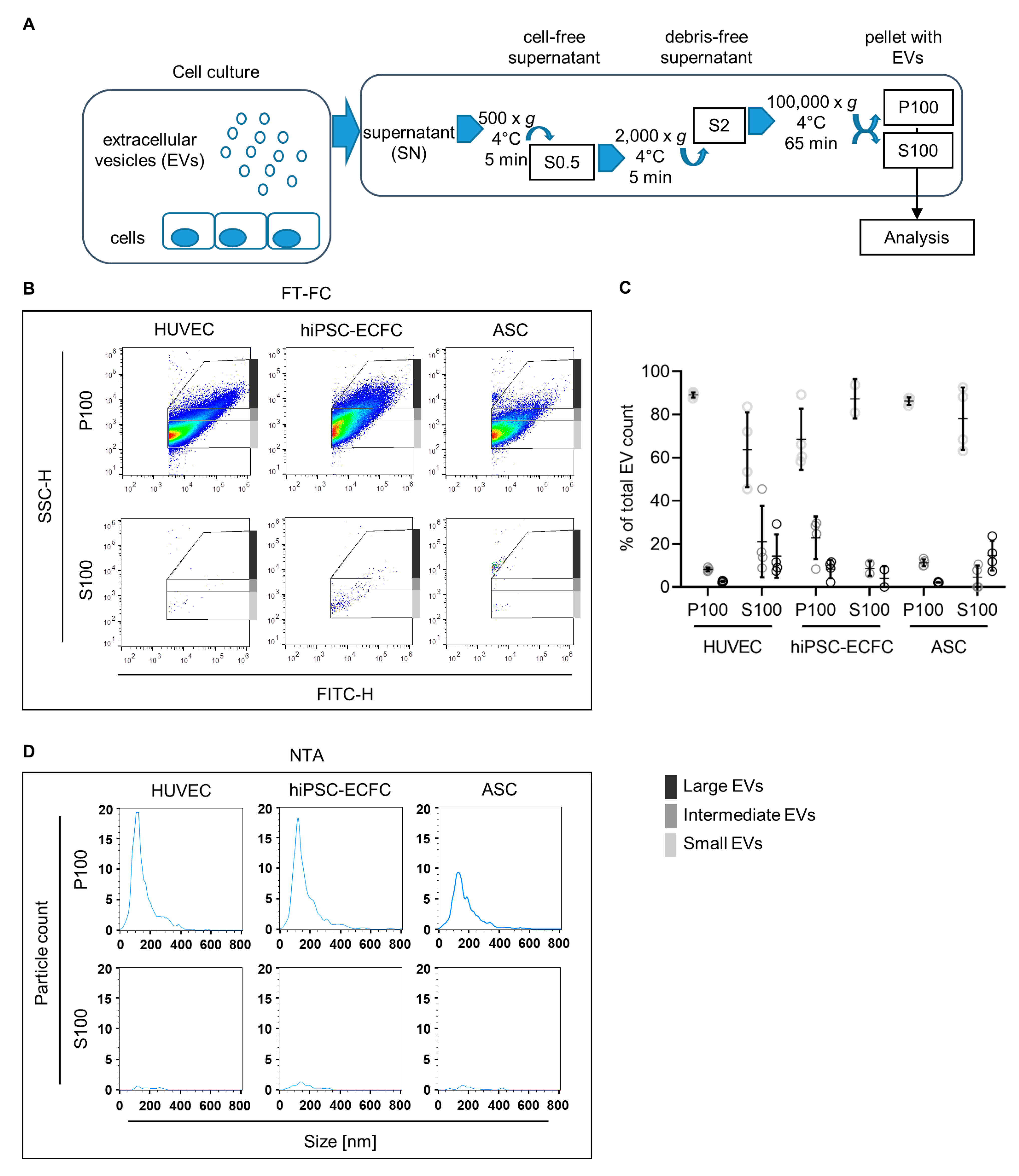
2.7. Characterization of EVs
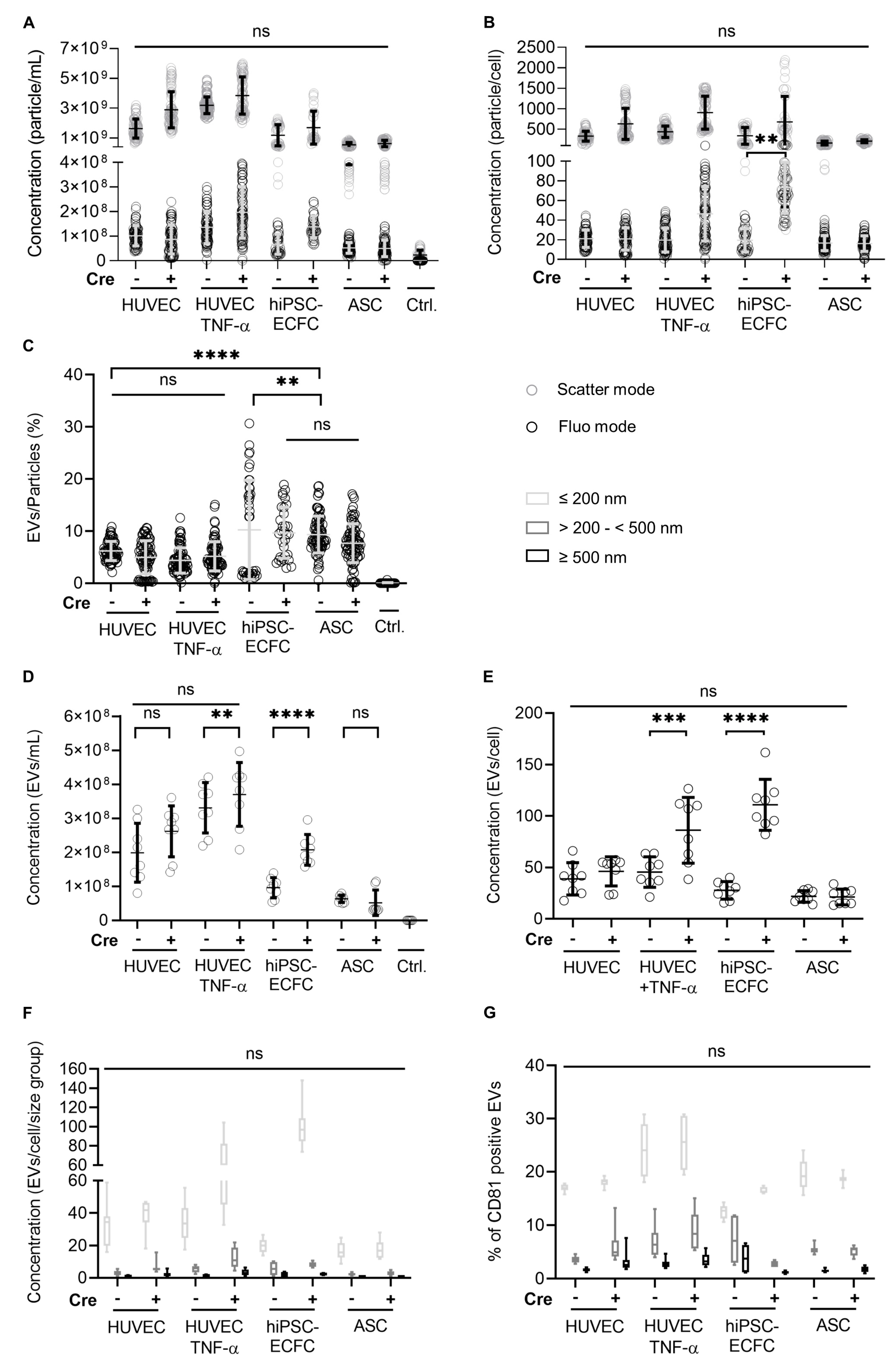
2.8. Investigation of Concentration and Size of EVs from HUVEC, hiPSC-ECFC, and ASC via NTA and FT-FC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Retroviral Infection of HUVEC, hiPSC-ECFC and ASC
4.3. Fluorescence Activated Cell Sorting
4.4. Imaging
4.5. Transfection of Reporter Cells
4.6. Supernatant Transfer from Cre-Expressing Cells to Reporter Cells
4.7. Direct Coculture of Cre-Expressing Cells with Reporter Cells
4.8. Supernatant Transfer from TNF-α Activated HUVEC to ASC/HUVEC Reporter Cells
4.9. Analyzing Reporter Cells for Cre Mediated Color Switch via Flow Cytometry
4.10. Enrichment of EVs via Ultracentrifugation
4.11. Nanoparticle Tracking Analysis
4.12. Analysis of EVs via Fluorescence-Triggered Flow Cytometry
4.13. Analysis of Cre Expression
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASC | Adipose tissue-derived MSC |
| CMG | Cell Mask Green |
| EBM-2 | Endothelial Basal Medium |
| EGM-2 | Endothelial Growth Medium |
| EVs | Extracellular Vesicles |
| ESCRT | Endosomal Sorting Complex Required for Transport |
| FACS | Fluorescence Activated Cell Sorting |
| FCS | Fetal Calf Serum |
| FT-FC | Fluorescence Trigger Flow Cytometry |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| hiPSC-ECFC | Human induced Pluripotent Stem Cell derived Endothelial Colony Forming Cells |
| HUVEC | Human Umbilical Vein Endothelial Cells |
| MSC | Mesenchymal Stromal/Stem Cells |
| MV | Microvesicles |
| MVB | Multivesicular Body |
| NTA | Nano Particle Tracking Analysis |
| NTC | Non-Template Control |
| RBP | RNA Binding Protein |
| TNF-α | Tumor Necrosis Factor-α |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor 2 |
References
- Rouwkema, J.; Rivron, N.C.; Van Blitterswijk, C.A. Vascularization in tissue engineering. Trends Biotechnol. 2008, 26, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Holnthoner, W.; Hohenegger, K.; Husa, A.M.; Muehleder, S.; Meinl, A.; Peterbauer-Scherb, A.; Redl, H. Adipose-derived stem cells induce vascular tube formation of outgrowth endothelial cells in a fibrin matrix: Vascular tube formation by endothelial and adipose-derived stem cells. J. Tissue Eng. Regen. Med. 2015, 9, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Pill, K.; Melke, J.; Mühleder, S.; Pultar, M.; Rohringer, S.; Priglinger, E.; Redl, H.R.; Hofmann, S.; Holnthoner, W. Microvascular networks from endothelial cells and mesenchymal stromal cells from adipose tissue and bone marrow: A comparison. Front. Bioeng. Biotechnol. 2018, 6, 156. [Google Scholar] [CrossRef] [PubMed]
- Carrion, B.; Kong, Y.P.; Kaigler, D.; Putnam, A.J. Bone marrow-derived mesenchymal stem cells enhance angiogenesis via their α6β1 integrin receptor. Exp. Cell Res. 2013, 319, 2964–2976. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, Y.K.; Yin, R.T.; Shang, M.R.; Shirure, V.S.; Moya, M.L.; George, S.C. Human induced pluripotent stem cell-derived endothelial cells for three-dimensional microphysiological systems. Tissue Eng. C Methods 2017, 23, 474–484. [Google Scholar] [CrossRef]
- Rohringer, S.; Hofbauer, P.; Schneider, K.H.; Husa, A.M.; Feichtinger, G.; Peterbauer-Scherb, A.; Redl, H.; Holnthoner, W. Mechanisms of vasculogenesis in 3D fibrin matrices mediated by the interaction of adipose-derived stem cells and endothelial cells. Angiogenesis 2014, 17, 921–933. [Google Scholar] [CrossRef]
- Gökçinar-Yagci, B.; Uçkan-Çetinkaya, D.; Çelebi-Saltik, B. Pericytes: Properties, functions and applications in tissue engineering. Stem Cell Rev. Rep. 2015, 11, 549–559. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosar, J.P.; Witwer, K.; Cayota, A. Revisiting extracellular RNA release, processing, and function. Trends Biochem. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hromada, C.; Mühleder, S.; Grillari, J.; Redl, H.; Holnthoner, W. Endothelial extracellular vesicles—Promises and challenges. Front. Physiol. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International society for extracellular vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbiano, F.; Corsi, J.; Gurrieri, E.; Trevisan, C.; Notarangelo, M.; D’Agostino, V.G. RNA packaging into extracellular vesicles: An orchestra of RNA-binding proteins? J. Extracell. Vesicles 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and micropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; Milito, D.A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell. Biochem. 2010, 111, 488–496. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; Von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Manikowski, D.; Andrée, B.; Samper, E.; Saint-Marc, C.; Olmer, R.; Vogt, P.; Strauß, S.; Haverich, A.; Hilfiker, A. Human adipose tissue-derived stromal cells in combination with exogenous stimuli facilitate three-dimensional network formation of human endothelial cells derived from various sources. Vasc. Pharmacol. 2018, 106, 28–36. [Google Scholar] [CrossRef]
- Fitzsimmons, R.E.; Mazurek, M.S.; Soos, A.; Simmons, C.A. Mesenchymal stromal/stem cells in regenerative medicine and tissue engineering. Stem Cells Int. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Todorova, D.; Simoncini, S.; Lacroix, R.; Sabatier, F.; Dignat-George, F. Extracellular vesicles in angiogenesis. Circ. Res. 2017, 120, 1658–1673. [Google Scholar] [CrossRef]
- Shabbir, A.; Cox, A.; Rodriguez-Menocal, L.; Salgado, M.; Badiavas, E.V. Mesenchymal stem cell exosomes induce proliferation and migration of normal and chronic wound fibroblasts and enhance angiogenesis in vitro. Stem Cells Dev. 2015, 24, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zhang, L.; Wang, S.; Han, Q.; Zhao, R.C. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J. Cell Sci. 2016, 129, 2182–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.; Steenbeek, S.C.; Maynard, C.; Van Rheenen, J. Studying extracellular vesicle transfer by a Cre-loxP method. Nat. Protoc. 2016, 11, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; Wit, D.E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letsiou, E.; Bauer, N. Endothelial Extracellular Vesicles in Pulmonary Function and Disease. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, pp. 197–256. [Google Scholar]
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y.; et al. Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes. Sci. Rep. 2015, 5, 8505. [Google Scholar] [CrossRef] [PubMed]
- Rajashekhar, G.; Willuweit, A.; Patterson, C.E.; Sun, P.; Hilbig, A.; Breier, G.; Helisch, A.; Clauss, M. Continuous endothelial cell activation increases angiogenesis: Evidence for the direct role of endothelium linking angiogenesis and inflammation. J Vasc. Res. 2006, 43, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Mateescu, B.; Kowal, E.J.; Van Balkom, B.W.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; Candia, D.P.; Chow, F.W.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridder, K.; Sevko, A.; Heide, J.; Dams, M.; Rupp, A.K.; Macas, J.; Starmann, J.; Tjwa, M.; Plate, K.H.; Sültmann, H.; et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. OncoImmunology 2015, 4, e1008371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, S.G.; Cormier, M.T.; Clayton, A.; Doucette, A.A. Differential proteome analysis of extracellular vesicles from breast cancer cell lines by chaperone affinity enrichment. Proteomes 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Cuesta, D.F.; Passalacqua, I.; Rodor, J.; Bhushan, R.; Denby, L.; Baker, A.H. Extracellular vesicle cross-talk between pulmonary artery smooth muscle cells and endothelium during excessive TGF-β signalling: Implications for PAH vascular remodelling. Cell Commun. Signal 2019, 17, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Company of Biologists Ltd. Cre-recombinase-associated toxicity highlights limitations of genome editing. Dis. Model. Mech. 2013, 6, 1299–1300. [Google Scholar]
- Mühleder, S.; Fuchs, C.; Basílio, J.; Szwarc, D.; Pill, K.; Labuda, K.; Slezak, P.; Siehs, C.; Pröll, J.; Priglinger, E.; et al. Purinergic P2Y2 receptors modulate endothelial sprouting. Cell. Mol. Life Sci. 2020, 77, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Oesterreicher, J.; Pultar, M.; Schneider, J.; Mühleder, S.; Zipperle, J.; Grillari, J.; Holnthoner, W. Fluorescence-based nanoparticle tracking analysis and flow cytometry for characterization of endothelial extracellular vesicle release. IJMS 2020, 21, 9278. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Source/Manufacturer |
|---|---|---|
| Cre sense Cre antisense Cre in sense Cre in antisense Cre sense new Cre antisense new hGAPDH sense hGAPDH antisense | CGACCAGGTTCGTTCACTCA AACACCCTGTTACGTATAGCC CGACCAGGTTCGTTCACTCA AACACCCTGTTACGTATAGCC ATGCTTCTGTCCGTTTGCC CCTGTTTTGCACGTTCACC GAGTCAACGGATTTGGTC GACAAGCTTCCCGTTCTC | Microsynth (Balgach, Switzerland) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, J.; Pultar, M.; Oesterreicher, J.; Bobbili, M.R.; Mühleder, S.; Priglinger, E.; Redl, H.; Spittler, A.; Grillari, J.; Holnthoner, W. Cre mRNA Is Not Transferred by EVs from Endothelial and Adipose-Derived Stromal/Stem Cells during Vascular Network Formation. Int. J. Mol. Sci. 2021, 22, 4050. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084050
Schneider J, Pultar M, Oesterreicher J, Bobbili MR, Mühleder S, Priglinger E, Redl H, Spittler A, Grillari J, Holnthoner W. Cre mRNA Is Not Transferred by EVs from Endothelial and Adipose-Derived Stromal/Stem Cells during Vascular Network Formation. International Journal of Molecular Sciences. 2021; 22(8):4050. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084050
Chicago/Turabian StyleSchneider, Jaana, Marianne Pultar, Johannes Oesterreicher, Madhusudhan Reddy Bobbili, Severin Mühleder, Eleni Priglinger, Heinz Redl, Andreas Spittler, Johannes Grillari, and Wolfgang Holnthoner. 2021. "Cre mRNA Is Not Transferred by EVs from Endothelial and Adipose-Derived Stromal/Stem Cells during Vascular Network Formation" International Journal of Molecular Sciences 22, no. 8: 4050. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084050