
Disulfiram Abrogates Morphine Tolerance—A Possible Role of µ-Opioid Receptor-Related G-Protein Activation in the Striatum

 ,
,
Abstract
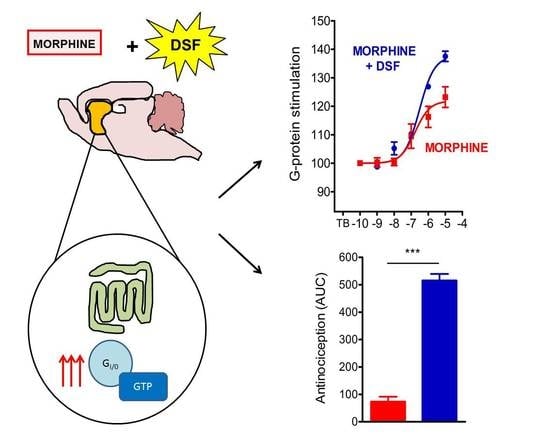
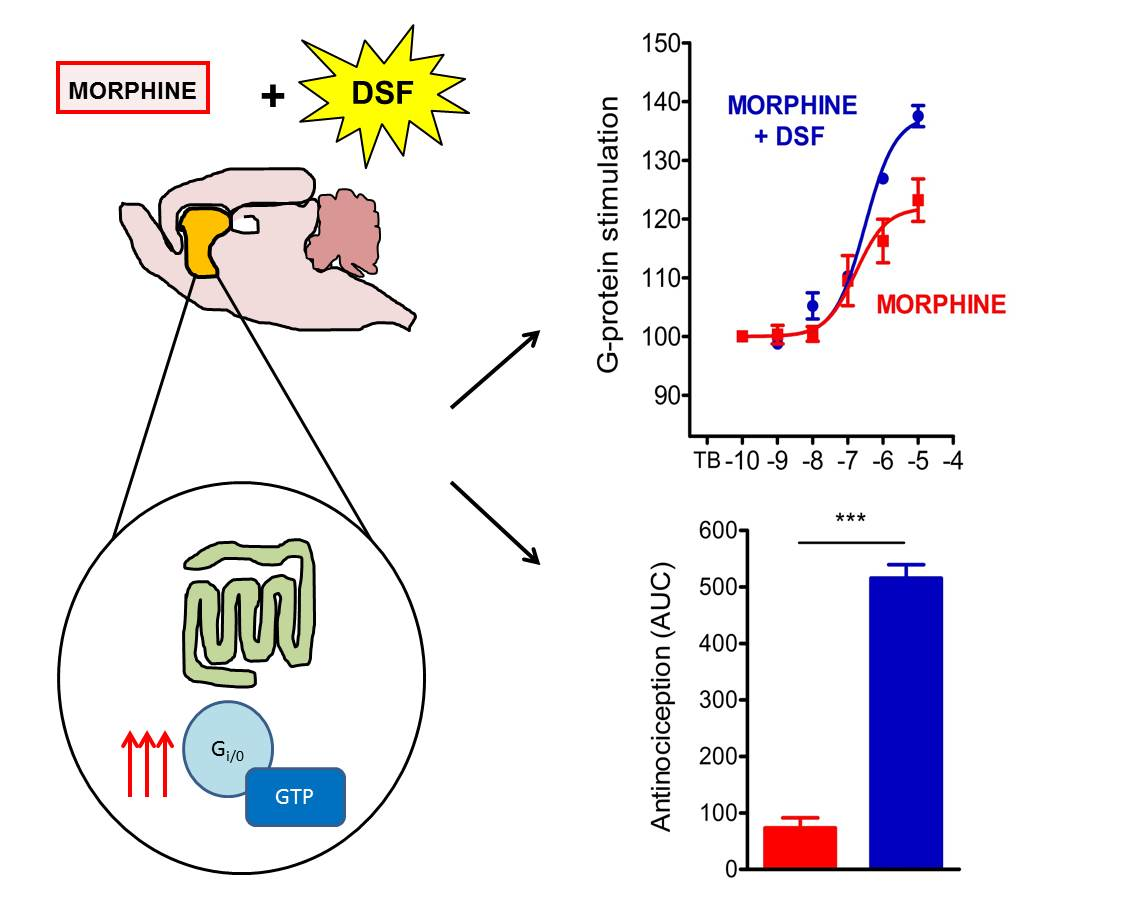
:
1. Introduction
2. Results
2.1. The Effect of Disulfiram on Mechanical and Thermal Thresholds
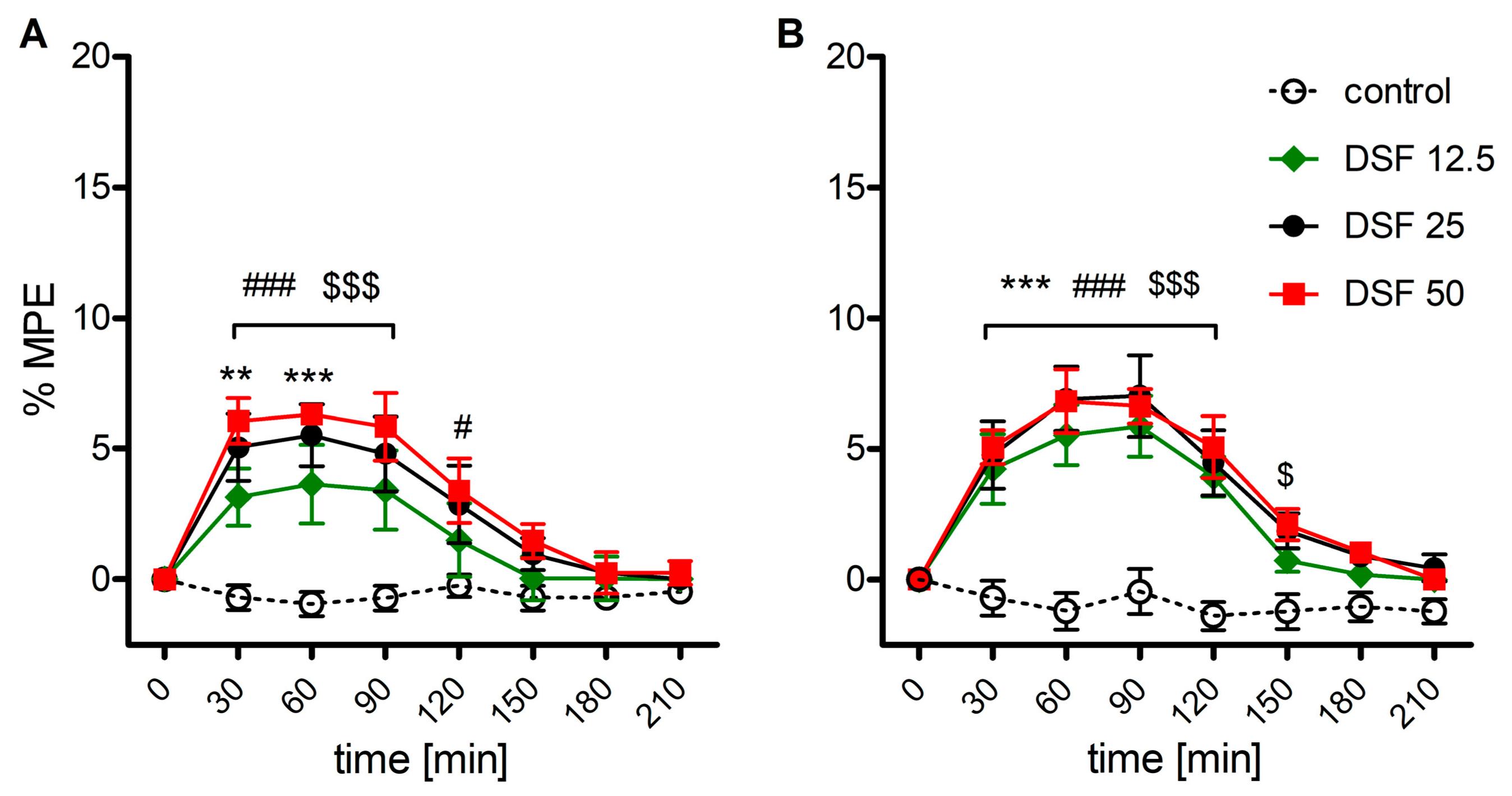
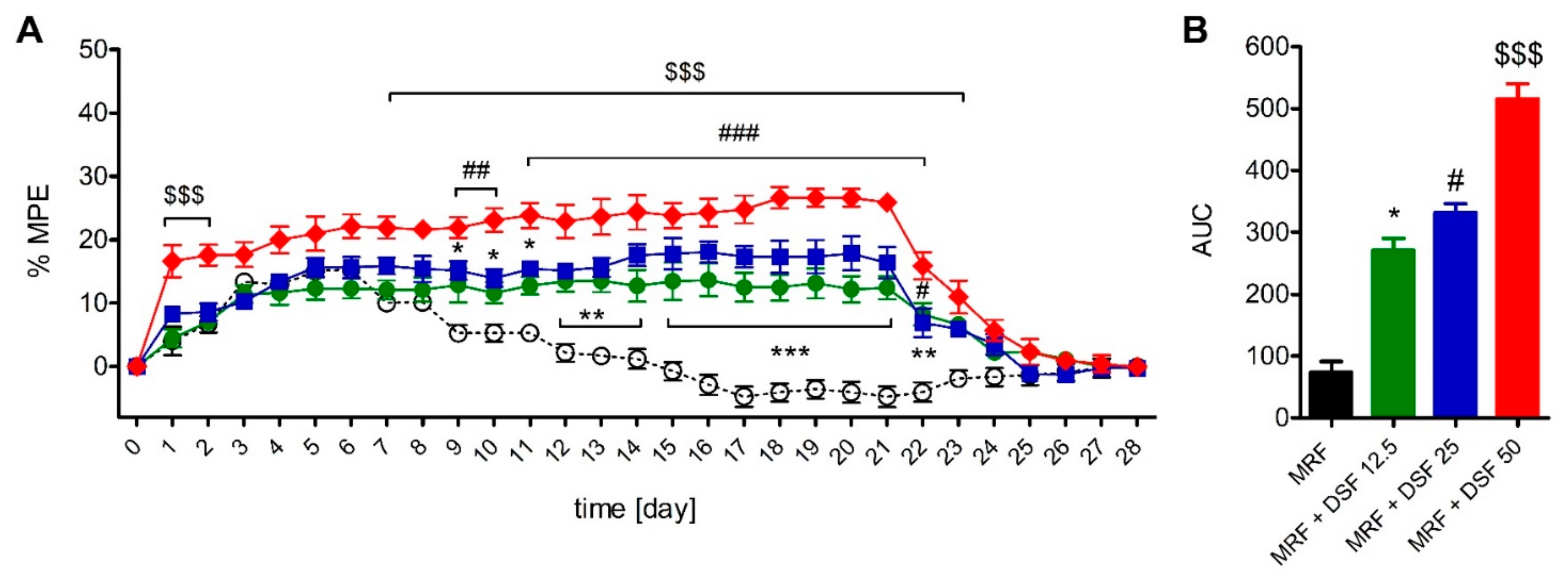
2.2. The Effect of Disulfiram on Morphine-Induced Antinociception
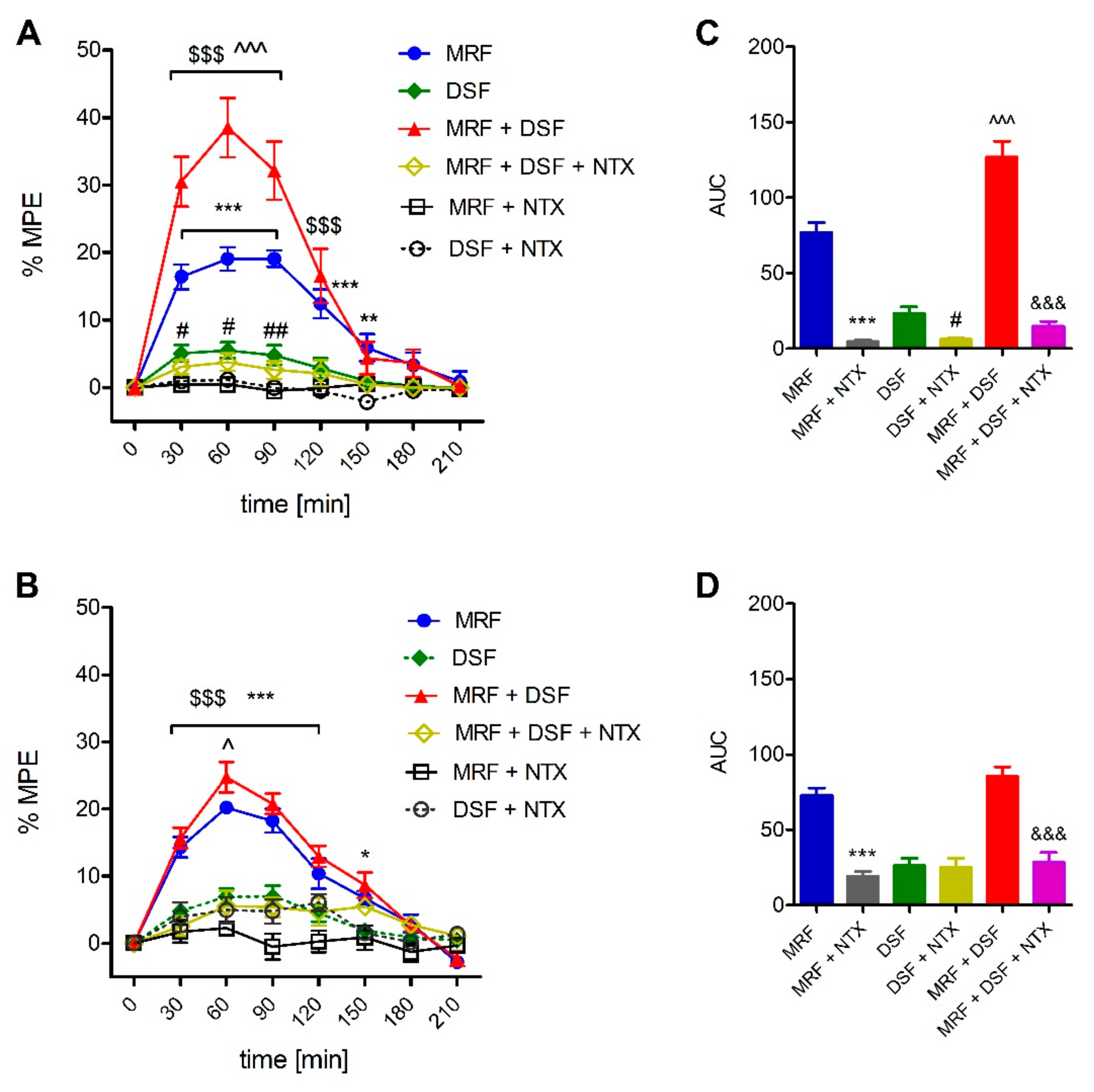
2.3. Involvement of the Opioid System in the Effect of Disulfiram (DSF)
2.4. The Effect of Disulfiram on Morphine Tolerance
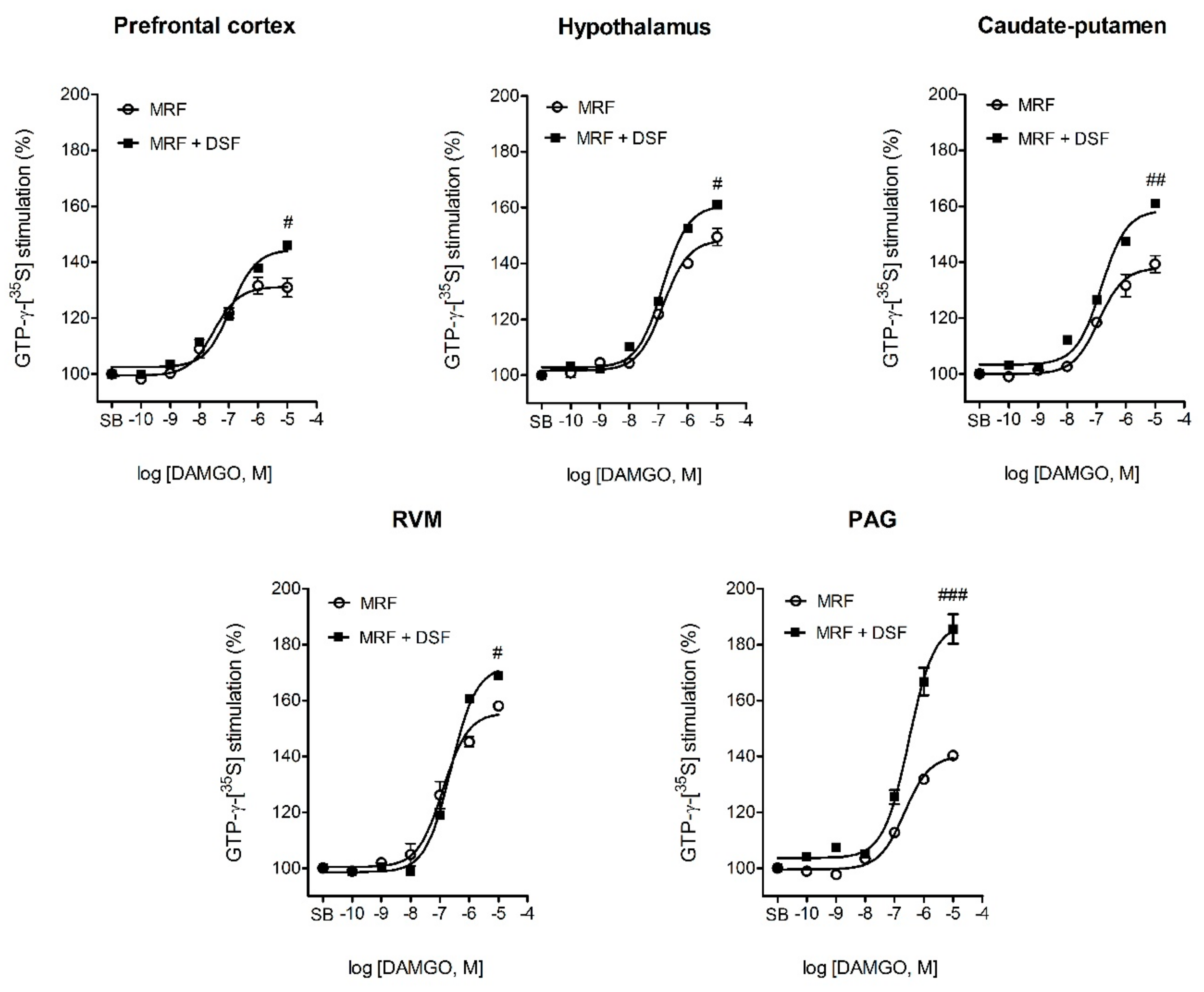
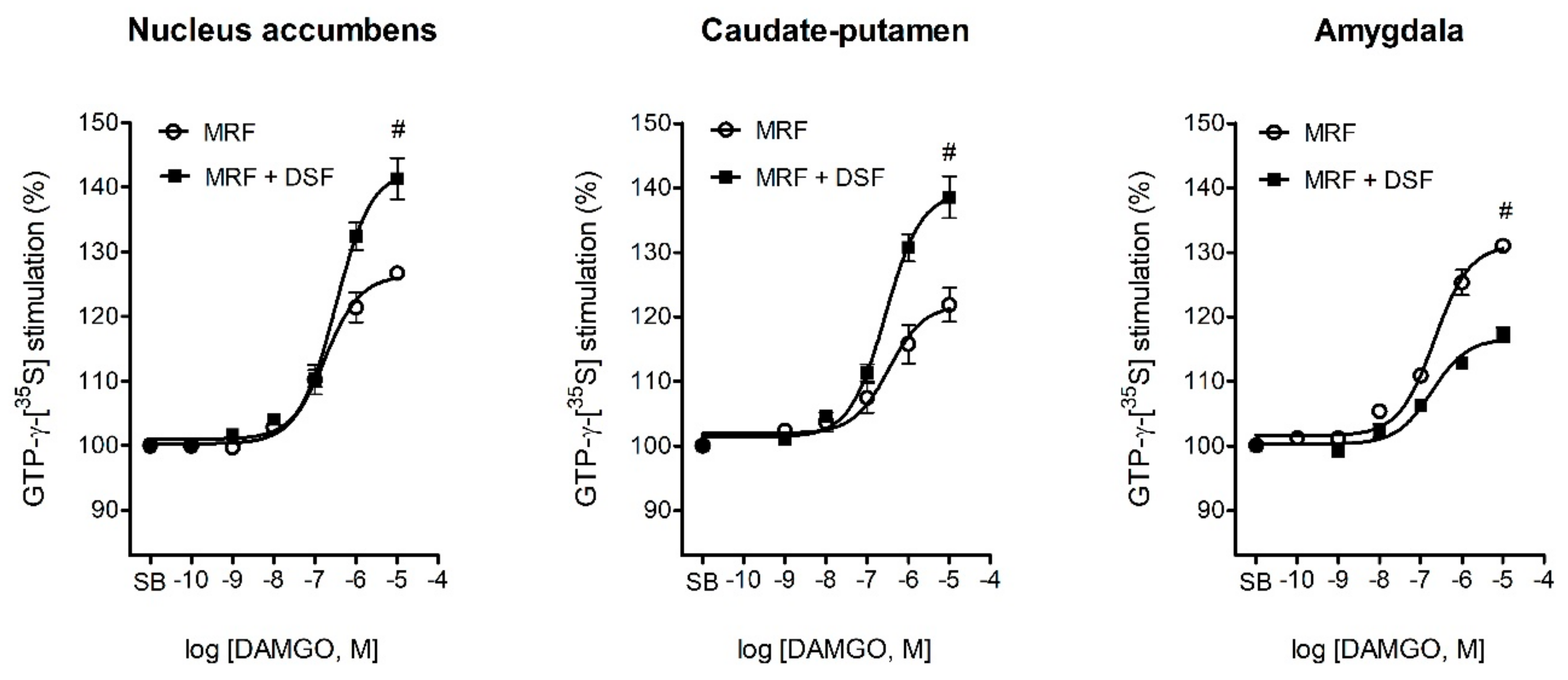
2.5. The Effect of Disulfiram on µ-Opioid G-Protein Activation in Morphine-Treated Rats
3. Discussion
4. Materials and Methods
4.1. Animals and Husbandry
4.2. Chemicals and Supplies
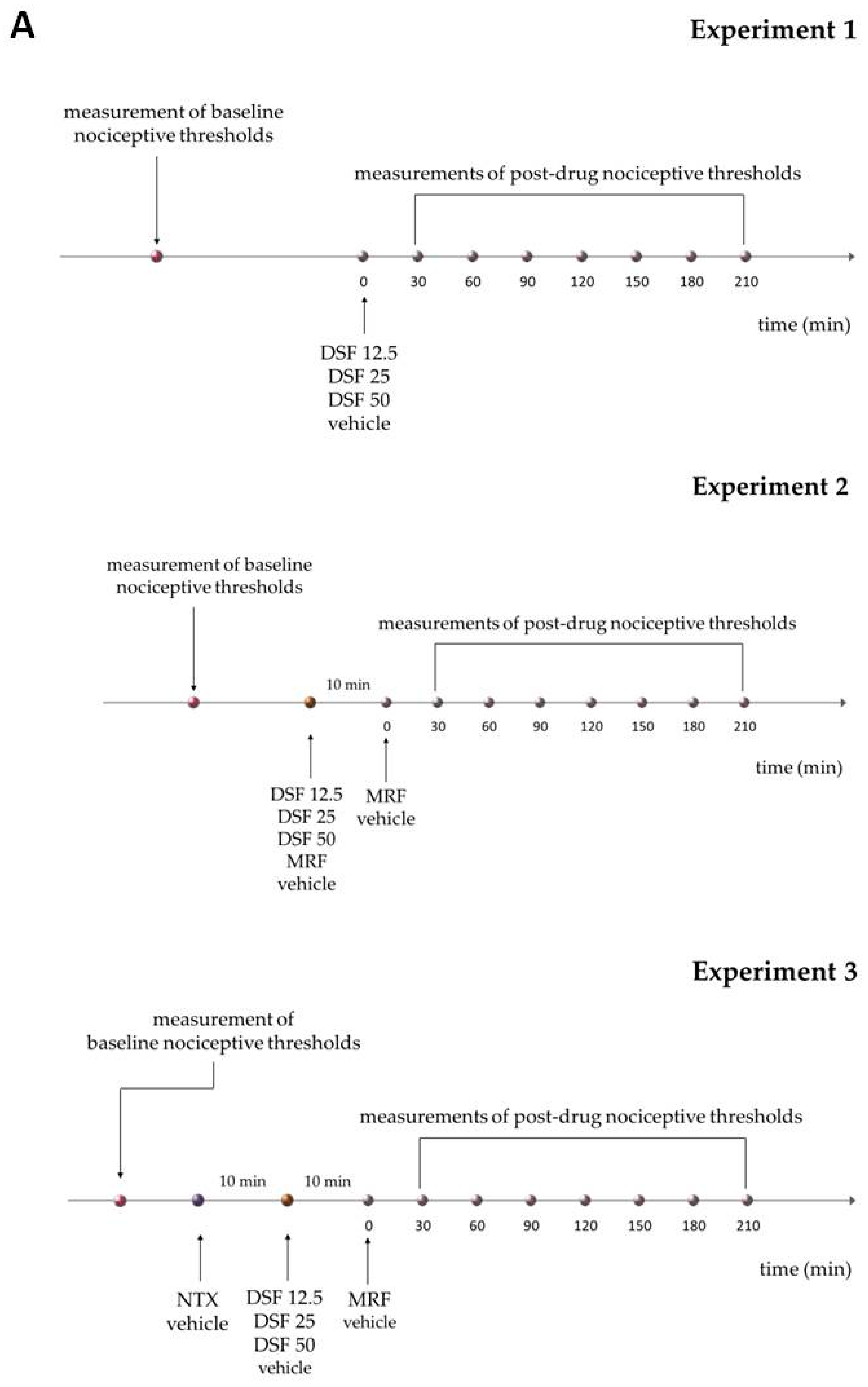
4.3. Drug Administration
4.4. Behavioral Testing
4.5. Measurement of Thermal Thresholds
4.6. Measurement of Mechanical Thresholds
4.7. Homogenate Preparation
4.8. [35. S]GTPγS Binding Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenblum, A.; Marsch, L.A.; Joseph, H.; Portenoy, R.K. Opioids and the treatment of chronic pain: Controversies, current status, and future directions. Exp. Clin. Psychopharmacol. 2008, 16, 405–416. [Google Scholar] [CrossRef]
- Kahan, M.; Srivastava, A.; Wilson, L.; Mailis-Gagnon, A.; Midmer, D. Opioids for managing chronic non-malignant pain: Safe and effective prescribing. Can. Fam. Physician 2006, 52, 1091–1096. [Google Scholar]
- Wiffen, P.J.; Wee, B.; Derry, S.; Bell, R.F.; Moore, R.A. Opioids for cancer pain—An overview of Cochrane reviews. Cochrane Database Syst. Rev. 2017, 7, CD012592. [Google Scholar] [CrossRef] [PubMed]
- Adriaensen, H.; Vissers, K.; Noorduin, H.; Meert, T. Opioid tolerance and dependence: An inevitable consequence of chronic treatment? Acta Anaesthesiol. Belg. 2003, 54, 37–47. [Google Scholar] [PubMed]
- McHugh, R.K.; Weiss, R.D.; Cornelius, M.; Martel, M.O.; Jamison, R.N.; Edwards, R.R. Distress Intolerance and Prescription Opioid Misuse among Patients with Chronic Pain. J. Pain 2016, 17, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, R.; Gordon, D.B.; de Leon-Casasola, O.A.; Rosenberg, J.M.; Bickler, S.; Brennan, T.; Carter, T.; Cassidy, C.L.; Chittenden, E.H.; Degenhardt, E.; et al. Management of Postoperative Pain: A Clinical Practice Guideline from the American Pain Society, the American Society of Regional Anesthesia and Pain Medicine, and the American Society of Anesthesiologists’ Committee on Regional Anesthesia, Executive Committee, and Administrative Council. J. Pain 2016, 17, 131–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedin, A.; Bedin, R.A.C.; Vieira, J.E.; Ashmawi, H.A. Duloxetine as an Analgesic Reduces Opioid Consumption After Spine Surgery: A Randomized, Double-Blind, Controlled Study. Clin. J. Pain 2017, 33, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Keskinbora, K.; Pekel, A.F.; Aydinli, I. Gabapentin and an Opioid Combination versus Opioid Alone for the Management of Neuropathic Cancer Pain: A Randomized Open Trial: A randomized open trial. J. Pain Symptom Manag. 2007, 34, 183–189. [Google Scholar] [CrossRef]
- Xiao, X.; Zhang, Q.; Ouyang, Z.; Guo, X. Comparison of perioperative flurbiprofen axetil or celecoxib administration for pain management after total-knee arthroplasty. Medicine 2018, 97, e12391. [Google Scholar] [CrossRef]
- Pereira, I.T.; Prado, W.A.; Dos Reis, M.P. Enhancement of the epidural morphine-induced analgesia by systemic nifedipine. Pain 1993, 53, 341–345. [Google Scholar] [CrossRef]
- Barreveld, A.M.; Correll, D.J.; Liu, X.; Max, B.; McGowan, J.A.; Shovel, L.; Wasan, A.D.; Nedeljkovic, S.S. Ketamine Decreases Postoperative Pain Scores in Patients Taking Opioids for Chronic Pain: Results of a Prospective, Randomized, Double-Blind Study. Pain Med. 2013, 14, 925–934. [Google Scholar] [CrossRef] [Green Version]
- Gawel, K.; Gibula-Bruzda, E.; Dziedzic, M.; Jenda-Wojtanowska, M.; Marszalek-Grabska, M.; Silberring, J.; Kotlinska, J.H. Cholinergic activation affects the acute and chronic antinociceptive effects of morphine. Physiol. Behav. 2017, 169, 22–32. [Google Scholar] [CrossRef]
- Gawel, K.; Jenda-Wojtanowska, M.; Gibula-Bruzda, E.; Kedzierska, E.; Filarowska, J.; Marszalek-Grabska, M.; Wojtanowski, K.; Komsta, L.; Talarek, S.; Kotlinska, J. The influence of AMN082, metabotropic glutamate receptor 7 (mGlu7) allosteric agonist on the acute and chronic antinociceptive effects of morphine in the tail-immersion test in mice: Comparison with mGlu5 and mGlu2/3 ligands. Physiol. Behav. 2018, 185, 112–120. [Google Scholar] [CrossRef]
- Garimella, V.; Cellini, C. Postoperative Pain Control. Clin. Colon Rectal Surg. 2013, 26, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Priano, J.; Faley, B.; Procopio, G.; Hewitt, K.; Feldman, J. Adjunct Analgesic Use for Acute Pain in the Emergency Department. Hosp. Pharm. 2017, 52, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.D.; Lahmek, P.; Pham, H.; Aubin, H.-J. Disulfiram Efficacy in the Treatment of Alcohol Dependence: A Meta-Analysis. PLoS ONE 2014, 9, e87366. [Google Scholar] [CrossRef] [Green Version]
- Lipsky, J.J.; Shen, M.L.; Naylor, S. In Vivo inhibition of aldehyde dehydrogenase by disulfiram. Chem. Interact. 2001, 130, 93–102. [Google Scholar] [CrossRef]
- Hosobuchi, Y.; Wemmer, J. Disulfiram inhibition of development of tolerance to analgesia induced by central gray stimulation in humans. Eur. J. Pharmacol. 1977, 43, 385–387. [Google Scholar] [CrossRef]
- Vaccari, A.; Saba, P.; Ruiu, S.; Collu, M.; Devoto, P. Disulfiram and Diethyldithiocarbamate Intoxication Affects the Storage and Release of Striatal Dopamine. Toxicol. Appl. Pharmacol. 1996, 139, 102–108. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G.; Saba, P.; Cadeddu, R.; Gessa, G.L. Disulfiram stimulates dopamine release from noradrenergic terminals and potentiates cocaine-induced dopamine release in the prefrontal cortex. Psychopharmacology 2011, 219, 1153–1164. [Google Scholar] [CrossRef]
- Karamanakos, P.N.; Pappas, P.; Stephanou, P.; Marselos, M. Differentiation of disulfiram effects on central catecholamines and hepatic ethanol metabolism. Pharmacol. Toxicol. 2001, 88, 106–110. [Google Scholar] [CrossRef]
- Kubota, Y.; Inagaki, S.; Kito, S.; Takagi, H.; Smith, A. Ultrastructural evidence of dopaminergic input to enkephalinergic neurons in rat neostriatum. Brain Res. 1986, 367, 374–378. [Google Scholar] [CrossRef]
- Sesack, S.; Pickel, V. Dual ultrastructural localization of enkephalin and tyrosine hydroxylase immunoreactivity in the rat ventral tegmental area: Multiple substrates for opiate-dopamine interactions. J. Neurosci. 1992, 12, 1335–1350. [Google Scholar] [CrossRef] [Green Version]
- Puopolo, M. The hypothalamic-spinal dopaminergic system: A target for pain modulation. Neural Regen. Res. 2019, 14, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Brousse, G.; Arnaud, B.; Vorspan, F.; Richard, D.; Dissard, A.; Dubois, M.; Pic, D.; Geneste, J.; Xavier, L.; Authier, N.; et al. Alteration of Glutamate/GABA Balance During Acute Alcohol Withdrawal in Emergency Department: A Prospective Analysis. Alcohol Alcohol. 2012, 47, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cordé, A.; Krząścik, P.; Wolińska, R.; Kleczkowska, P.; Filip, M.; Bujalska-Zadrożny, M. Disulfiram attenuates morphine or methadone withdrawal syndrome in mice. Behav. Pharmacol. 2018, 29, 393–399. [Google Scholar] [CrossRef]
- Faiman, M.D.; Kaul, S.; Latif, S.A.; Williams, T.D.; Lunte, C.E. S-(N, N-diethylcarbamoyl)glutathione (carbamathione), a disulfiram metabolite and its effect on nucleus accumbens and prefrontal cortex dopamine, GABA, and glutamate: A microdialysis study. Neuropharmacology 2013, 75, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Popik, P.; Kozela, E. Clinically available NMDA antagonist, memantine, attenuates tolerance to analgesic effects of morphine in a mouse tail flick test. Pol. J. Pharmacol. 1999, 51, 223–231. [Google Scholar]
- Gintzler, A.R.; Chakrabarti, S. Opioid Tolerance and the Emergence of New Opioid Receptor-Coupled Signaling. Mol. Neurobiol. 2000, 21, 021–034. [Google Scholar] [CrossRef]
- Hosobuchi, Y.; Adams, J.E.; Linchitz, R. Pain relief by electrical stimulation of the central gray matter in humans and its reversal by naloxone. Science 1977, 197, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.W.; Ingram, S.L.; Connor, M.A.; Christie, M.J. How opioids inhibit GABA-mediated neurotransmission. Nat. Cell Biol. 1997, 390, 611–614. [Google Scholar] [CrossRef]
- Vaughan, C.W.; Christie, M.J. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey In Vitro. J. Physiol. 1997, 498, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Loyd, D.R.; Murphy, A.Z. Forebrain Modulation of the Periaqueductal Gray and Its Role in Pain. In Encyclopedia of Pain; Gebhart, G.F., Schmidt, R.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1297–1303. [Google Scholar]
- Cavanaugh, D.J.; Lee, H.; Lo, L.; Shields, S.D.; Zylka, M.J.; Basbaum, A.I.; Anderson, D.J. Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc. Natl. Acad. Sci. USA 2009, 106, 9075–9080. [Google Scholar] [CrossRef] [Green Version]
- François, A.; Low, S.A.; Sypek, E.I.; Christensen, A.J.; Sotoudeh, C.; Beier, K.T.; Ramakrishnan, C.; Ritola, K.D.; Sharif-Naeini, R.; Deisseroth, K.; et al. A Brainstem-Spinal Cord Inhibitory Circuit for Mechanical Pain Modulation by GABA and Enkephalins. Neuron 2017, 93, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Medbak, S.; Mason, D.F.J.; Rees, L.H. Effects of ethanol and inhibitors of its metabolism on the circulating levels of Met-enkephalin in greyhounds. J. Endocrinol. 1989, 120, 473–480. [Google Scholar] [CrossRef]
- Duncan, G.H.; Kupers, R.C.; Marchand, S.; Villemure, J.-G.; Gybels, J.M.; Bushnell, M.C. Stimulation of Human Thalamus for Pain Relief: Possible Modulatory Circuits Revealed by Positron Emission Tomography. J. Neurophysiol. 1998, 80, 3326–3330. [Google Scholar] [CrossRef]
- Shiraishi, T.; Onoe, M.; Kojima, T.; Sameshima, Y.; Kageyama, T. Effects of hypothalamic paraventricular nucleus: Electrical stimulation produce marked analgesia in rats. Neurobiology 1995, 3, 393–403. [Google Scholar]
- Sims-Williams, H.; Matthews, J.C.; Talbot, P.S.; Love-Jones, S.; Brooks, J.C.; Patel, N.K.; Pickering, A.E. Deep brain stimulation of the periaqueductal gray releases endogenous opioids in humans. NeuroImage 2017, 146, 833–842. [Google Scholar] [CrossRef]
- Poznański, P.; Lesniak, A.; Bujalska-Zadrozny, M.; Strzemecka, J.; Sacharczuk, M. Bidirectional selection for high and low stress-induced analgesia affects G-protein activity. Neuropharmacology 2019, 144, 37–42. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nat. Cell Biol. 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Sim-Selley, L.J.; Scoggins, K.L.; Cassidy, M.P.; Smith, L.A.; Dewey, W.L.; Smith, F.L.; Selley, D.E. Region-dependent attenuation of μ opioid receptor-mediated G-protein activation in mouse CNS as a function of morphine tolerance. Br. J. Pharmacol. 2007, 151, 1324–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, D.E.; Murray, S.R.; Zaki, P.A.; Chu, P.C.; Lissin, D.V.; Kang, L.; Evans, C.J.; von Zastrow, M. Morphine Activates Opioid Receptors without Causing Their Rapid Internalization. J. Biol. Chem. 1996, 271, 19021–19024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, S.; Mayer, D.; Pfeiffer, M.; Stumm, R.; Koch, T.; Höllt, V. Morphine induces terminal μ-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004, 23, 3282–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, T.; Schulz, S.; Schröder, H.; Wolf, R.; Raulf, E.; Höllt, V. Carboxyl-terminal Splicing of the Rat μ Opioid Receptor Modulates Agonist-mediated Internalization and Receptor Resensitization. J. Biol. Chem. 1998, 273, 13652–13657. [Google Scholar] [CrossRef] [Green Version]
- Allouche, S.; Polastron, J.; Hasbi, A.; Homburger, V.; Jauzac, P. Differential G-protein activation by alkaloid and peptide opioid agonists in the human neuroblastoma cell line SK-N-BE. Biochem. J. 1999, 342 Pt 1, 71–78. [Google Scholar] [CrossRef]
- Sim, L.; Selley, D.; Dworkin, S.; Childers, S. Effects of chronic morphine administration on mu opioid receptor- stimulated [35S] GTPgammaS autoradiography in rat brain. J. Neurosci. 1996, 16, 2684–2692. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zeng, J.; Li, Q.; Huang, J.; Couture, R.; Hong, Y. Contribution of adrenomedullin to the switch of G protein-coupled μ-opioid receptors from Gi to Gs in the spinal dorsal horn following chronic morphine exposure in rats. Br. J. Pharmacol. 2016, 173, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Avidor-Reiss, T.; Bayewitch, M.; Levy, R.; Matus-Leibovitch, N.; Nevo, I.; Vogel, Z. Adenylylcyclase Supersensitization in μ-Opioid Receptor-transfected Chinese Hamster Ovary Cells Following Chronic Opioid Treatment. J. Biol. Chem. 1995, 270, 29732–29738. [Google Scholar] [CrossRef] [Green Version]
- Fabian, G.; Bozó, B.; Szikszay, M.; Horvath, G.; Coscia, C.J.; Szucs, M. Chronic Morphine-Induced Changes in μ-Opioid Receptors and G Proteins of Different Subcellular Loci in Rat Brain. J. Pharmacol. Exp. Ther. 2002, 302, 774–780. [Google Scholar] [CrossRef]
- Harris, H.N.; Peng, Y.B. Evidence and explanation for the involvement of the nucleus accumbens in pain processing. Neural Regen. Res. 2020, 15, 597–605. [Google Scholar] [CrossRef]
- Brown, C.A.; Matthews, J.; Fairclough, M.; McMahon, A.; Barnett, E.; Al-Kaysi, A.; El-Deredy, W.; Jones, A.K. Striatal opioid receptor availability is related to acute and chronic pain perception in arthritis: Does opioid adaptation increase resilience to chronic pain? Pain 2015, 156, 2267–2275. [Google Scholar] [CrossRef] [Green Version]
- Hagelberg, N.; Aalto, S.; Tuominen, L.; Pesonen, U.; Någren, K.; Hietala, J.; Scheinin, H.; Pertovaara, A.; Martikainen, I.K. Striatal μ-opioid receptor availability predicts cold pressor pain threshold in healthy human subjects. Neurosci. Lett. 2012, 521, 11–14. [Google Scholar] [CrossRef]
- Mamatha, R.K.; Nagendra, S.N. Effect of disulfiram administration on glutamate uptake by synaptosomes in the rat brain. Eur. J. Pharmacol. Environ. Toxicol. Pharmacol. 1994, 292, 89–94. [Google Scholar] [CrossRef]
- Noble, F.; Cox, B.M. Differential desensitization of μ- and δ-opioid receptors in selected neural pathways following chronic morphine treatment. Br. J. Pharmacol. 1996, 117, 161–169. [Google Scholar] [CrossRef]
- Sim-Selley, L.J.; Selley, D.E.; Vogt, L.J.; Childers, S.R.; Martin, T.J. Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain. J. Neurosci. 2000, 20, 4555–4562. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.L.; Tambeli, C.H.; Barletta, J.; Luo, L.; Green, P.; Levine, J.D.; Gear, R.W. Altered Nucleus Accumbens Circuitry Mediates Pain-Induced Antinociception in Morphine-Tolerant Rats. J. Neurosci. 2002, 22, 6773–6780. [Google Scholar] [CrossRef]
- Goldstein, M.; Nakajima, K. The effect of disulfiram on catecholamine levels in the brain. J. Pharmacol. Exp. Ther. 1967, 157, 96–102. [Google Scholar] [PubMed]
- Meyer, P.J.; Morgan, M.M.; Kozell, L.B.; Ingram, S.L. Contribution of dopamine receptors to periaqueductal gray-mediated antinociception. Psychopharmacology 2009, 204, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobaldini, G.; Reis, R.A.; Sardi, N.F.; Lazzarim, M.K.; Tomim, D.H.; Lima, M.M.; Fischer, L. Dopaminergic mechanisms in periaqueductal gray-mediated antinociception. Behav. Pharmacol. 2018, 29, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Azaryan, A.V.; Clock, B.J.; Cox, B.M. Mu opioid receptor mRNA in nucleus accumbens is elevated following dopamine receptor activation. Neurochem. Res. 1996, 21, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Gratton, A. Opioid modulation and sensitization of dopamine release elicited by sexually relevant stimuli: A high speed chronoamperometric study in freely behaving rats. Brain Res. 1991, 551, 20–27. [Google Scholar] [CrossRef]
- Schad, C.A.; Justice, J.B.; Holtzman, S.G. Naloxone reduces the neurochemical and behavioral effects of amphetamine but not those of cocaine. Eur. J. Pharmacol. 1995, 275, 9–16. [Google Scholar] [CrossRef]
- Weidenauer, A.; Bauer, M.; Sauerzopf, U.; Bartova, L.; Nics, L.; Pfaff, S.; Philippe, C.; Berroterán-Infante, N.; Pichler, V.; Meyer, B.M.; et al. On the relationship of first-episode psychosis to the amphetamine-sensitized state: A dopamine D2/3 receptor agonist radioligand study. Transl. Psychiatry 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Luo, F.; Ge, X.-C.; Fu, A.-H.; Han, J.-S. Effects of lesions of various brain areas on drug priming or footshock-induced reactivation of extinguished conditioned place preference. Brain Res. 2002, 950, 1–9. [Google Scholar] [CrossRef]
- Befort, K.; Filliol, D.; Ghate, A.; Darcq, E.; Matifas, A.; Muller, J.; Lardenois, A.; Thibault, C.; Dembele, D.; Le Merrer, J.; et al. Mu-opioid receptor activation induces transcriptional plasticity in the central extended amygdala. Eur. J. Neurosci. 2008, 27, 2973–2984. [Google Scholar] [CrossRef]
- Gadd, C.A.; Murtra, P.; De Felipe, C.; Hunt, S.P. Neurokinin-1 Receptor-Expressing Neurons in the Amygdala Modulate Morphine Reward and Anxiety Behaviors in the Mouse. J. Neurosci. 2003, 23, 8271–8280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, J.E.; Hershman, K.; Kumar, M.S.; Thompson, M.L.; Kream, R.M. Disulfiram administration affects substance P-like immunoreactive and monoaminergic neural systems in rodent brain. J. Biol. Chem. 1990, 265, 264–273. [Google Scholar] [CrossRef]
- Poulsen, H.E.; Loft, S.; Andersen, J.R.; Andersen, M. Disulfiram therapy—Adverse drug reactions and interactions. Acta Psychiatr. Scand. 1992, 86, 59–66. [Google Scholar] [CrossRef]
- Frączek, K.; Kowalczyk, A.; Pekala, M.; Kasarello, K.; Sygitowicz, G.; Sulejczak, D.; Zaremba, M.; Konop, M.; Frankowska, M.; Filip, M.; et al. The Positive and Negative Outcome of Morphine and Disulfiram Subacute Co-Administration in Rats in the Absence of Ethanol Challenge. Pharmaceutics 2020, 13, 29. [Google Scholar] [CrossRef]
- Bujalska-Zadrożny, M.; Wolińska, R.; Gąsińska, E.; Nagraba, Ł. Tapentadol and nitric oxide synthase systems. Behav. Pharmacol. 2015, 26, 282–288. [Google Scholar] [CrossRef]
- Amour, F.E.; Smith, D.L. A Method for Determining Loss of Pain Sensation. J. Pharmacol. Exp. Ther. 1941, 72, 74. [Google Scholar]
- Randall, L.O.; Selitto, J.J. A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 1957, 111, 409–419. [Google Scholar] [PubMed]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Combination | Randal–Selitto Test | Tail-Flick Test |
|---|---|---|
| MRF + DSF 12.5 | 0.69 | 1.12 |
| MRF + DSF 25 | 0.61 | 1.04 |
| MRF + DSF 50 | 0.66 | 0.9 |
| Emax ± SEM (%) | |||||
|---|---|---|---|---|---|
| CNS Structure | Control | DAY 1 | DAY 14 | ||
| DSF + MRF | MRF | DSF + MRF | MRF | ||
| frontal cortex | 140 ± 2.9 | 140 ± 2.4 | 134 ± 3.4 | 121 ± 2.9 ** | 129 ± 2.1* |
| thalamus | 180 ± 3.4 | 157 ± 2.2 *** | 155 ± 2.2 *** | 136 ± 1.9 *** | 139 ± 3.0 *** |
| hippocampus | 139 ± 2.8 | 131 ± 2.6 | 134 ± 2.9 | 120 ± 1.2 ** | 120 ± 2.4 ** |
| prefrontal cortex | 133 ± 3.0 | 142 ± 1.9 * # | 131 ± 2.5 | 122 ± 2.3 * | 118 ± 2.6 * |
| nucleus accumbens | 171 ± 3.6 | 153 ± 1.8 ** | 155 ± 2.5 * | 139 ± 2.6 # | 120 ± 1.8 *** |
| hypothalamus | 177 ± 4.2 | 158 ± 2.24 ** # | 148 ± 2.2 ** | 113 ± 1.0 *** | 116 ± 2.0 *** |
| caudate-putamen | 147 ± 2.6 | 156 ± 2.3 * ## | 138 ± 2.6 * | 138 ± 1.9 # | 120 ± 1.6 *** |
| RVM | 156 ± 2.0 | 168 ± 1.8 ** # | 159 ± 2.3 | 134 ± 2.5 ** | 132 ± 2.3 *** |
| amygdala | 151 ± 3.4 | 145 ± 2.9 | 141 ± 2.4 | 116 ± 1.5 *** | 130 ± 4.8 * # |
| PAG | 162 ± 3.1 | 186 ± 5.5 ** ### | 141 ± 1.5 ** | 133 ± 3.3 *** | 136 ± 3.5 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Corde-Skurska, A.; Krzascik, P.; Lesniak, A.; Sacharczuk, M.; Nagraba, L.; Bujalska-Zadrozny, M. Disulfiram Abrogates Morphine Tolerance—A Possible Role of µ-Opioid Receptor-Related G-Protein Activation in the Striatum. Int. J. Mol. Sci. 2021, 22, 4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084057
de Corde-Skurska A, Krzascik P, Lesniak A, Sacharczuk M, Nagraba L, Bujalska-Zadrozny M. Disulfiram Abrogates Morphine Tolerance—A Possible Role of µ-Opioid Receptor-Related G-Protein Activation in the Striatum. International Journal of Molecular Sciences. 2021; 22(8):4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084057
Chicago/Turabian Stylede Corde-Skurska, Anna, Pawel Krzascik, Anna Lesniak, Mariusz Sacharczuk, Lukasz Nagraba, and Magdalena Bujalska-Zadrozny. 2021. "Disulfiram Abrogates Morphine Tolerance—A Possible Role of µ-Opioid Receptor-Related G-Protein Activation in the Striatum" International Journal of Molecular Sciences 22, no. 8: 4057. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084057