
Chromeno[3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors


 ,
,
Abstract
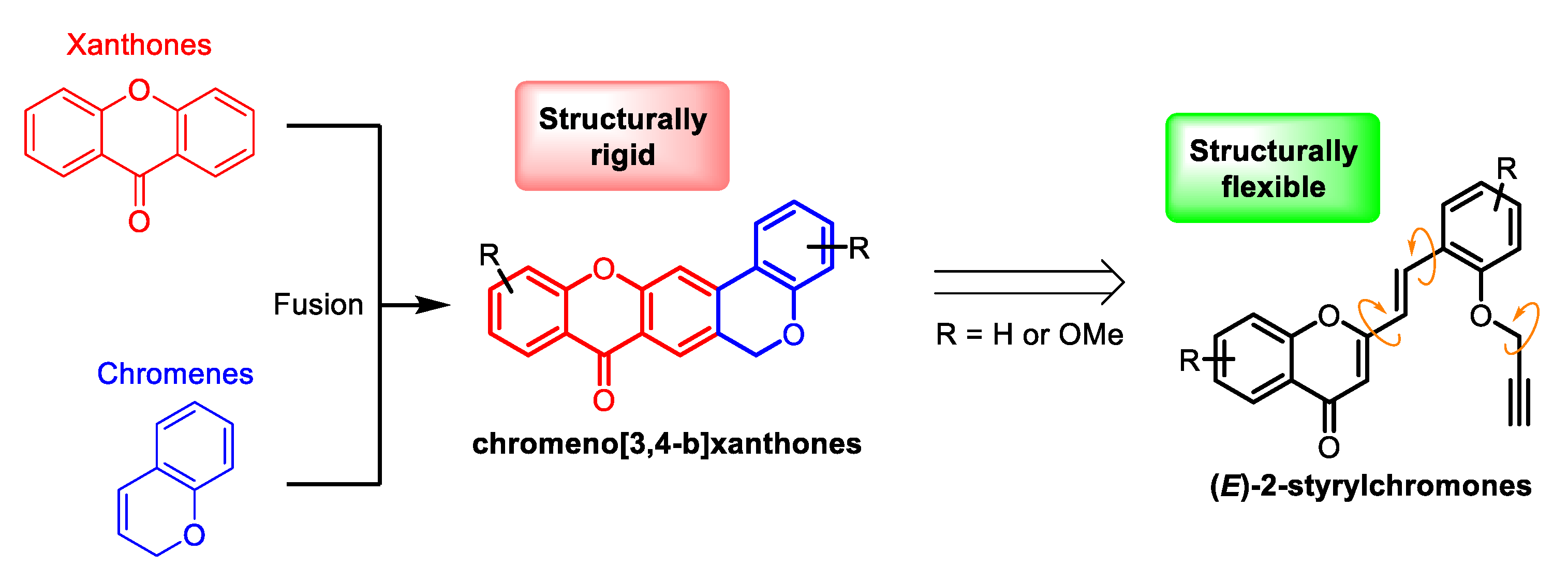
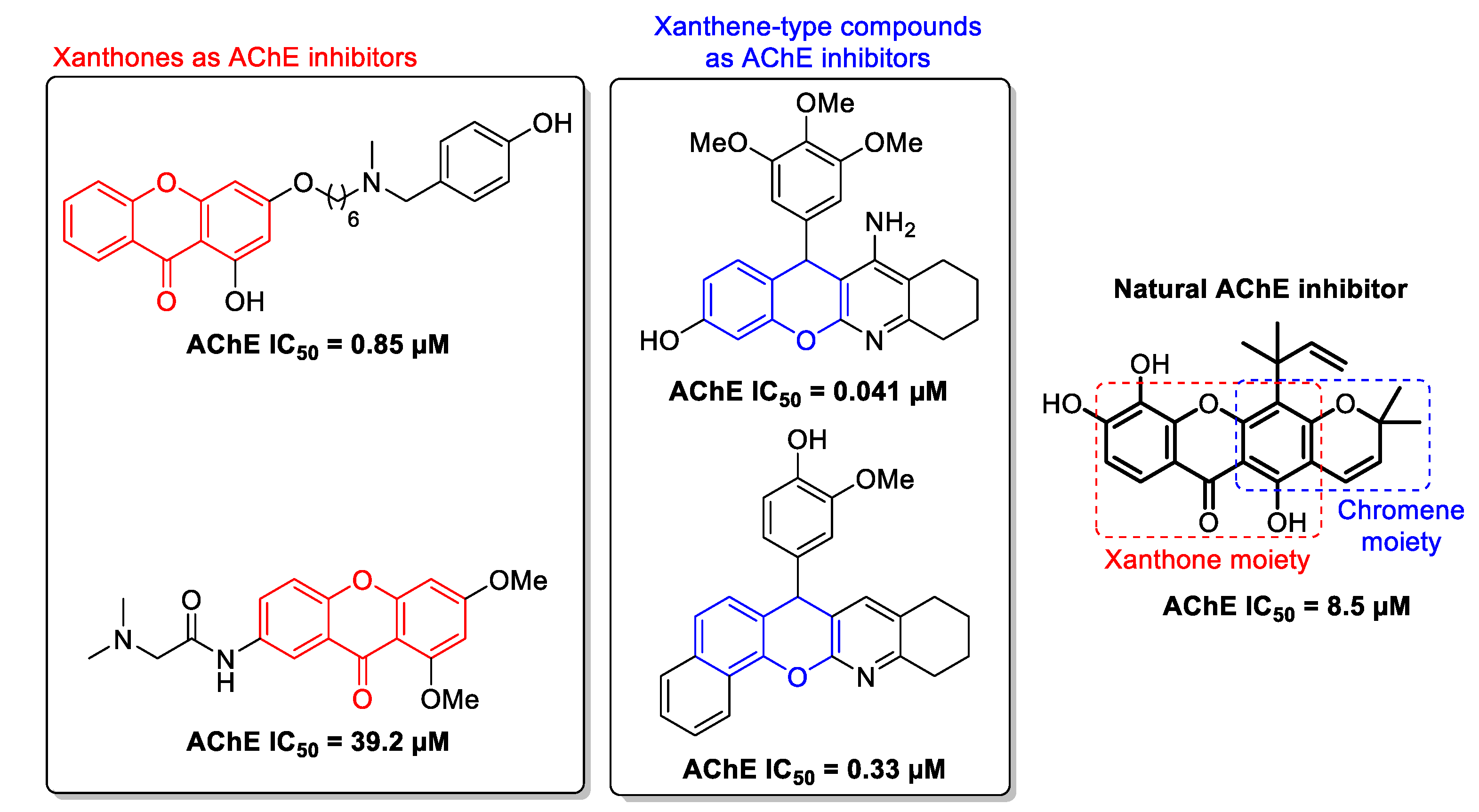
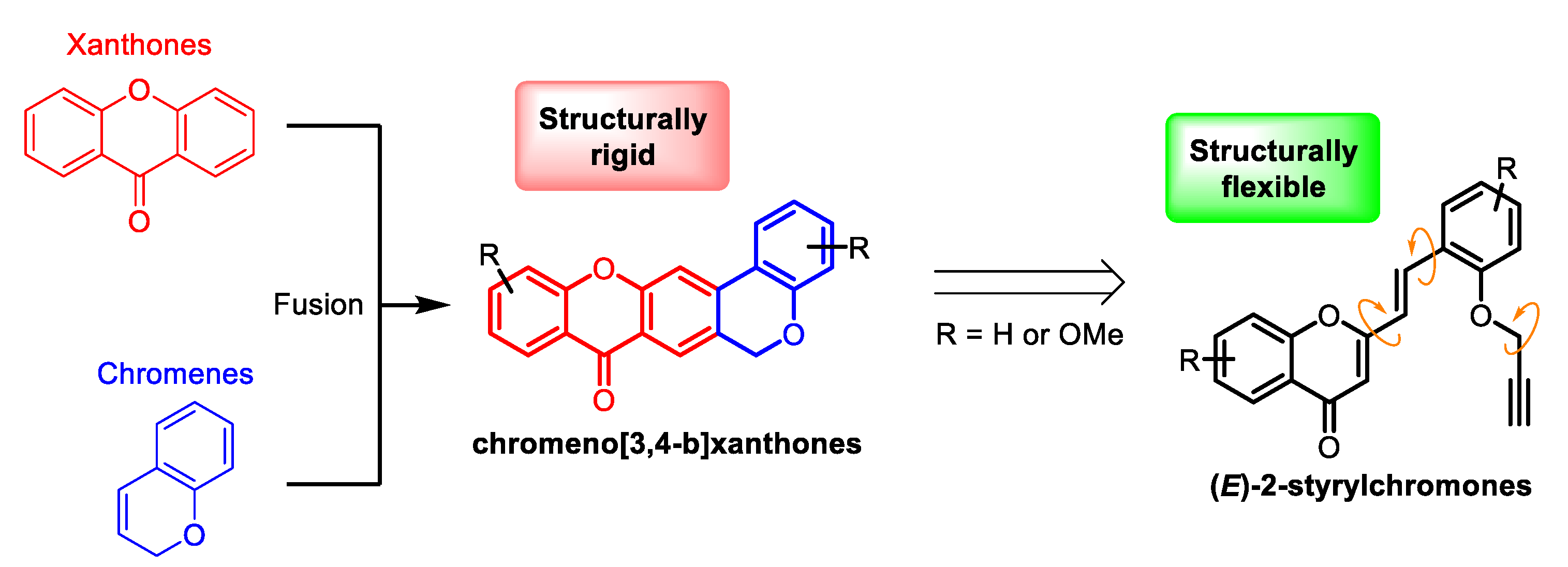
:1. Introduction
2. Results and Discussion
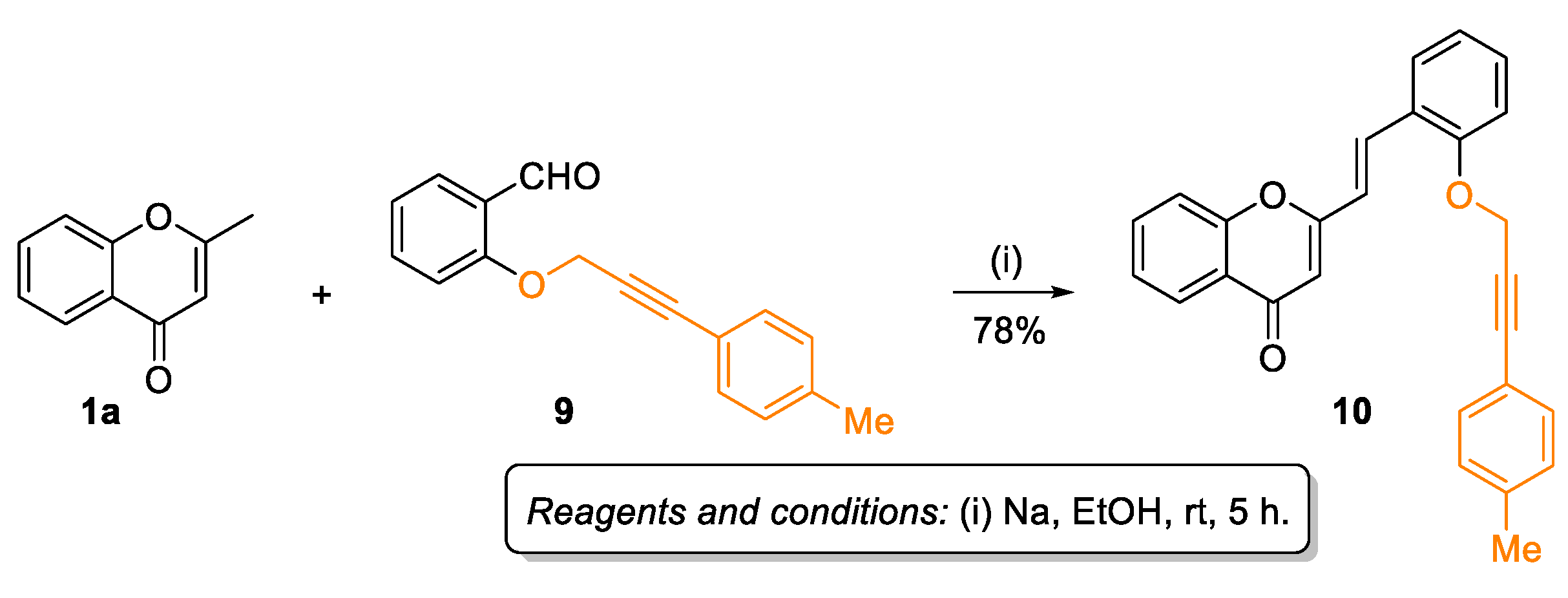
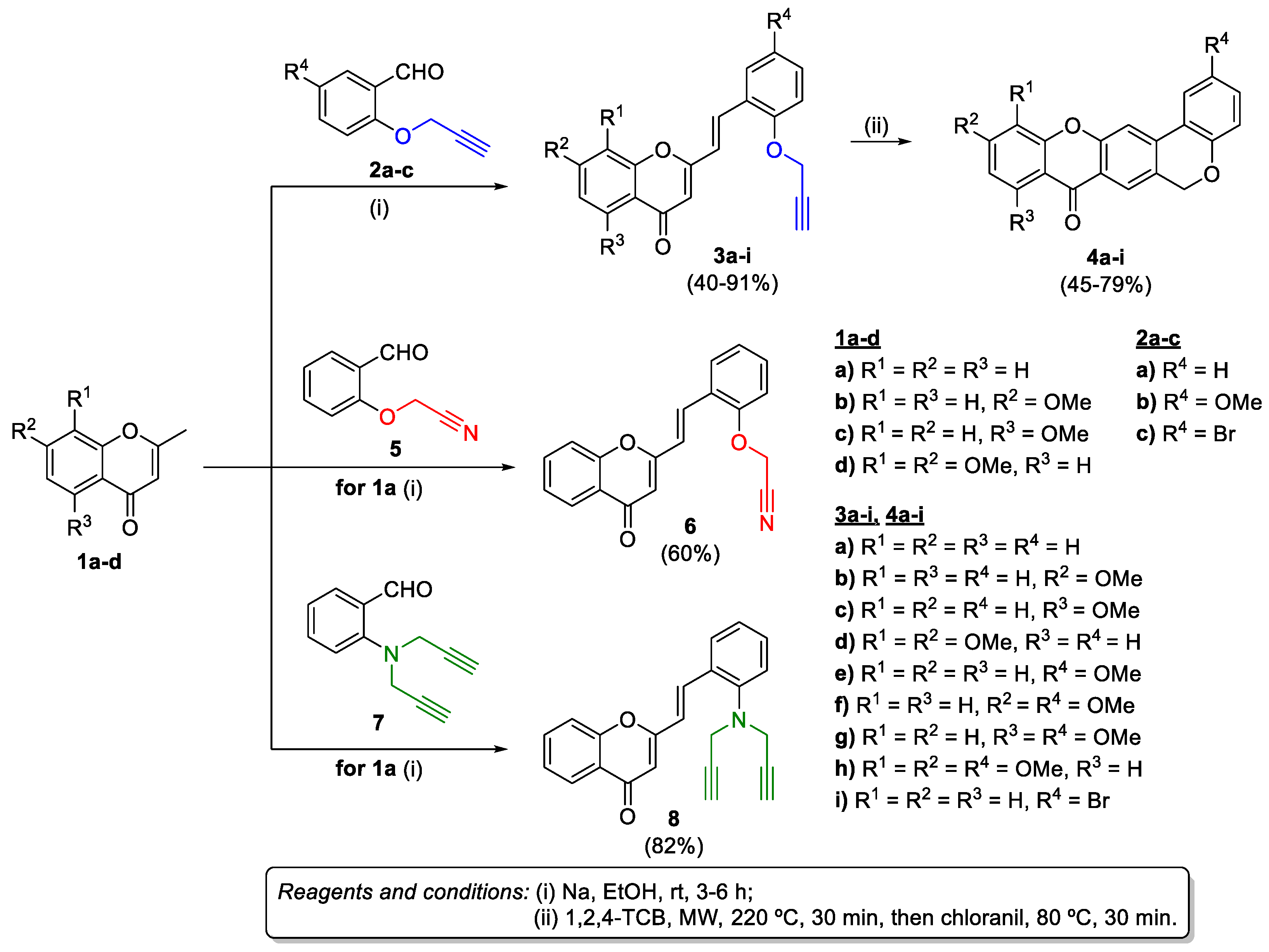
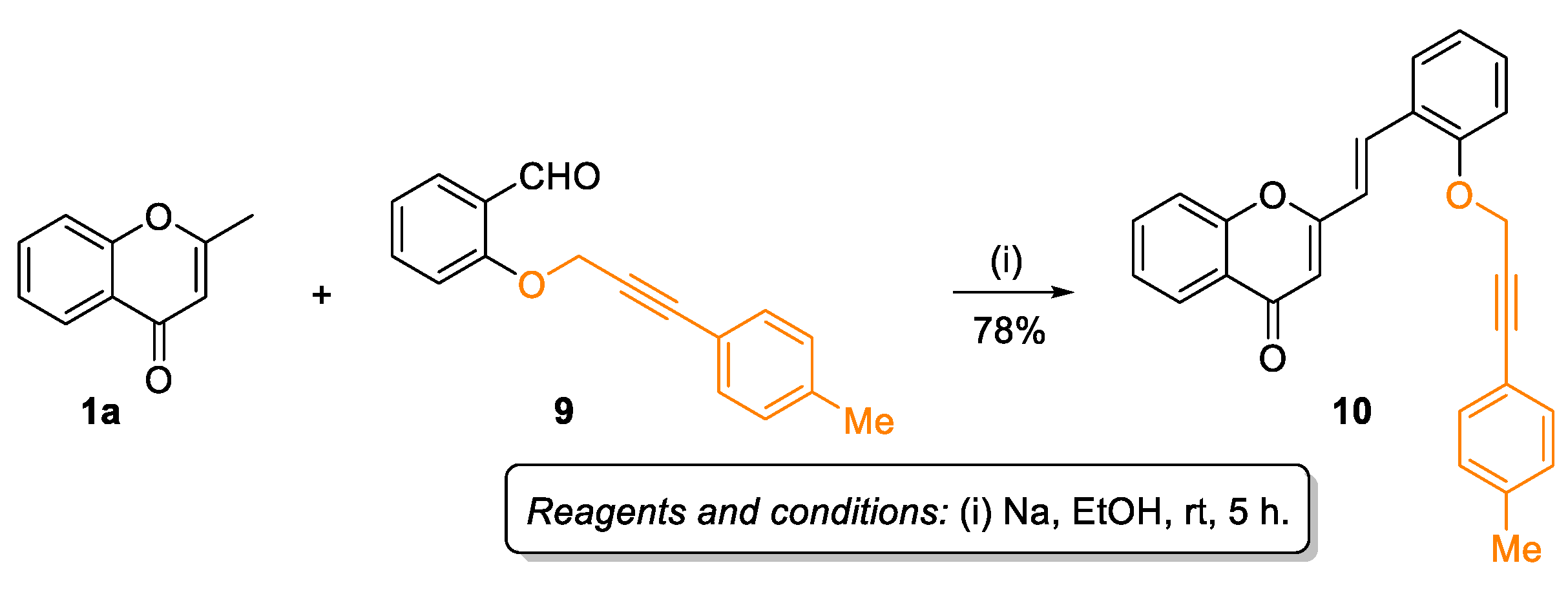
2.1. Synthesis of Target Compounds
2.2. Biological Evaluation
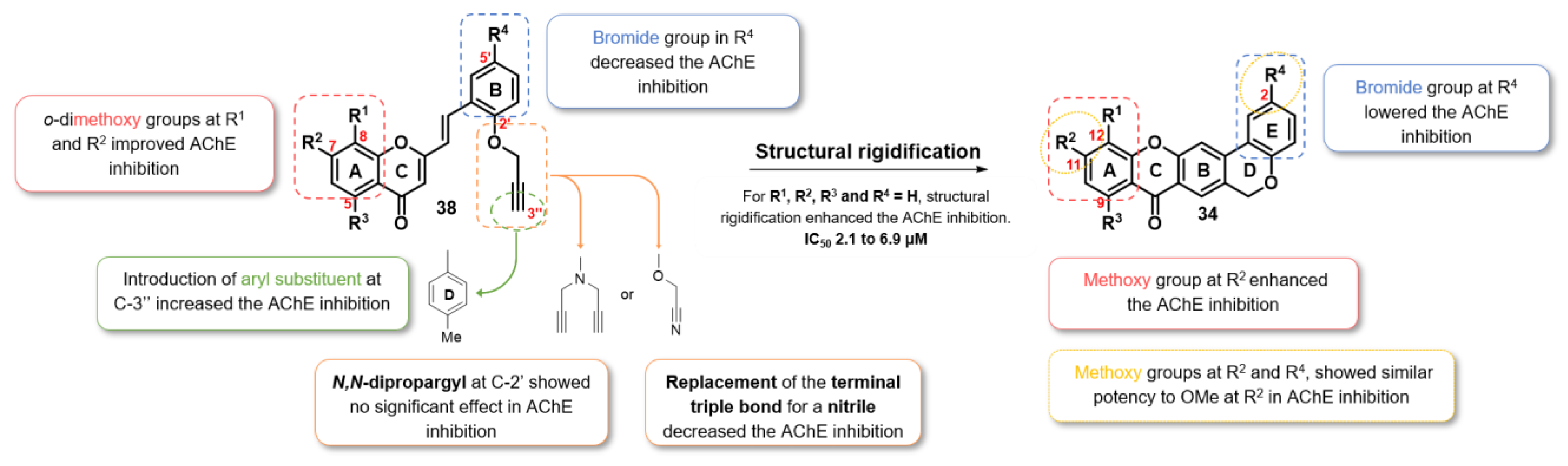
2.2.1. AChE Inhibition
2.2.2. Molecular Docking
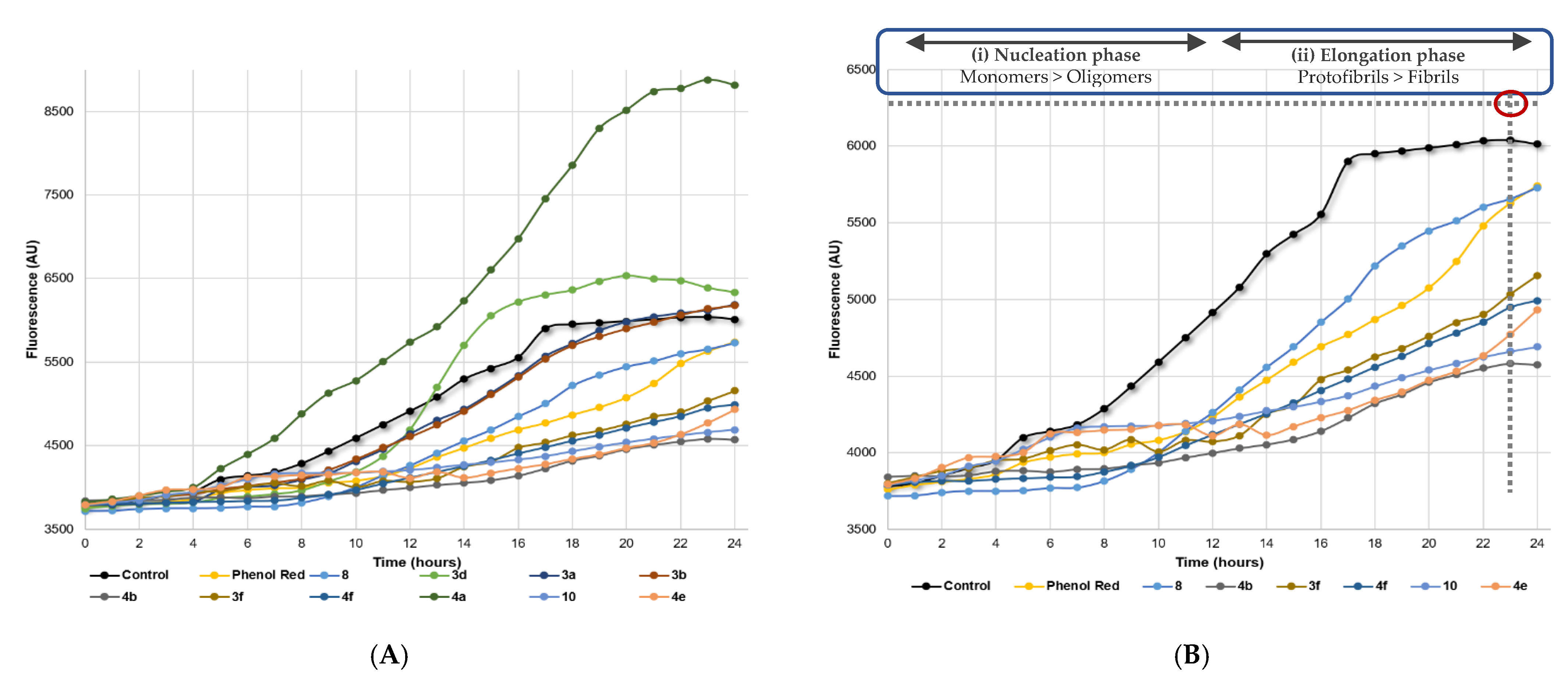
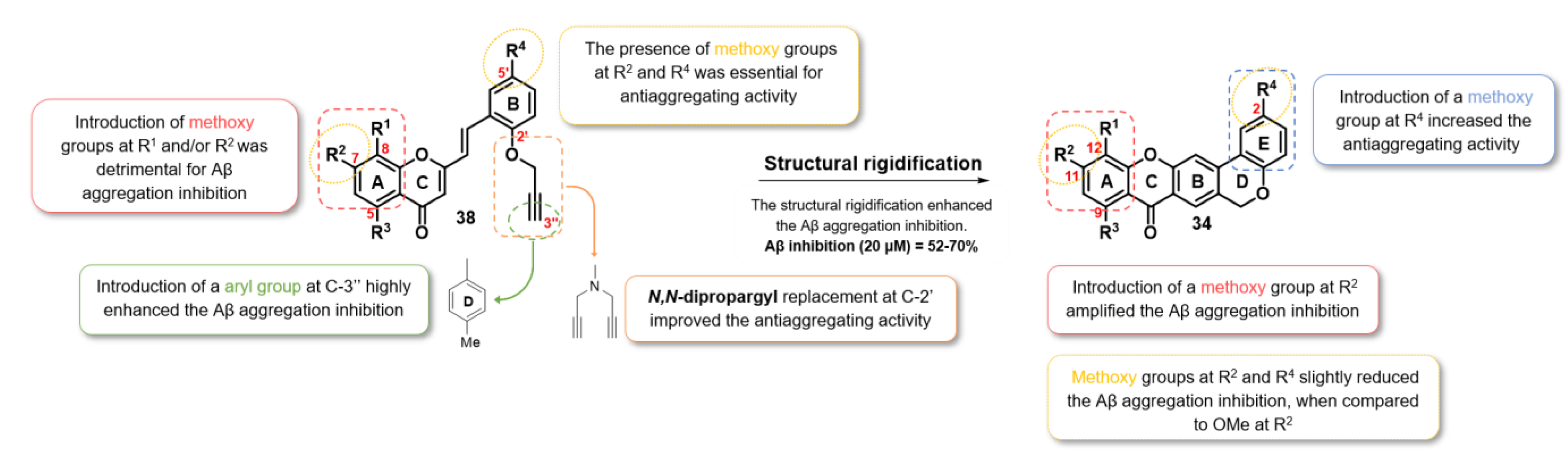
2.2.3. Aβ Aggregation Inhibition
2.2.4. Dual-Target Activity Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Remarks
3.1.2. Synthesis of (E)-2-Styrylchromones 3a-i, 6, and 10
3.1.3. Synthesis of Chromeno[3,4-b]xanthones (4a-i)
3.2. AChE Inhibition
3.3. Molecular Docking
3.4. Aβ Aggregation Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal. Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H. Reconsideration of Anticholinesterase Therapeutic Strategies against Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Association, A.S. 2019 Alzheimer’s disease facts and figures. Alzheimer Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Blaikie, L.; Kay, G.; Kong Thoo Lin, P. Current and emerging therapeutic targets of alzheimer’s disease for the design of multi-target directed ligands. Med. Chem. Commun. 2019, 10, 2052–2072. [Google Scholar] [CrossRef]
- Soreq, H.; Seidman, S. Acetylcholinesterase—new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Panek, D.; Wichur, T.; Godyń, J.; Pasieka, A.; Malawska, B. Advances toward multifunctional cholinesterase and β-amyloid aggregation inhibitors. Future Med. Chem. 2017, 9, 1835–1854. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Squire, L.R. Encyclopedia of Neuroscience; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Aminoff, M.J.; Daroff, R.B. Encyclopedia of the Neurological Sciences; Academic Press: Cambridge, MA, USA, 2014; pp. 1–4740. [Google Scholar]
- Hansson, O.; Zetterberg, H.; Vanmechelen, E.; Vanderstichele, H.; Andreasson, U.; Londos, E.; Wallin, A.; Minthon, L.; Blennow, K. Evaluation of plasma Aβ40 and Aβ42 as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol. Aging 2010, 31, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Bulic, B.; Pickhardt, M.; Schmidt, B.; Mandelkow, E.-M.; Waldmann, H.; Mandelkow, E. Development of Tau Aggregation Inhibitors for Alzheimer’s Disease. Angew. Chem. Int. Ed. 2009, 48, 1740–1752. [Google Scholar] [CrossRef]
- Majdi, A.; Sadigh-Eteghad, S.; Rahigh Aghsan, S.; Farajdokht, F.; Vatandoust, S.M.; Namvaran, A.; Mahmoudi, J. Amyloid-β, tau, and the cholinergic system in Alzheimer’s disease: Seeking direction in a tangle of clues. Rev. Neurosci. 2020, 31, 391–413. [Google Scholar] [CrossRef]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- González, J.F.; Alcántara, A.R.; Doadrio, A.L.; Sánchez-Montero, J.M. Developments with multi-target drugs for Alzheimer’s disease: An overview of the current discovery approaches. Expert Opin. Drug Discov. 2019, 14, 879–891. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, J.; Cheng, M.; Zhou, W.; Wan, Y.; Wang, R.; Fang, Y.; Jin, Y.; Liu, J.; Xie, S.-S. Design, synthesis, and biological evaluation of novel xanthone-alkylbenzylamine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 213, 113154. [Google Scholar] [CrossRef] [PubMed]
- Jalili-Baleh, L.; Babaei, E.; Abdpour, S.; Nasir Abbas Bukhari, S.; Foroumadi, A.; Ramazani, A.; Sharifzadeh, M.; Abdollahi, M.; Khoobi, M. A review on flavonoid-based scaffolds as multi-target-directed ligands (MTDLs) for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 152, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.I.; Cidade, H.; Pinto, M. Dual/multitargeted xanthone derivatives for Alzheimer’s disease: Where do we stand? Future Med. Chem. 2017, 9, 1611–1630. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Dias, T.A.; Brito, A.; Proença, F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016, 123, 487–507. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.H.; Orhan, I.; Şenol, F.S.; Kartal, M.; Şener, B.; Dvorská, M.; Šmejkal, K.; Šlapetová, T. Cholinesterase inhibitory activities of some flavonoid derivatives and chosen xanthone and their molecular docking studies. Chem. Biol. Interact. 2009, 181, 383–389. [Google Scholar] [CrossRef]
- Malafaia, D.; Albuquerque, H.M.T.; Silva, A.M.S. Amyloid-β and tau aggregation dual-inhibitors: A synthetic and structure-activity relationship focused review. Eur. J. Med. Chem. 2021, 214, 113209. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Levine III, H. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef]
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides—a trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2011, 3, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, H.M.T.; Santos, C.M.M.; Cavaleiro, J.A.S.; Silva, A.M.S. First intramolecular Diels–Alder reactions using chromone derivatives: Synthesis of chromeno[3,4-b]xanthones and 2-(benzo[c]chromenyl)chromones. New J. Chem. 2018, 42, 4251–4260. [Google Scholar] [CrossRef] [Green Version]
- Bourne, Y.; Grassi, J.; Bougis, P.E.; Marchot, P. Conformational Flexibility of the Acetylcholinesterase Tetramer Suggested by X-ray Crystallography*. J. Biol. Chem. 1999, 274, 30370–30376. [Google Scholar] [CrossRef] [Green Version]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | R1 | R2 | R3 | R4 | eeAChE [IC50 (μM)] [a] |
|---|---|---|---|---|---|---|
| 3a |  | H | H | H | H | 9.5 ± 0.3 |
| 3b | H | OMe | H | H | >20 | |
| 3c | H | H | OMe | H | >20 | |
| 3d | OMe | OMe | H | H | 2.4 ± 0.1 | |
| 3e | H | H | H | OMe | >20 | |
| 3f | H | OMe | H | OMe | >20 | |
| 3g | H | H | OMe | OMe | >20 | |
| 3h | OMe | OMe | H | OMe | >20 | |
| 3i | H | H | H | Br | >20 | |
| 6 |  | - | - | - | - | >20 |
| 8 |  | - | - | - | - | 8.7 ± 0.2 |
| 10 |  | - | - | - | - | 2.9 ± 0.1 |
| 4a |  | H | H | H | H | 2.1 ± 0.2 |
| 4b | H | OMe | H | H | 3.9 ± 0.1 | |
| 4c | H | H | OMe | H | >20 | |
| 4d | OMe | OMe | H | H | >20 | |
| 4e | H | H | H | OMe | 6.9 ± 0.3 | |
| 4f | H | OMe | H | OMe | 4.8 ± 0.1 | |
| 4g | H | H | OMe | OMe | >20 | |
| 4h | OMe | OMe | H | OMe | >20 | |
| 4i | H | H | H | Br | >20 | |
| Donepezil | 0.0156 ± 0.0016 |
| Compound | Structure | R1 | R2 | R3 | R4 | Aβ1-42 Aggregation (% Inhib. at 20 μM) |
|---|---|---|---|---|---|---|
| 3a |  | H | H | H | H | <10 [a] |
| 3b | H | OMe | H | H | <10 [a] | |
| 3d | OMe | OMe | H | H | <10 [a] | |
| 3f | H | OMe | H | OMe | 48 | |
| 8 |  | - | - | - | - | 18 |
| 10 |  | H | H | H | H | 66 |
| 4a |  | H | H | H | H | <10 [a] |
| 4b | H | OMe | H | H | 70 | |
| 4e | H | H | H | OMe | 61 | |
| 4f | H | OMe | H | OMe | 52 | |
| Phenol Red | 20 |
| Compound | Structure | R1 | R2 | R3 | R4 | eeAChE (IC50 [μM]) [a] | Aβ1-42 Aggregation (% Inhib. at 20 μM) |
|---|---|---|---|---|---|---|---|
| 8 |  | - | - | - | - | 8.7 ± 0.2 | 18 |
| 10 |  | - | - | - | - | 2.9 ± 0.1 | 66 |
| 4b |  | H | OMe | H | H | 3.9 ± 0.1 | 70 |
| 4e | H | H | H | OMe | 6.9 ± 0.3 | 61 | |
| 4f | H | OMe | H | OMe | 4.8 ± 0.1 | 52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malafaia, D.; Oliveira, A.; Fernandes, P.A.; Ramos, M.J.; Albuquerque, H.M.T.; Silva, A.M.S. Chromeno[3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors. Int. J. Mol. Sci. 2021, 22, 4145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084145
Malafaia D, Oliveira A, Fernandes PA, Ramos MJ, Albuquerque HMT, Silva AMS. Chromeno[3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors. International Journal of Molecular Sciences. 2021; 22(8):4145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084145
Chicago/Turabian StyleMalafaia, Daniela, Ana Oliveira, Pedro A. Fernandes, Maria J. Ramos, Hélio M. T. Albuquerque, and Artur M. S. Silva. 2021. "Chromeno[3,4-b]xanthones as First-in-Class AChE and Aβ Aggregation Dual-Inhibitors" International Journal of Molecular Sciences 22, no. 8: 4145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084145