
Functional Analysis of the PCCA and PCCB Gene Variants Predicted to Affect Splicing

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Variant Selection and Bioinformatic Analysis
2.2. Minigene Assay
2.3. Variant Calling and Classification
3. Results
3.1. The PCCA and PCCB Variant Selection
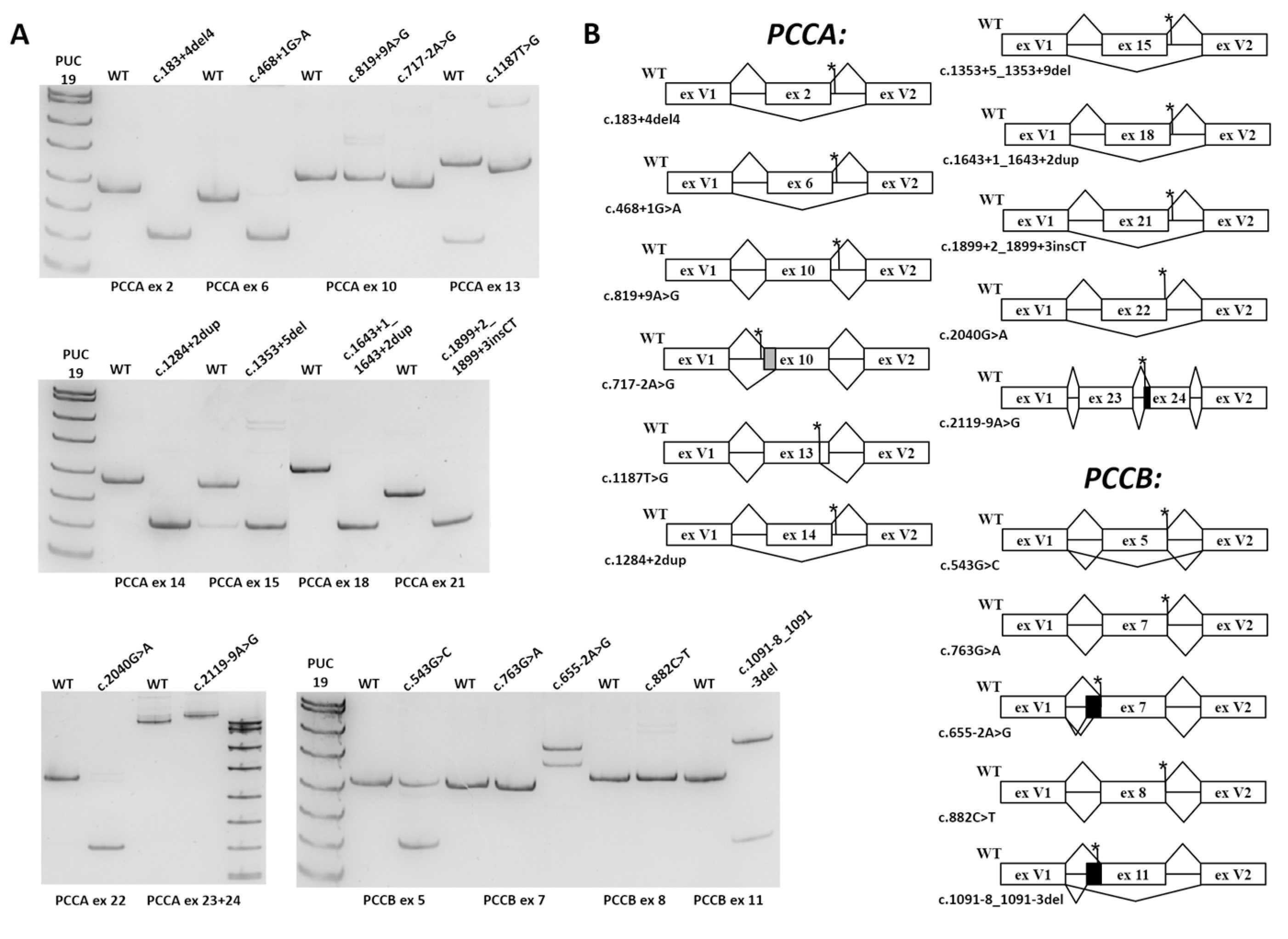
3.2. Minigene Assay
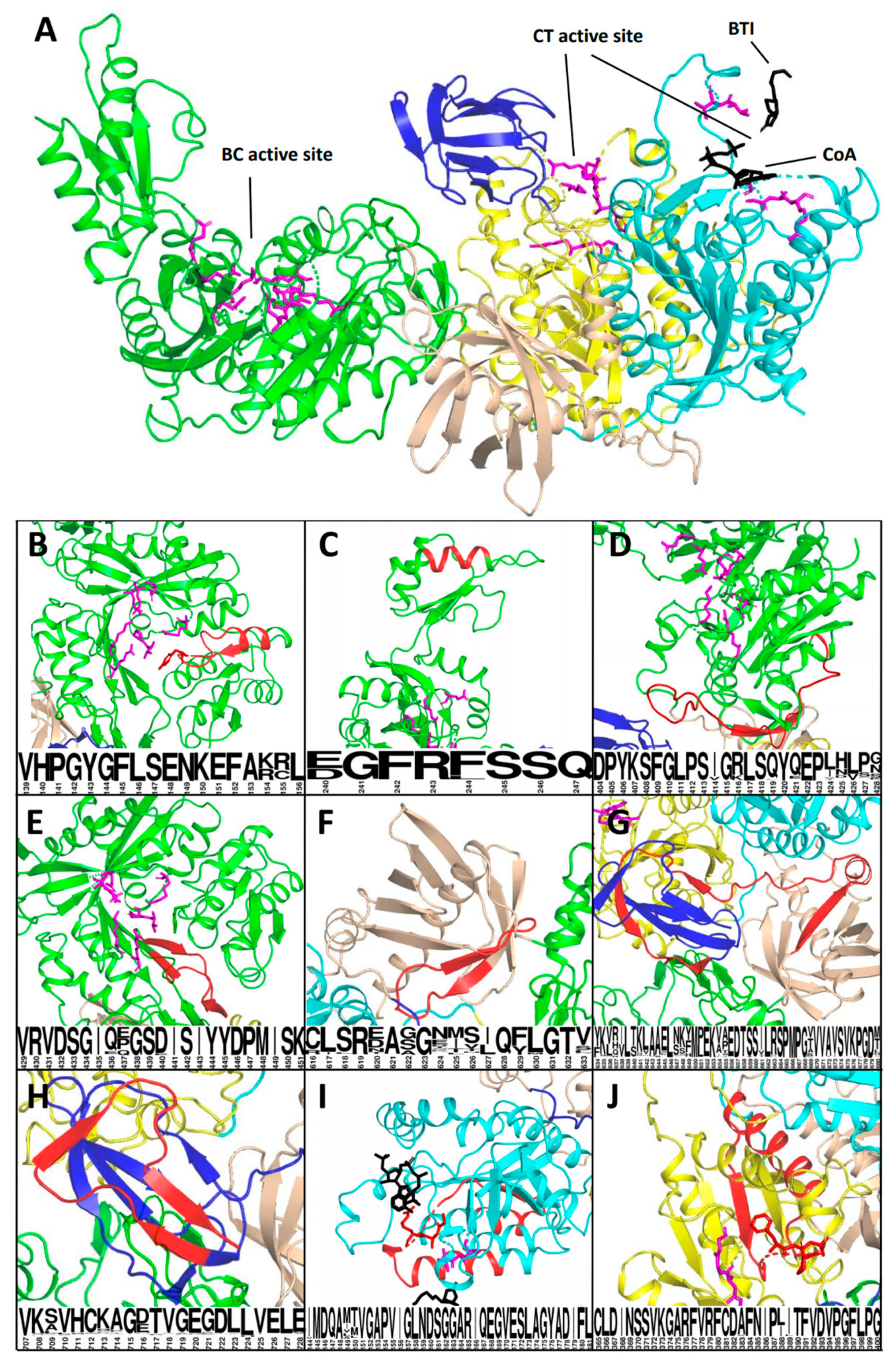
3.3. Analysis of the Affected Protein Structures
3.4. Classification of the Studied Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalousek, F.; Darigo, M.D.; Rosenberg, L.E. Isolation and characterization of propionyl-CoA carboxylase from normal human liver. Evidence for a protomeric tetramer of nonidentical subunits. J. Biol. Chem. 1980, 255, 60–65. [Google Scholar] [CrossRef]
- Shchelochkov, O.A.; Venditti, C. Propionic Acidemia. GeneReviews®, 17 May 2012 [Updated 6 October 2016]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK92946/ (accessed on 15 April 2021).
- Chapman, K.A.; Gropman, A.; MacLeod, E.; Stagni, K.; Summar, M.L.; Ueda, K.; Mew, N.A.; Franks, J.; Island, E.; Matern, D. Acute management of propionic acidemia. Mol. Genet. Metab. 2012, 105, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, I.K.; Hsia, Y.E.; Clement, D.H.; Provence, S.A. Propionicacidemia (ketotic hyperglycinemia): Dietary treatment resulting in normal growth and development. Pediatrics 1974, 53, 391–395. [Google Scholar]
- Jurecki, E.; Ueda, K.; Frazier, D.; Rohr, F.; Thompson, A.; Hussa, C.; Obernolte, L.; Reineking, B.; Roberts, A.; Yannicelli, S. Nutrition management guideline for propionic acidemia: An evidence-and consensus-based approach. Mol. Genet. Metab. 2019, 126, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Wongkittichote, P.; Ah Mew, N.; Chapman, K.A. Propionyl-CoA carboxylase—A review. Mol. Genet. Metab. 2017, 122, 145–152. [Google Scholar] [CrossRef]
- Kraus, J.P.; Spector, E.; Venezia, S.; Estes, P.; Chiang, P.W.; Creadon-Swindell, G.; Mullerleile, S.; de Silva, L.; Barth, M.; Walter, M.; et al. Mutation analysis in 54 propionic acidemia patients. J. Inherit. Metab. Dis. 2012, 35, 51–63. [Google Scholar] [CrossRef]
- Rivera-Barahona, A.; Navarrete, R.; Garcia-Rodriguez, R.; Richard, E.; Ugarte, M.; Perez-Cerda, C.; Perez, B.; Gamez, A.; Desviat, L.R. Identification of 34 novel mutations in propionic acidemia: Functional characterization of missense variants and phenotype associations. Mol. Genet. Metab. 2018, 125, 266–275. [Google Scholar] [CrossRef]
- Sánchez-Alcudia, R.; Pérez, B.; Pérez-Cerdá, C.; Ugarte, M.; Desviat, L.R. Overexpression of adapted U1snRNA in patients’ cells to correct a 5′ splice site mutation in propionic acidemia. Mol. Genet. Metab. 2011, 102, 134–138. [Google Scholar] [CrossRef]
- Rincon, A.; Aguado, C.; Desviat, L.; Sanchez-Alcudia, R.; Ugarte, M.; Perez, B. Propionic and methylmalonic acidemia: Antisense therapeutics for intronic variations causing aberrantly spliced messenger RNA. Am. J. Hum. Genet. 2007, 81, 1262–1270. [Google Scholar] [CrossRef] [Green Version]
- Pagenstecher, C.; Wehner, M.; Friedl, W.; Rahner, N.; Aretz, S.; Friedrichs, N.; Sengteller, M.; Henn, W.; Buettner, R.; Propping, P. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum. Genet. 2006, 119, 9–22. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A. Use of minigene systems to dissect alternative splicing elements. Methods 2005, 37, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Tournier, I.; Vezain, M.; Martins, A.; Charbonnier, F.; Baert-Desurmont, S.; Olschwang, S.; Wang, Q.; Buisine, M.P.; Soret, J.; Tazi, J. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum. Mutat. 2008, 29, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Acedo, A.; Velasco, E.A. Identification of eight spliceogenic variants in BRCA2 Exon 16 by minigene assays. Front. Genet. 2018, 9, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Brnich, S.E.; Abou Tayoun, A.N.; Couch, F.J.; Cutting, G.R.; Greenblatt, M.S.; Heinen, C.D.; Kanavy, D.M.; Luo, X.; McNulty, S.M.; Starita, L.M.; et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Yunin, M.; Kurkina, M.; Bychkov, I.; Melikyan, L.; Itkis, Y.; A, L.; Krylova, T.; Baydakova, G. Molecular Genetic Characterization of Fifteen Patients with Propionic Acidemia. 2020. Available online: https://www.researchgate.net/publication/339740135_Molecular_genetic_characterization_of_fifteen_patients_with_propionic_acidemia (accessed on 15 April 2021).
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Nguyen, T.Y.D.; Cygan, K.J.; Çelik, M.H.; Fairbrother, W.G.; Avsec, Ž.; Gagneur, J. Modular modeling improves the predictions of genetic variant effects on splicing. bioRxiv 2018, 438986. [Google Scholar] [CrossRef]
- Leman, R. Development of Bioinformatics and Biostatistics Tools to Predict and Analyze Splicing Defects: Use Case about Genes Involved in Hereditary Breast and Ovarian Cancers. 2019. Available online: https://www.theses.fr/2019NORMC418.pdf (accessed on 15 April 2021).
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkelenz, S.; Theiss, S.; Otte, M.; Widera, M.; Peter, J.O.; Schaal, H. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014, 42, 10681–10697. [Google Scholar] [CrossRef] [Green Version]
- Tubeuf, H.; Charbonnier, C.; Soukarieh, O.; Blavier, A.; Lefebvre, A.; Dauchel, H.; Frebourg, T.; Gaildrat, P.; Martins, A. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mutat. 2020, 41, 1811–1829. [Google Scholar] [CrossRef] [PubMed]
- Grodecká, L.; Buratti, E.; Freiberger, T. Mutations of pre-mRNA splicing regulatory elements: Are predictions moving forward to clinical diagnostics? Int. J. Mol. Sci. 2017, 18, 1668. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatova, A.Y.; Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Zinchenko, R.A.; Skoblov, M.Y. Functional reassessment of PAX6 single nucleotide variants by in vitro splicing assay. Eur. J. Hum. Genet. 2019, 27, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Calcium Phosphate-Mediated Transfection of Eukaryotic Cells with Plasmid DNAs. Cold Spring Harb. Protoc. 2019, 2019, pdb-prot095430. [Google Scholar] [CrossRef]
- Lee, E.H.; Ko, J.M.; Kim, J.M.; Yoo, H.W. Genotype and clinical features of Korean patients with methylmalonic aciduria and propionic aciduria. Korean J. Pediatr. 2008, 51, 964–970. [Google Scholar] [CrossRef]
- Hu, Y.H.; Han, L.S.; Ye, J.; Qiu, W.J.; Zhang, Y.F.; Yang, Y.L.; Liu, L.; Ma, H.W.; Gao, X.L.; Gu, X.F. Gene mutation analysis in patients with propionic acidemia. Chin. J. Pediatr. 2008, 46, 416–420. [Google Scholar]
- Huang, C.S.; Sadre-Bazzaz, K.; Shen, Y.; Deng, B.; Zhou, Z.H.; Tong, L. Crystal structure of the alpha(6)beta(6) holoenzyme of propionyl-coenzyme A carboxylase. Nature 2010, 466, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Diacovich, L.; Mitchell, D.L.; Pham, H.; Gago, G.; Melgar, M.M.; Khosla, C.; Gramajo, H.; Tsai, S.-C. Crystal structure of the β-subunit of acyl-CoA carboxylase: Structure-based engineering of substrate specificity. Biochemistry 2004, 43, 14027–14036. [Google Scholar] [CrossRef] [PubMed]
- Arabolaza, A.; Shillito, M.E.; Lin, T.-W.; Diacovich, L.; Melgar, M.; Pham, H.; Amick, D.; Gramajo, H.; Tsai, S.-C. Crystal structures and mutational analyses of acyl-CoA carboxylase β subunit of Streptomyces coelicolor. Biochemistry 2010, 49, 7367–7376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, D.; Bijarnia-Mahay, S.; Kohli, S.; Saxena, R.; Puri, R.D.; Shigematsu, Y.; Yamaguchi, S.; Sakamoto, O.; Gupta, N.; Kabra, M.; et al. Seventeen Novel Mutations in PCCA and PCCB Genes in Indian Propionic Acidemia Patients, and Their Outcomes. Genet. Test. Mol. Biomark. 2016, 20, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frebourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Variant Description | Bioinformatic Analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene/Exon | Variant | Source | Classified in ClinVar | Predicted Effect | HSF3.0 Matrices/MaxEntScan | SpliceAI DS | MMSplice Result | SPiP (Splicing Disruption Risk) |
| PCCA 2 | c.183+4_ 183+7del | Clinvar ID 554669, [19] | US | Alteration of WT DSS | −23.87%/−91.15% for WT DS | 0.9 for DL | D | Alter by SPiCE 85.82% |
| PCCA 6 | c.468+1G>A | [19] | N/A | Alteration of WT DSS | −28.72%/−78.13% for WT DS | 0.99 for DL | D | Alter by SPiCE 85.82% |
| PCCA 10 | c.819+9A>G | Clinvar ID 568750 | US | Activation of intronic cryptic DSS | +16.82%/+548.07% for CR DS | 0.57 for DG | N | Alter by creating de novo splice site, 16.88% |
| PCCA 10 | c.717−2A>G | Clinvar ID 850378 | LP | Alteration of WT ASS | −30.7%/−69.37% for WT AS | 1.0 for AL | D | Alter by SPiCE 98.67% |
| PCCA 13 | c.1187T>G (p.Val396Gly) | [31] | N/A | Activation of exonic CR DSS | +44.85%/+447.37% for CR DS | 0.96 for DG | N | Alter by creating de novo splice site, 30.18% |
| PCCA 14 | c.1284+2dup | [19] | N/A | Alteration of WT DSS | −46.61%/−376.08% for WT DS | 0.98 for DL | D | Alter by SPiCE 97.46% |
| PCCA 15 | c.1353+5_ 1353 + 9del | Clinvar ID 254165 | P based only on bioinformatic analysis (PMID: 27227689) | Alteration of WT DSS | −14.57%/−162.03% for WT DS | 0.62 for DL | D | Alter by SPiCE 97.46% |
| PCCA 18 | c.1643+1_ 1643+2dup | Clinvar ID 553799 | US | Alteration of WT DSS | −13.13%/−70.73% for WT DS | 0.74 for DL | D | Alter by SPiCE 85.82% |
| PCCA 21 | c.1899+2_ 1899 + 3insCT | [32] | N/A | Alteration of WT DSS | −32.63%/−202.22% for WT DS | 1.0 for DL | D | Alter by SPiCE 97.46% |
| PCCA 22 | c.2040G>A (p.Ala680=) | Clinvar ID 218256 | US, P (based only on bioinformatic analysis (PMID: 27227689) | Alteration of WT DSS | −11.56%/−51.3% for WT DS | 0.63 for DL | D | Alter by SPiCE 85.82% |
| PCCA 24 | c.2119-9A>G | Clinvar ID 459938 | LP | Activation of intronic cryptic ASS | +50.57/+723.95 for CR AS | 0.99 for AG | D | Alter by creating de novo splice site, 66.21% |
| PCCB 5 | c.543G>C (p.Leu181=) | Clinvar ID 658152 | US | Alteration of WT DSS | −12.01/−57.03% for WT DS | 0.15 for DL | D | Alter by SPiCE 85.82% |
| PCCB 7 | c.763G>A (p.Gly255Ser) | Clinvar ID 557375 | US | Alteration of WT DSS | −14.8/−42.95% for WT DS | 0.08 for DL | D | Alter by SPiCE 85.82% |
| PCCB 7 | c.655-2A>G | [19] | N/A | Alteration of WT ASS | −30.97%/−121.41% for WT DS | 1.0 for AL | D | Alter by SPiCE 98.67% |
| PCCB 8 | c.882C>T (p.Pro294=) | Clinvar ID 343472 | US/LB | Alteration of WT DSS | −2.19/−35.05% for WT DS | 0.06 for DL | N | Alter by SPiCE 85.82% |
| PCCB 11 | c.1091 -8_1091-3del | Clinvar ID 802010 | P, without evidence | Alteration of WT ASS | −5.08%/−71.79% for WT AS | 0.86 for AL | D | Alter by SPiCE 66.21% |
| Gene/Exon | Variant | Effect in Minigene/Potential Effect at the PCCA/PCCB mRNA Level | Potential Effect at the Protein Level | Affected mRNA/Protein Properties | Classified in ClinVar | New Classification, According to ACMG |
|---|---|---|---|---|---|---|
| PCCA 2 | c.183+4_183+7del | exon skipping/ r.106_183del | p.His36_Lys61del | Mitochondrial target peptide | US | P (PS3, PM2, PM3, PP3, PP4) |
| PCCA 6 | c.468+1G>A | exon skipping/ r.415_468del | p.Val139_Leu156del | BC domain active site | N/A | P (PVS1, PM2, PM3, PP4) |
| PCCA 10 | c.819+9A>G | no significant effect | unknown | unknown | US | US (PM2, BS3) |
| PCCA 10 | c.717-2A>G | 24 b.p. deletion/ r.717_741del | p.Asp240_Gln247del | BC/B subdomain | LP | P (PVS1, PM2, PP4) |
| PCCA 13 | c.1187T>G (p.Val396Gly) | 23 b.p. deletion/ r.1066_1089del | p.Val396Glyfs*37 | NMD | N/A | P (PS3, PM2, PM3, PP3, PP4) |
| PCCA 14 | c.1284+2dupT | exon skipping/ r.1210_1284del | p.Asp404_Gly428del | BC domain | N/A | P (PS3, PM2, PM3, PP3, PP4) |
| PCCA 15 | c.1353+5_ 1353+9del | exon skipping/ r.1285_1353del | p.Val429_Lys451del | BC domain | P* | P (PS3, PM2, PM3_supportive, PP3, PP4) |
| PCCA 18 | c.1643+1_ 1643+2dup | exon skipping/ r.1541_1643del | p.Gly514Glufs*9 | NMD | US | LP (PS3, PM2, PP3) |
| PCCA 21 | c.1899+2_ 1899+3insCT | exon skipping/ r.1846_1899del | p.Cys616_Val633del | BT domain | N/A | LP (PS3, PM2, PP3, PP4) |
| PCCA 22 | c.2040G>A (p.Ala680=) | exon skipping/ r.1900_2040del | p.Tyr634_Ala680del | BT and BCCP domain | US, P* | P (PS3, PM2, PM3_supportive, PP3, PP4) |
| PCCA 24 | c.2119-9A>G | 8 b.p. insertion/ r.2118_2119ins8 | p.Val707Asnfs*4 | BCCP domain | LP | P (PS3, PM2, PM3, PP3, PP4) |
| PCCB 5 | c.543G>C (p.Leu181=) | exon skipping with significant amount of the full-length mRNA isoform/ r.430_543del | p.Ile144_Leu181del | CT domain active site | US | US (PM2, PP3) |
| PCCB 7 | c.763G>A (p.Gly255Ser) | no significant effect | unknown | unknown | US | LP (PM1, PM2, PM5, PP2, PP3) |
| PCCB 7 | c.655-2A>G | 107 b.p. and 56 b.p. insertions/ r.654_655ins107 and r.654_655ins56 | p.Asp219Profs*13 and p.Asp219Alafs*32 | NMD | N/A | P (PVS1, PM2, PM3, PP4) |
| PCCB 8 | c.882C>T (p.Pro294=) | no significant effect | unknown | unknown | US/LB | US (PM2, BS3) |
| PCCB 11 | c.1091-8_1091-3del | exon skipping and 96 b.p. insertion/ r.1091_1198del and r.1090_1091ins96 | p.Cys365_Gly400del and p.Gly364Glufs*16 | CT domain active site and NMD | P* | LP (PS3, PM2, PP3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bychkov, I.; Galushkin, A.; Filatova, A.; Nekrasov, A.; Kurkina, M.; Baydakova, G.; Ilyushkina, A.; Skoblov, M.; Zakharova, E. Functional Analysis of the PCCA and PCCB Gene Variants Predicted to Affect Splicing. Int. J. Mol. Sci. 2021, 22, 4154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084154
Bychkov I, Galushkin A, Filatova A, Nekrasov A, Kurkina M, Baydakova G, Ilyushkina A, Skoblov M, Zakharova E. Functional Analysis of the PCCA and PCCB Gene Variants Predicted to Affect Splicing. International Journal of Molecular Sciences. 2021; 22(8):4154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084154
Chicago/Turabian StyleBychkov, Igor, Artur Galushkin, Alexandra Filatova, Andrey Nekrasov, Marina Kurkina, Galina Baydakova, Alexandra Ilyushkina, Mikhail Skoblov, and Ekaterina Zakharova. 2021. "Functional Analysis of the PCCA and PCCB Gene Variants Predicted to Affect Splicing" International Journal of Molecular Sciences 22, no. 8: 4154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084154