
Role of Insulin Resistance in MAFLD

1
Department of Diabetes and Metabolic Diseases, Graduate School of Medicine, The University of Tokyo, Tokyo 113-8655, Japan
2
Department of Clinical Nutrition Therapy, The University of Tokyo, Tokyo 113-8655, Japan
3
Clinical Nutrition Program, National Institutes of Biomedical Innovation, Health and Nutrition, Osaka 567-0085, Japan
4
Toranomon Hospital, Tokyo 105-8470, Japan
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(8), 4156; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084156
Submission received: 7 April 2021
/
Accepted: 14 April 2021
/
Published: 16 April 2021
(This article belongs to the Special Issue Metabolic Associated Fatty Liver Disease: A New Definition)
Abstract
:Many studies have reported that metabolic dysfunction is closely involved in the complex mechanism underlying the development of non-alcoholic fatty liver disease (NAFLD), which has prompted a movement to consider renaming NAFLD as metabolic dysfunction-associated fatty liver disease (MAFLD). Metabolic dysfunction in this context encompasses obesity, type 2 diabetes mellitus, hypertension, dyslipidemia, and metabolic syndrome, with insulin resistance as the common underlying pathophysiology. Imbalance between energy intake and expenditure results in insulin resistance in various tissues and alteration of the gut microbiota, resulting in fat accumulation in the liver. The role of genetics has also been revealed in hepatic fat accumulation and fibrosis. In the process of fat accumulation in the liver, intracellular damage as well as hepatic insulin resistance further potentiates inflammation, fibrosis, and carcinogenesis. Increased lipogenic substrate supply from other tissues, hepatic zonation of Irs1, and other factors, including ER stress, play crucial roles in increased hepatic de novo lipogenesis in MAFLD with hepatic insulin resistance. Herein, we provide an overview of the factors contributing to and the role of systemic and local insulin resistance in the development and progression of MAFLD.
1. Introduction
The term non-alcoholic fatty liver disease (NAFLD) was coined by Ludwig in 1980 to describe fatty liver disease of unknown cause in subjects without a history of excessive alcohol intake [1]. NAFLD has been categorized histologically into non-alcoholic fatty liver (NAFL) or non-alcoholic steatohepatitis (NASH); the former is defined as the presence of ≥5% hepatic steatosis without evidence of hepatocellular injury in the form of hepatocyte ballooning, while the latter is defined as the presence of ≥5% hepatic steatosis, associated with inflammation and hepatocyte injury (e.g., hepatocyte ballooning), with or without fibrosis [2]. The global prevalence of NAFLD is estimated at 24%; the highest rates are reported from South America and the Middle East, followed by Asia, the USA, and Europe [3]. Whereas it has been known that the vast majority of patients diagnosed as having isolated fatty liver (NAFL) on initial biopsy are at a low risk for the development of more advanced liver disease [4], a recent prospective cohort study with a mean follow-up time of 19.8 years showed that 16% and 9% of patients with NAFLD developed advanced fibrosis and end-stage liver disease, respectively [5], suggesting that the prognosis of NAFLD cannot be underestimated at all. Although NASH with cirrhosis is thought to predispose to hepatocellular carcinoma (HCC), hepatocarcinogenesis has also come to be increasingly recognized in NASH patients without cirrhosis [6]. In regard to the apparently complex mechanism involved in the progression from NAFLD to NASH/HCC, inflammatory mediators derived from various tissues could play a central role in the cascade of inflammation, fibrosis, and finally tumor development [7], and metabolic dysfunction is considered to be closely associated with these kinds of inflammatory changes.
It has been reported that patients with NAFLD are among those at the greatest risk for the development of type 2 diabetes mellitus (T2DM). A systematic review and meta-analysis showed that NAFLD (diagnosed based on either elevation of the serum levels of liver enzymes or the findings of ultrasonography) is associated with an approximately twofold increased risk of incident T2DM and metabolic syndrome over a median period of 5 years [8]. Another meta-analysis reported that metabolic comorbidities were frequently associated with NAFLD, including obesity (51.34%), T2DM (22.51%), dyslipidemia (69.16%), hypertension (39.34%), and metabolic syndrome (42.54%) [9].
Another study showed that almost all patients with T2DM have NASH, regardless of the liver enzyme levels, suggesting that steatohepatitis may represent the sole feature of liver damage in patients with type 2 diabetes mellitus [10]. Besides metabolic dysfunction, NAFLD may also coexist with other liver diseases. The prevalence of NAFLD is similar between groups of individuals with and without suspected liver disease (elevated serum levels of liver enzymes, hepatitis B surface antigen, or hepatitis C virus RNA positivity) [11]. Whereas the impact of hepatitis B virus (HBV) infection on hepatic steatosis still remains controversial, chronic liver infection with hepatitis C virus (HCV) is known to be directly associated with hepatic steatosis, suggesting that coexistence of chronic HCV hepatitis and metabolic comorbidities could increase the risk of development of NAFLD [12]. Since the common pathophysiological abnormality underlying obesity, T2DM, and metabolic syndrome is insulin resistance, NAFLD is regarded as one of the phenotypes of the pathophysiological conditions associated with insulin resistance.
Certain medications may also adversely affect the risk of progression of NAFLD through exacerbating insulin resistance. While altered morphine metabolism and distribution has been reported in patients with liver disease [13], opioid use is more common in NAFLD patients with cirrhosis [14] as well as other liver diseases [15]. Since opioid peptides have been reported to play roles in some metabolic events associated with the development of obesity [16], use of some drugs such as opioids might be associated with development of NAFLD in patients with other liver diseases, including viral hepatitis and autoimmune diseases.
In addition to reports of the increased incidence of cardiovascular diseases (CVDs) in insulin-resistant patients with pre-diabetes or normal glucose tolerance [17,18], NAFLD itself has also been reported to be strongly associated with an elevated risk of CVD [19]. Moreover, the coexistent NAFLD and T2DM is known to be associated with an increased risk of development of chronic macrovascular and microvascular complications via several mechanisms, such as increased insulin resistance, dyslipidemia, upregulated proinflammatory cytokines, dysbiosis, and alterations of hemostatic–fibrinolytic factors [20]. Based on the above, NAFLD is still considered as being the hepatic component of metabolic syndrome from the perspective of the risk of CVD.
2. Renaming NAFLD to MAFLD
The current definition of NAFLD requires the exclusion of other causes of liver diseases and of a history of excessive alcohol intake. However, the precise definition of excessive alcohol consumption in patients with suspected NAFLD is uncertain. A consensus meeting recommended that for the purposes of eligibility for NASH clinical trials, significant alcohol consumption be defined as >21 standard drinks per week for men and >14 standard drinks per week for women over a 2-year period preceding the baseline liver histology [2]. Moderate alcohol consumption has also been demonstrated to be significantly and independently associated with worsening of hepatic fibrosis, suggesting that even moderate alcohol consumption might be harmful [21]. In the clinical setting, it seems difficult to grasp the exact amount of drinking by patients. Based on the fact that NAFLD could coexist with other liver diseases, including viral hepatitis and autoimmune diseases, the expression of “non-alcoholic” does not appear to accurately reflect the pathogenesis of this heterogeneous liver disease. Recently, experts reached the consensus that “metabolic dysfunction-associated fatty liver disease or MAFLD” may be a more appropriate and inclusive definition [22]. The criterion for the diagnosis of MAFLD would be: evidence of hepatic steatosis (detected by imaging, blood biomarkers, or liver histology), associated with any one or more of the following: overweight/obesity, T2DM, evidence of metabolic dysregulation [23].
3. Factors Underlying the Pathogenesis of MAFLD
Various factors are recognized to be involved in the development and progression of MAFLD. Several gene variants are reported to serve as genetic susceptibility genes for MAFLD. In addition, pathological alterations of the gut microbiota and increased insulin resistance in the adipose tissue and skeletal muscle are known to affect hepatic lipid metabolism, promoting further hepatic fat accumulation and inflammation.
3.1. Genetic Background
Many genome-wide association studies have identified novel loci associated with disease severity phenotypes of NAFLD and NASH [24,25] (Figure 1). While PNPLA3 (patatin-like phospholipase domain-containing protein 3), which hydrolyzes glycerolipids, is known to be expressed in a variety of tissues, particularly high expression levels have been reported in the liver and kidneys [26]. Romeo et al. identified an allele in the PNPLA3 (rs738409; I148M) gene that was strongly associated with increased hepatic fat levels and hepatic inflammation [27]. Several studies have confirmed that the I148M variant predisposes to progression along the full spectrum of liver damage associated with fatty liver, from simple steatosis to steatohepatitis and progressive fibrosis, and could promote fibrogenesis in other liver diseases, including alcoholic liver diseases, viral hepatitis, and autoimmune liver diseases [28]. The PNPLA3 rs738409 C > G polymorphism has also been reported to be associated with an elevated risk of development of HCC [29]. The I148M substitution was reported to cause loss-of-function of PNPLA3 [30]. Using knock-in mice of PNPLA3-I148M, it was demonstrated that this variant disrupts ubiquitylation and proteasomal degradation of PNPLA3, resulting in the accumulation of PNPLA3-I148M and impaired mobilization of TG from lipid droplets [31].
TM6SF2, reported to be expressed mainly in the intestine and liver, seems to be related to VLDL-TG secretion in the liver [26]. The TM6SF2-E167K variant has also been identified as a loss-of-function variant associated with hepatic steatosis. Knockdown of Tm6sf2 in mice increased the liver TG content and decreased VLDL secretion [32], suggesting that TM6SF2 is related to hepatic secretion of TG. While subjects with the TM6SF2-E167K variant are more susceptible to progressive NASH, the incidence of cardiovascular events in these patients is reported to be lower, because of the lower levels of circulating lipids [33].
Variants of the MBOAT7 gene, which has been reported to be the risk locus for alcohol-related cirrhosis [34], have also been shown to be associated with the development and severity of NAFLD [35]. The rs641738 polymorphism of MBOAT7 is associated with increased hepatic fat content, lower MBOAT7 protein expression in the liver, and changes in the plasma phosphatidylinositol species [36]. Mammalian membrane-bound O-acyltransferase 7 (MBOAT7), also known as lysophosphatidylinositol acyltransferase 1, plays a major role in regulating arachidonic acid availability for eicosanoid biosynthesis [37]. It was reported that mice with hepatocyte-specific deletion of MBOAT7 showed spontaneous steatosis with increased hepatic cholesterol ester content, hepatic fibrosis with minimal inflammation after receiving a high-fat, low-methionine, choline-deficient diet, and increased plasma total lysophosphatidylinositol levels, suggesting that Mboat7 deficiency may trigger an inflammation-independent pathway of liver fibrosis [38]. Recently, another group reported that direct metabolic flow from phosphatidylinositol to TG caused by hepatic MBOAT7 depletion resulted in an increase in the total hepatic TG content [39].
Besides the above, variations of the glucokinase regulator (GCKR) gene locus have been reported to be associated with NAFLD, and various studies have demonstrated the roles of other genetic variations in the regulation of lipid metabolism (LYPLAL1, APOB, MTP, LPIN1, UCP2), innate immunity (IL28B, MERTK), insulin signaling (ENPP1, IRS1), oxidative stress (SOD2), and fibrogenesis (KLF6) in relation to the progression of NAFLD [24] (Figure 1).
3.2. Alterations of Lipid Metabolism in the Liver
3.2.1. Increase in De Novo Lipogenesis
Liver triglyceride (TG) is synthesized from fatty acyl CoA. The hepatocellular concentration of fatty acyl CoA is determined from the balance between free fatty acid (FFA) formation (from circulating FFAs, de novo lipogenesis (DNL), lipoprotein uptake, and TG breakdown) and utilization (lipid synthesis and oxidation) [40]. In an experiment conducted with isotopes, a threefold increase of DNL and higher nocturnal plasma levels of FFAs were observed in patients with NAFLD [41]. In NAFLD and obese patients, the TG composition of the liver is reported to be 59% from NEFA, 26% from DNL, and 15% from the diet, suggesting that DNL is elevated in the fasting state in NAFLD and that the NEFA pool is the primary contributor to hepatic TG in NAFLD [42]. There are two major transcriptional factors for DNL: sterol regulatory element binding protein 1c (SREBP1c), regulated by insulin signaling, and carbohydrate response element binding protein (ChREBP), activated by glucose uptake [43]. Among the peroxisome proliferator-activated receptors (PPARs), PPARγ appears to upregulate the expressions of genes involved in lipogenesis (such as Acc1, Cd36, and Scd1), as well as triglyceride synthesis (Mogat1) and lipid droplet formation (Fsp27, Plin2, and Cidea) [44]. Although upregulation of hepatic PPARγ has been observed in mice with hepatic steatosis, previous studies using genetically modified mouse models identified the different roles of PPARγ in hepatocytes and non-liver parenchymal cells, indicating the complicated mechanism of hepatic lipogenesis mediated by PPARγ in the liver [45]. Moreover, PPARγ expression has been reported not to be correlated with the severity of NASH in humans [46]. The precise role of PPARγ in the development of MAFLD is not yet known.
3.2.2. Increased Mitochondrial Fatty Acid Oxidation
In response to lipid overload, oxidative and TCA cycle activities are increased, which potentiates oxidative stress and liver damage during chronic hepatic steatosis [47]. Koliaki et al., proposed that the “hepatic mitochondrial flexibility” could be a compensatory and protective response in the early stages of NAFLD. In their study, the maximal respiration rates of isolated mitochondria were higher in obese persons with or without NAFL than in lean persons in spite of similar mitochondrial contents, which was abolished in obese patients with NASH. Elevated hepatic insulin resistance, mitochondrial uncoupling, and leaking activity might result in augmented hepatic oxidative stress and inflammatory response [48]. Indeed, increased lipid delivery caused by either intralipid infusion or a high-fat diet (HFD) not only induced oxidative metabolism, but also caused a proportional increase in oxidative stress and inflammation [49]. Thus, the state of fatty acid oxidation in NAFLD seems to depend on the progression of the disease spectrum, and relatively impaired mitochondrial oxidative capacity is associated with hepatocyte damage through enhanced oxidative stress and inflammation. As many studies imply an important role of mitochondria in the pathogenesis of metabolic disorders such as obesity, metabolic syndrome, and T2DM [50], mitochondrial dysfunction in the liver is also considered to be a significant contributor to the pathogenesis of MAFLD.
3.2.3. Increased Uptake of FFAs
In regard to FFA uptake by the liver, many studies have shown that CD36, which facilitates intracellular FFA uptake and trafficking, might contribute to the progression to NASH [51]. In a mouse model, hepatocyte-specific CD36 overexpression was associated with increased hepatic TG storage and secretion [52]. In contrast, hepatocyte-specific disruption of CD36 attenuated fatty liver induced by an HFD and in a genetic model with increased circulating FFAs [53]. The expression of CD36 was found to be upregulated in patients with high hepatic fat content [54]. Hepatic CD36 upregulation was also significantly associated with insulin resistance, hyperinsulinemia, and increased steatosis in patients with NASH [55]. CD36 localization on the plasma membranes of hepatocytes was markedly increased in patients with NASH as compared to people with normal livers or simple hepatic steatosis, in which CD36 palmitoylation plays a key role via increased CD36 protein hydrophobicity [56].
3.2.4. Alterations in TG Secretion
As mentioned above in relation to TM6SF2, TG secretion is also involved in NAFLD progression. Obese patients with NAFLD show increased VLDL-TG secretion, although the increase in hepatic TG export is not sufficient to normalize the intrahepatic TG content [57]. In patients with NAFLD in which insulin-induced endogenous glucose production was suppressed, impaired insulin-induced suppression of VLDL-TG secretion was observed [58], suggesting that the increase in VLDL-TG secretion may be an early pathophysiological manifestation of NAFLD. Apolipoprotein B100 (ApoB100) and microsomal TG transfer protein (MTP) are required for the formation of mature VLDL particles. Hepatic steatosis, secondary to compromised TG export, is common in patients with genetic defects in the ApoB or MTP gene [59]. Whereas ApoB100 synthesis is a rate-determining step in hepatocyte lipid export, decreased synthesis of this protein was observed in patients with NASH [60], suggesting that impaired VLDL-TG secretion exacerbates NAFLD. Hepatic induction of MTP in a mouse model of hereditary fatty liver without obesity led to a reduction in hepatic TG accumulation, improvement of VLDL export, amelioration of NASH-like lesions, as well as glucose intolerance [61]. Despite a greater degree of impairment of VLDL synthesis and secretion in cases of NASH than in those of NAFL, the hepatic TG content was found to be similar between NAFL and NASH, suggesting that impairment of VLDL synthesis might result in increased lipid oxidation and DNA oxidative damage, resulting in the progression to NASH [62] (Figure 2).
3.3. Insulin Resistance in Adipose Tissue
During the development of obesity, various changes are observed in the microenvironment of the adipose tissue. While under physiological conditions, adipose tissue expands through effective recruitment of adipogenic precursor cells along with an adequate angiogenic response and appropriate remodeling of the extracellular matrix, pathological adipose tissue expansion consists of massive enlargement of existing adipocytes, limited angiogenesis, and hypoxia, resulting in the activation of HIF-1α, induction of a fibrotic program, and systemic insulin resistance, accompanied by activation of proinflammatory macrophages [63]. As insulin inhibits lipolysis in the adipocytes [64], insulin resistance in the adipose tissue causes increased release of FFAs. Mice with adipose tissue-specific knockout of the insulin receptor exhibited lipodystrophy and severe HFD-induced fatty liver [65]. Humans with congenital generalized lipodystrophy develop extreme insulin resistance and its complications, such as T2DM, hyperlipidemia, and fatty liver [66]. Indeed, in humans, visceral adipose tissue lipolysis accounts for an increasing proportion of hepatic FFA delivery as the visceral fat increases [67]. NAFLD patients have significantly higher serum FFA levels, and serum FFA level has been identified as an independent predictor of advanced fibrosis in NAFLD patients [68]. Circulating FFA increases activation of the proinflammatory nuclear factor-kappa B (NF-kappa B) pathway in the liver [69]. Thus, the release of fatty acids from dysfunctional and insulin-resistant adipocytes results in lipotoxicity, leading to the accumulation of triglyceride-derived toxic metabolites in ectopic tissues, including the liver [70].
Activation of proinflammatory macrophages during pathological adipose tissue expansion leads to upregulation of the proinflammatory cytokine tumor necrosis factor-α (TNF-α), which not only promotes adipocyte lipolysis [71] but also inhibits insulin-stimulated tyrosine phosphorylation of insulin receptor substrate1 (Irs1) by enhancing phosphorylation of Ser307 on Irs1 [72]. These effects were mediated by the activated IKKβ-NF-κB pathway [73]. TNF-α is also known to play an important role in the development of systemic insulin resistance [74], and overexpression of TNF-α mRNA has been reported in the liver and adipose tissue of NASH patients [75], suggesting that upregulated TNF-α in the adipose tissue might be involved in the progression of NAFLD via increasing systemic insulin resistance, as well as promoting inflammation in various tissues. Enhanced secretion of interleukin-6 (IL-6) from the visceral adipose tissue has been observed in obese individuals [76]. JNK1-dependent secretion of IL-6 in the adipose tissue caused increased expression of hepatic SOCS3, which induced degradation of molecules involved in the insulin signaling, resulting in hepatic insulin resistance [77]. Serum IL-6 levels are reported to be significantly increased in NAFLD and NASH patients as compared to healthy individuals [78]. Although IL-6 exerts both pro- and anti-inflammatory activities depending on the context [79], chronic low-grade inflammation mediated by IL-6 in the adipose tissue could be related to the progression of NAFLD.
Besides the adipocytes and the stromal vascular fraction secreting various proinflammatory cytokines, adipocytes also secrete more specialized adipokines, such as leptin and adiponectin [80]. Leptin is known to be associated with food intake, energy homeostasis, metabolism, cognition, immune functions, and bone metabolism [81]. Although circulating leptin levels are directly proportional to the amount of body fat, leptin resistance is also seen in obese people with decreased leptin-induced suppression of appetite and weight gain [81]. Circulating leptin levels have been reported to be higher in patients with NAFLD than in controls, and higher levels of circulating leptin have been shown with increasing severity of NAFLD [82]. In addition to leptin promoting profibrogenic responses via TGF-β signaling [83,84], obesity-induced leptin exacerbates NASH via enhancing the responsiveness to endotoxins [85], suggesting that leptin plays a key role in proinflammatory, profibrogenic processes involved in the progression of NAFLD. On the other hand, adiponectin has the effect of ameliorating NAFLD. Adiponectin has two major receptors, AdipR1 and AdipoR2. In the liver, AdipoR1 is involved in the activation of AMP-activated kinase (AMPK), and AdipoR2 is involved in the activation of PPARα, both of which lead to increased insulin sensitivity [86]. As obesity is associated with reduced adiponectin levels via multiple mechanisms, including decreased levels of SirT1, hyperinsulinemia, oxidative stress, and inflammation [87], decreased levels of circulating adiponectin in NAFLD are related to a decrease in hepatic insulin sensitivity and increase in the amount of hepatic fat content [88]. Administration of recombinant adiponectin to either mice fed high-fat ethanol-containing food or non-alcoholic obese ob/ob mice alleviated steatosis and attenuated inflammation [89]. Recently, it was reported that treatment with a dual AdipoR1/AdipoR2 agonist improved both NASH and fibrosis in mice models via reduced hepatic stellate cell activation [90] (Figure 2). While several observational studies have reported elevated serum levels of other obesity-related adipokines, such as resistin, visfatin, and RBP4 in NAFLD patients, some contradictory results have also been reported, suggesting that the causal relationship between these adipokines and NAFLD remains under debate [91,92].
3.4. Insulin Resistance in the Skeletal Muscle
Skeletal muscle plays a major role in the regulation of blood glucose via insulin-stimulated glucose transport into the skeletal muscle mediated by GLUT4 [93]. The glucose taken up is used for glycogen synthesis and glycolysis. Many studies have indicated that skeletal muscle insulin resistance is the primary defect in T2DM [94]. Excess fatty acid supply promotes skeletal muscle insulin resistance together with an increase in intramyocellular lipids, leading to impaired insulin signaling induced by lipid-derived metabolites, such as diacylglycerols (DAGs) [70]. The muscle-specific insulin receptor-knockout (MIRKO) mice were normoglycemic, but exhibited elevated fat mass, and elevated serum levels of TG and FFA [95]. In MIRKO mice, insulin-stimulated muscle glucose transport and glycogen synthesis were decreased, whereas insulin-stimulated glucose transport in the adipose tissue was increased, suggesting that selective inactivation of muscle insulin signaling resulted in redistribution of substrates from the muscle to adipose tissue [96]. Glucose not utilized in the skeletal muscle could also serve as a substrate for hepatic DNL. In humans, insulin-resistant subjects exhibited reduced muscle glycogen synthesis and increased hepatic DNL [97]. A comparison of healthy, normal-weight, sedentary elderly subjects with younger subjects revealed that hepatic DNL occurred at a more than twofold higher level in the elderly than in the younger subjects, and that the postprandial hepatic TG content was approximately threefold higher in the former than in the latter group [98]. Conversely, improved muscle insulin responsiveness with exercise resulted in an increase in postprandial net muscle glycogen synthesis, leading to reduced hepatic TG synthesis, accompanied by a decrease in hepatic DNL [99]. Using the specific index of skeletal muscle insulin sensitivity (Matsuda index), which is calculated from the plasma glucose and insulin concentrations in the fasting state and during the OGTT [100], it was found that liver steatosis was associated with insulin resistance in the skeletal muscle rather than in the liver in patients with NAFLD [101]. In relation to skeletal muscle insulin resistance, sarcopenia, defined as reduced muscle quantity or quality, is a progressive and generalized skeletal muscle disorder that is associated with increased likelihood of certain adverse outcomes, such as falls, fractures, physical disability, and mortality [102]. In a prospective observational cohort study, individuals with a low muscle mass were found to be at an increased risk of development of NAFLD, even after adjusting for confounding factors, including insulin resistance and inflammation [103]. In a cohort of biopsy-proven NAFLD, sarcopenia was significantly associated with NASH and significant fibrosis, independent of obesity, inflammation, and insulin resistance [104]. As there seems to be an overlap in the pathophysiology of NAFLD and sarcopenia, it remains unclear if sarcopenia is the cause or consequence of progression of NAFLD [105] (Figure 2).
3.5. Gut Microbiota
Changes in the gut microbiota have been shown to be associated with metabolic syndrome, characterized by chronic low-grade inflammation, obesity, hyperglycemia, and dyslipidemia, as well as with intestinal diseases and neurological diseases [106]. Many studies have provided evidence to suggest that changes in the gut microbiota contribute to the pathogenesis of NAFLD via various mechanisms (Figure 2).
3.5.1. Dysbiosis
Dysbiosis is defined as an imbalance in the microbiota composition. Since the microbiota respond rapidly to large changes in the diet, diet is thought to be as an important modulator of the gut microbiota [107]. There are several reports of associations between the severity of NAFLD and qualitative changes in the intestinal bacterial flora [108,109,110,111]. Although findings in relation to specific changes in the microbiota associated with the progression of NAFLD are not consistent, due to factors such as differences in the diets and ethnicities of the study populations, dysbiosis of the gut microbiota is considered to be involved in the development of NAFLD.
3.5.2. Increased Gut Permeability
Dysbiosis can disrupt the tight junctions in the intestinal endothelial cells, leading to increased mucosal permeability and translocation of bacterial LPS from Gram-negative bacteria [112]. Actually, patients with NAFLD had significantly increased gut permeability, which was correlated with the severity of steatosis, but not with the presence of NASH [113]. Thus, dysbiosis is frequently associated with increased production of endotoxins from bacteria. In addition to the increase in the proportion of an LPS-containing microbiota in the gut following HFD feeding, mice with endotoxemia induced by continuous subcutaneous infusion of LPS showed exacerbation of glucose intolerance, accompanied by increased adipose tissue inflammation and hepatic TG content [114]. An increase in the plasma endotoxin activity was also observed after a Western-style diet for 1 month in healthy human subjects [115]. The endotoxin levels were significantly increased in NAFLD patients, with marked elevation noted in the early stages of fibrosis [116]. Activation of Toll-like receptors (TLRs) by bacterial LPS, especially TLR-4, in the Kupffer cells leads to a proinflammatory response, resulting in hepatic fibrogenesis [117]. As described above, increased leptin in people with obesity enhanced responsiveness to endotoxin, and activated STAT3 signaling through CD14, which is co-receptor of TLR4 [85].
3.5.3. Small Intestinal Bacterial Overgrowth (SIBO)
In previous experiments using animal models, small intestinal bacterial overgrowth (SIBO), characterized by excessive bacteria in the small intestine, was shown to induce hepatic injury [118]. The prevalence of SIBO is reported to be higher in morbidly obese patients than in healthy subjects [119]. A recent meta-analysis confirmed a significant association between NAFLD and SIBO [120]. SIBO has been reported to be associated with systemic endotoxemia in patients with cirrhosis [121], and NAFLD patients with SIBO have also been demonstrated to exhibit significantly higher serum endotoxin levels [122]; thus, SIBO may affect the pathogenesis of NAFLD through causing endotoxemia.
3.5.4. Gut-Derived Metabolites
The increased abundance of alcohol-producing bacteria in NASH microbiomes has been reported to promote liver inflammation [123]. Indeed, higher breath ethanol concentrations have been reported in obese individuals as compared to leaner individuals, among both mice [124] and human [125], independent of ethanol ingestion. Since ethanol is known to be associated with the increased gut permeability and endotoxemia in alcohol-induced liver disease [126,127], it is speculated that endogenous ethanol production by some kinds of gut microbiota might contribute to the pathogenesis of NAFLD. In addition, increased choline metabolism by the gut microbiota, altered bile acid metabolism, and short chain fatty acids (SCFAs) (acetate, propionate, and butyrate) formed during bacterial fermentation might also affect the pathogenesis of NAFLD, although the precise underlying mechanisms are not yet fully understood [128].
4. Role of Insulin Signaling in Hepatic De Novo Lipogenesis
After insulin binds to the insulin receptor, autophosphorylation of tyrosine residues in the intracellular subunit of each of the receptors causes docking and phosphorylation of the insulin receptor substrates, followed by activation of downstream kinase cascades, such as the phosphatidylinositol-3 kinase (PI3K)/Akt pathway [129]. Activated AKT phosphorylates FoxO1, which subsequently reduces the transcriptional activity of various genes, including those encoding G6Pase and PEPCK, that are involved in gluconeogenesis. While activated Akt suppresses hepatic gluconeogenesis, it mediates hepatic lipid metabolism via activation of SREBP1c. Sterol-regulatory element binding protein-1c (SREBP1c) is regulated at both the transcriptional and proteolytic processing levels by mTORC1, one of the downstream molecules in the Akt signaling pathway [130] (Figure 3). SREBP cleavage-activating protein (SCAP) is a polytopic membrane protein that forms a complex with SREBPs in the ER and transports them to the Golgi apparatus for cleavage. After the NH2-terminal domain of SREBP reaches the nucleus and acts as a transcription factor, activated SREBP1c upregulates various lipogenic genes, including genes encoding acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), ATP citrate lyase, and genes encoding the enzymes of the fatty acid elongase complex, leading to activation of the fatty acid biosynthetic pathway [131]. Nuclear SREBP1c protein levels were significantly elevated in the livers of ob/ob and aP2-SREBP1c mice [132].
SCAP-deficient mice show a reduction in the basal rates of cholesterol and fatty acid synthesis in the liver [133]. Liver-specific deletion of SCAP in ob/ob mice also prevented fatty liver despite persistent obesity, hyperinsulinemia, and hyperglycemia [134], suggesting that SREBP1c plays a key role on hepatic DNL. A number of studies have attempted to identify the lipogenic roles of molecules involved in hepatic insulin signaling using genetically modified mouse models of insulin signaling (Table 1).
4.1. Insulin Receptor
To directly evaluate the metabolic effect of the insulin signaling in the liver, liver-specific insulin receptor knockout (LIRKO) mice were generated [135]. These mice exhibited marked insulin resistance, severe glucose intolerance, impaired glycogen storage, and a failure of insulin-induced suppression of hepatic glucose production. The metabolic phenotype became less severe with age-dependent progression of liver dysfunction. Impaired glucose tolerance, impaired suppression of hepatic glucose production, and failure of induction of lipogenic genes were reproduced in inducible insulin receptor-knockout mice [136]. In addition, LIRKO mice also showed high circulating levels of the leptin receptor, with normal leptin sensitivity, suggesting that insulin signaling in the liver also plays an important role in the control of leptin receptor expression and shedding [137]. Another group reported that LIRKO mice showed a decrease in the plasma levels of insulin-like growth factor 1, delayed growth during the first 2 months of life, and derangements in fatty acid metabolism, with decreased expression levels of the lipogenic enzymes, hepatic DAG, and plasma TG [138]. In regard to the lipoprotein profile of the LIRKO mice, these mice exhibited reduced plasma levels of HDL cholesterol and increased plasma levels of cholesterol-enriched ApoB containing lipoprotein particles, coupled with decreased TG secretion, resulting in an elevated risk of atherosclerosis [139].
While insulin-independent induction of SREBP1c through mTORC1 can occur after feeding, induction of lipogenic gene expressions in ob/ob mice or HFD-fed mice requires insulin signaling. Indeed, LIRKO mice, as well as inducible LIRKO mice generated with antisense oligonucleotides, showed reduction in the nuclear SREBP1 levels, lipogenic gene expressions, and hepatic TG content [140]. Although additional deletion of FoxO1 in the LIRKO mice rescued glucose tolerance and allowed for normal suppression of gluconeogenic gene expression after feeding or systemic administration of insulin, induction of lipogenic gene expressions by feeding was still suppressed [136,138], suggesting that hepatic insulin signaling other than via the IR-Akt-FoxO1 pathway is required for SREBP1c induction and DNL in vivo.
4.2. Insulin Receptor Substrate
Stimulation of insulin receptors with increased tyrosine kinase activity leads to autophosphorylation and phosphorylation of receptor substrates (Irs). Like the LIRKO mice, liver-specific Irs1/Irs2 double-knockout (LIrs1/2DKO) mice also exhibited impaired glucose tolerance with hyperinsulinemia and impaired suppression of hepatic glucose production [141,142]. Liver-specific Irs1-knockout mice (LIrs1KO) failed to exhibit insulin resistance during fasting, but showed insulin resistance after refeeding; conversely, liver-specific Irs2-knockout (LIrs2KO) mice exhibited insulin resistance during fasting, but not after refeeding. In relation to the suppression of SERBP1c by insulin signaling, the expression levels of SREBP1c in LIrs1KO mice were significantly lower only after refeeding, whereas in the LIrs2KO mice, by contrast, the expression levels of SREBP1c were significantly reduced only in the fasting state [141]. While all of the LIrs1KO, LIrs2KO, and LIrs1/2DKO mice exhibited insulin resistance and glucose intolerance under an HFD diet, only the LIrs2KO mice, and not the LIrs1KO or LIrs1/2DKO mice, showed hepatic steatosis [143].
4.3. PI3K
Class I phosphatidylinositol 3-kinase (PI3K), consisting of the p85 regulatory subunit and p110 catalytic subunit, with eight identified isoforms of the regulatory subunit, is encoded by three genes that undergo alternative splicing [144]. Phosphorylated Irs serves as a docking protein for the PI3K p85 subunit, and the docking leads to activation of the p110 catalytic subunit. To evaluate the role of the regulatory subunit of PI3K in the liver, mice lacking any isoform of the PI3K regulatory subunit in the liver (L-p85DKO mice) were generated by crossing the hepatocyte-specific Pik3r1 gene (producing p85α, p55α, and p50α) knockout mice with Pik3r2-knockout mice lacking the other major p85 isoform, p85β, in all the tissues [145]. L-p85DKO mice failed to show activation of PI3K and Akt by insulin stimulation, resulting in increased expressions of the gluconeogenic genes, impaired glucose tolerance, and hyperinsulinemia. In addition, defective activation of PKCλ by insulin was associated with hypolipidemia and decreased transcription of SREBP1c. In these mice, suppressed activity of PKCλ, but not of Akt, might be associated with low levels of hepatic SREBP1c expression. Similar phenotypes, including hyperinsulinemia, glucose intolerance, upregulated expressions of gluconeogenic genes, and decreased lipogenesis were observed when the hepatic PI3K activity was inhibited with an adenoviral vector encoding a dominant-negative mutant of p85 [146]. Mice with hepatic knockout of p110α also showed reduced insulin sensitivity, impaired glucose tolerance, increased gluconeogenesis, and decreased expression levels of several lipogenic genes in the fed state [147]. While the loss of p110β had no significant effect on liver lipid accumulation, hepatic TG levels in HFD-fed liver-specific p110α-knockout mice were lower than in the HFD-fed control mice [148]. Conversely, enhanced PI3K activity resulted in hypoglycemia and increased hepatic TG levels when miR-378, which can inhibit hepatic p110α expression via epigenetic modifications, was knocked out in the liver [149].
4.4. PDK1
PI3K phosphorylates and converts phosphatidylinositol-3,4-diphosphate (PIP2) into PIP3, which recruits the serine/threonine kinase phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane through its PH domain, and phosphorylates Thr308 on Akt [144]. Liver-specific PDK1-knockout (LPDK1KO) mice showed impaired Akt activation by insulin, lower hepatic glycogen contents, impaired suppression of gluconeogenesis, and impaired induction of lipogenic genes following refeeding [150]. It was thought that the reason for the death of these mice between 4 and 16 weeks of age due to liver failure was the use of the AFP promoter-driven Cre-recombinase. When a promoter of the albumin gene was used, LPDK1KO mice exhibited marked hyperinsulinemia, postprandial hyperglycemia, and lower expression levels of SREBP1c in the randomly fed condition, without liver damage or shortening of the life span [151].
4.5. Akt
Although Akt1, one of the three Akt isoforms, is important for cell survival and growth, Akt2 appears to play a more prominent role in the liver [144]. Reduced TG contents of the liver, a decrease in the expressions of lipogenic genes, and DNL were observed in both ob/ob mice and HFD-induced obese mice with loss of hepatic Akt2, although these changes were not observed under the condition of a normal chow diet [152]. Mice with hepatic deletion of Akt1 and Akt2 showed glucose intolerance, insulin resistance, and defects in the transcriptional response to feeding in the liver, including lipogenic gene expressions, all of which were normalized with concomitant liver-specific deletion of FoxO1 [153].
4.6. PTEN
Since PTEN dephosphorylates PIP3, inactivation of PTEN results in enhanced activation of the PI3K pathway. Hepatocyte-specific PTEN-knockout (LPTENKO) mice exhibited massive hepatomegaly and steatohepatitis with TG accumulation, accompanied by elevated lipogenesis gene expression levels and β-oxidation [154,155,156]. Whereas enhanced hepatocarcinogenesis was also observed, the LPTENKO mice showed not only improved glucose tolerance, but also improved systemic insulin sensitivity, caused by redistribution of the body fat from the fatty tissue to the liver, and/or increased hepatic FGF21 production. Based on a series of these results in genetically modified mouse models of insulin signaling (Table 1), hepatic insulin signaling via at least the IR/Irs/PI3K/Akt pathway is definitely important for the development of fatty liver.
5. Pathogenesis of Hepatic Insulin Resistance
While insulin signaling is known to be positively correlated with hepatic fat accumulation, hepatic insulin signaling is thought to be disturbed in NAFLD. As in the adipose tissue and skeletal muscle, proinflammatory cytokines, endoplasmic reticulum (ER) stress, and reactive oxygen species (ROS) cause JNK overactivation in hepatocytes, leading to hepatic insulin resistance, in part through inhibitory serine phosphorylation of the insulin signaling molecules Irs1 and Irs2 [157]. After three days of high-fat feeding with hepatic fat accumulation and hepatic insulin resistance in the absence of significant peripheral fat accumulation or peripheral insulin resistance, hepatic insulin resistance could be attributed to impaired insulin-stimulated Irs1 and Irs2 tyrosine phosphorylation, associated with activation of PKC-epsilon (PKCε) and JNK1 [158].
It is well known that Irs2 expression is suppressed by insulin signaling itself at the transcriptional level through the insulin response element via inactivated FoxO or activated SREBP proteins [159,160,161,162,163], suggesting that hyperinsulinemia, which is common in obesity, metabolic syndrome, T2DM, and NAFLD, could cause hepatic insulin resistance by downregulation of Irs2. In addition to this regulatory circuit, epigenetic regulation of Irs2 has also been reported recently. According to one study, decreased Irs2 transcription in obese patients with T2DM was associated with elevated miRNA let-7e-5p levels and altered Irs2 DNA methylation at the SP1- and SREBF1-binding sites [164]. Since hepatic Irs2 mRNA levels have been reported to be actually lower in NAFLD patients [165], downregulated Irs2 might be involved in the pathogenesis of hepatic insulin resistance.
In addition, a large number of studies have indicated that accumulation of DAG in the liver plays a crucial role in mediating hepatic insulin resistance [166]. In humans with obesity, it was found that intrahepatic TG, rather than the visceral adipose tissue area, was a better marker of the metabolic derangements associated with obesity [167]. The DAG content in cytoplasmic lipid droplets of the hepatocytes was found to be the best predictor of insulin resistance [168]. DAG is a lipid intermediate in TG synthesis. In the physiological state, an increase in TG is accompanied by an increase in DAG. DAG-mediated activation of PKCε induces inhibition of the insulin receptor (INSR) kinase activity due to INSR Thr1160 phosphorylation [169]. Subcellular distributions of DAG and PKCε seem important for INSR Thr1160 phosphorylation, based on the findings of studies using comparative gene identification 58 (CGI-58), which is a lipid droplet-associated protein that promotes the hydrolysis of TG by activating adipose triglyceride lipase (ATGL). Whereas mice with knockdown of CGI-58 showed hepatic steatosis as well as increased DAG content, they remained insulin sensitive and were protected from diet-induced obesity and glucose intolerance [170,171]. Knockdown of CGI-58 caused increased DAG content in hepatocyte lipid droplets and suppressed PKCε translocation to the plasma membrane, suggesting that intracellular compartmentalization of DAG is key to the interaction of DAG and PKCε [171]. Among the three known DAG stereoisomers, increased content of sn-1,2-DAG in the plasma membrane has been observed in both humans with hepatic insulin resistance and rats with hepatic insulin resistance induced by acute hepatic DGAT2 knockdown, which results in enhanced phosphorylation of INSR Thr1160 [172]. (Figure 3)
6. How Is Hepatic Lipogenesis Increased Even in the Presence of Hepatic Insulin Resistance?
While impaired suppression of hepatic gluconeogenesis is caused by hepatic insulin resistance, increased hepatic DNL is also observed in NAFLD. As hepatic insulin signaling positively regulates hepatic DNL, the condition in NAFLD wherein insulin fails to suppress gluconeogenesis but continues to activate DNL is referred to as “selective hepatic insulin resistance” [173]. A number of studies have attempted to identify the branch point in hepatic insulin signaling at which its effects on glucose and lipid metabolism diverge, and also other factors that induce hepatic lipogenesis, such as increased lipogenic substrate supply from other tissues, which are independent of hepatic insulin signaling (Figure 3).
6.1. Point of Divergence of Glucose and Lipid Metabolism in the Liver
As described above, SREBP1c is regulated at both the transcriptional level and at the level of proteolytic processing by mTORC1 [130]. Whereas inhibitors of PI3K and Akt blocked insulin-mediated increase in SREBP1c mRNA expression and suppression of gluconeogenic genes, rapamycin, an inhibitor of the mTORC1, blocked insulin-mediated induction of SREBP1c, while having no effect on insulin-mediated suppression of the gluconeogenic genes [174]. As the mTORC1 pathway senses nutrients such as amino acids through Rag GTPase, leading to the translocation of mTORC1 to the lysosomes [175], mTORC1 is considered as being one of the causes of selective hepatic insulin resistance. However, based on the findings of recent studies, activation of mTORC1 alone is not sufficient to induce DNL and development of steatosis. Stimulation of mTORC1 and SREBP1c is not sufficient to drive postprandial lipogenesis in the absence of Akt2 [176]. Akt activates mTORC1 through phosphorylation and inhibition of the TSC2 protein within the TSC1–TSC2 complex. Liver-specific deletion of TSC1 (LTsc1KO), which results in insulin-independent activation of mTORC1, protected against age- and diet-induced hepatic steatosis and led to hepatocyte-intrinsic defects in SREBP1c activation and DNL [177,178]. These phenotypes were thought to be due to decreased Akt activation through a negative feedback loop in LTsc1KO, especially impaired Akt-mediated suppression of Insig2a, a liver-specific transcript encoding the SREBP1c inhibitor INSIG2 [178]. From the results of experiments using mice lacking hepatocyte-specific Akt1, Akt2, FoxO1, and TSC1, the coincident inhibition of FoxO1 and activation of mTORC1 by insulin was reported to be both necessary and sufficient for activation of DNL [179].
6.2. Substrate-Driven Increase in Hepatic DNL
Since hepatic insulin resistance caused by the DAG–PKCε pathway occurs at the level of phosphorylation of the insulin receptor, it is reasonable to assume that insulin-independent DNL occurs in cases of MAFLD with nutrient oversupply. Carbohydrate-response element binding protein (ChREBP) is a transcription factor that is known to mediate the glucose responsive lipogenic pathway. ChREBP expression in the liver has been shown to be elevated in both patients with obesity [180] and NASH [181]. Transient overexpression of ChREBP in the liver results in hepatic steatosis without insulin resistance through induction of the lipogenic enzyme Scd1 [181]. Transient liver-specific inhibition of ChREBP in ob/ob mice markedly improved hepatic steatosis by decreasing DNL through suppression of the lipogenic enzymes ACC and FAS [182]. Similarly, hepatocyte-specific ChREBP-knockout mice were protected against carbohydrate-induced hepatic steatosis in spite of showing impaired glucose tolerance [183]. Thus, ChREBP seems to be a key factor for understanding the pathophysiological condition of increased DNL existing concomitantly with hepatic insulin resistance. Insulin-independent regulation of fatty acid esterification has also been reported. Using LC–MS/MS, it was found that the rate of fatty acid esterification into hepatic TG was dependent on the plasma fatty acid infusion rates, but independent of changes in the plasma insulin concentrations and hepatic insulin signaling [184]. Recently, Ter Horst et al. reported the results of clinical research in obese patients with and without NAFLD [185]. Obese patients with NAFLD not only showed impaired insulin-mediated suppression of glucose production and increased DNL after overnight fasting, but also attenuated glucose-stimulated/high-insulin lipogenesis. Fructose ingestion is also known to robustly stimulate hepatic DNL, and fructose-stimulated/low-insulin lipogenesis was thought to be intact in NFALD. Hepatic insulin signaling in liver biopsy samples collected during bariatric surgery was found to be attenuated at the level of insulin receptor phosphorylation after administration of glucose. Moreover, the expression levels of ChREBPβ (active isoform), but not ChREBPα, were found to be increased in patients with NAFLD, both after administration of glucose and in the fasting state. These data suggest that an increase in substrate-driven hepatic DNL via ChREBP may be one of the plausible mechanisms to explain increased hepatic DNL in MAFLD with hepatic insulin resistance.
6.3. Hepatic Zonation of Irs1
As increased hepatic DNL in obese NAFLD patients has recently been reported to be inversely correlated with hepatic and whole-body insulin sensitivity and directly correlated with the 24 h plasma glucose and insulin concentrations [186], it is still possible that enhanced hepatic insulin signaling resulting from hyperinsulinemia in cases of NAFLD directly promotes hepatic DNL. Among mouse models of diet-induced obesity (DIO) with hepatocyte-specific knockout of various genes related to insulin signaling (Table 1), LIrs2KO mice, but not LIrs1KO and LIrs1/2DKO mice, were not protected from hepatic steatosis in spite of the partial impairment of hepatic insulin signaling. We previously identified that hepatic zonation of Irs1 plays a crucial role in selective insulin resistance [143]. It is known that the hepatic periportal (PP) zone is the primary site of gluconeogenesis, whereas the hepatic perivenous (PV) zone is the primary site of lipogenesis. Among the molecules of the insulin signaling pathways, such as IR, Irs1, Irs2, Akt1, and Akt2, only the expression of Irs1 is twofold higher in the PV zone than in the PP zone. Under an HFD condition, the phosphorylation levels of Akt are decreased in the PP zone due to suppressed expression of Irs2 at the transcriptional level by the hyperinsulinemia associated with DIO. On the other hand, the phosphorylation level of Akt is increased in the PV zone, because of the intact expression levels of Irs1 in the absence of suppression by hyperinsulinemia. While insulin signaling in the PP and PV zones is impaired in both LIrs1KO and LIrs1/2DKO mice, it is impaired only in the PP zone in LIrs2KO mice. Due to the abundant expression of Irs1 in the PV zone, insulin signaling is considered to be rather enhanced in the presence of hyperinsulinemia despite the downregulation of Irs2, resulting in increased lipogenesis and development of steatosis. Wnt/β-catenin signaling is assumed to be involved in the hepatic zonation of Irs1. While the stabilized and active form of β-catenin is localized in the hepatocytes of the PV zone, a negative regulator of β-catenin, the adenomatous polyposis coli (APC) protein, is expressed in a complementary manner to inactivate β-catenin in the hepatocytes of the PP zone. Indeed, the β-catenin/TCF4 complex directly binds to the Irs1 promoter. Moreover, expression of Irs1 was upregulated in mouse primary hepatocytes stimulated with recombinant Wnt3a, which can activate canonical Wnt/β-catenin signaling [187]. In immunohistochemical staining of liver biopsies from NAFLD patients, the staining intensities of β-catenin and Irs1 seemed to be positively correlated [188]. While downregulation of Irs2 accompanied by decreased gluconeogenic gene expressions was observed in both patients with simple steatosis and NASH, expression of FAS, a lipogenic gene, was not decreased and was strongly correlated with Irs1 expression in NAFLD, suggesting that selective insulin resistance via downregulation of Irs2 is present in the early stages of NAFLD in humans [165]. All these data lend support to the notion that differential hepatic distribution of Irs1/Irs2 could be one of the mechanisms underlying the selective insulin resistance observed in MAFLD.
6.4. ER Stress
Enhanced inflammation, ROS, and ER stress observed in NAFLD directly or indirectly lead to subsequent steatosis and disease progression [7]. ER stress is caused as a response to the accumulation of unfolded proteins in the endoplasmic reticulum (ER). The unfolded protein response (UPR) gene is activated to varying degrees in the liver of patients with NAFLD [189]. ER stress has been reported to be related to the progression of NAFLD through causing dysregulated apoptosis, increased generation of ROS, and exacerbation of hepatic insulin resistance caused by IRE1α-dependent JNK activation [190]. Recently, Sasako et al. demonstrated that “ER stress response failure”, as well as enhanced ER stress, was associated with progression of insulin resistance and steatohepatitis [191]. Although the relationship between ER stress and hepatic DNL is not yet fully understood, Kim et al. identified the mechanism of ER stress-driven DNL [192]. Elevated caspase-2 expression was observed in ER-stressed mice and human with NASH, and caspase-2 ablation abolished SREBP1/2 activation in the liver of ER-stressed mice. In addition, pharmacological inhibition of caspase-2 also prevented hepatic steatosis, fibrosis, and inflammation. Activation of SREBP1/2 by increased caspase-2 activation was considered to be caused by increased cleavage of site 1 protease (S1P), leading to SCAP-independent SREBP1/2 activation in the ER. Thus, ER stress might contribute to the development of increased hepatic DNL in hepatic insulin resistance, at least in the advanced stages of NAFLD/NASH.
7. Conclusions
With a vast majority of studies reporting the etiological heterogeneity and pathogenesis of NAFLD, it has become clear that metabolic dysfunction is closely involved in the complex mechanism underlying the development of NAFLD, prompting a call to consider switching the term NAFLD to MAFLD. The metabolic dysfunction mentioned above consists of obesity, T2DM, hypertension, dyslipidemia, and metabolic syndrome, with insulin resistance as the common underlying pathophysiology. Imbalance between energy intake and expenditure leads to insulin resistance in various tissues and alterations of the gut microbiota, resulting in fat accumulation in the liver. Recent studies have also revealed the roles of some genetic factors involved in the development and progression of MAFLD. During the process of fat accumulation, intracellular damage, as well as hepatic insulin resistance, further potentiates inflammation, fibrosis, and carcinogenesis. Hepatic insulin resistance is aggravated by inhibition of physiological phosphorylation of the insulin receptor via the DAG–PKCε pathway and downregulation of hepatic Irs2 expression via hyperinsulinemia. As reasons for the impaired suppression of hepatic gluconeogenesis with increased hepatic DNL in the presence of hepatic insulin resistance, increased lipogenic substrate supply from other tissues, hepatic zonation of Irs1, and some other factors, including ER stress, may be important (Figure 3). Since MAFLD can be regarded as a hepatic phenotype of systemic insulin resistance, renaming NAFLD as MAFLD may enable more precise recognition and comprehension of fatty liver disease, with beneficial effects in basic research, clinical practice, and public health.
Author Contributions
Y.S. drafted the manuscript. N.K., T.Y., T.K. contributed to data discussion and confirmed the correction. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| CVD | Cardiovascular disease |
| CIDEA | Cell death-inducing DNA fragmentation factor alpha-like effector A |
| ChREBP | Carbohydrate-response element binding protein |
| DAG | Diacylglycerol |
| DIO | Diet-induced obesity |
| DNL | De novo lipogenesis |
| ER | Endoplasmic reticulum |
| FAS | Fatty acid synthase |
| FFA | Free fatty acid |
| FoxO1 | Forkhead box O1 |
| FSP27 | Fat specific protein 27 |
| GLUT | Glucose transporter |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HCC | Hepatocellular carcinoma |
| HFD | High-fat diet |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase subunit β |
| IL-6 | Interleukin-6 |
| INSR | Insulin receptor |
| Irs | Insulin receptor substrate |
| JNK | c-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| NAFL | Non-alcoholic fatty liver |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NEFA | Non-esterified fatty acid |
| NF-kappa | Nuclear factor-kappa B |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| miRNA | microRNA |
| MOGAT1 | Monoacylglycerol O-acyltransferase 1 |
| OGTT | Oral glucose tolerance test |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PI3K | Phosphatidylinositol-3 kinase |
| PKC | Protein kinase C |
| PLIN2 | Perilipin 2 |
| PPAR | Peroxisome proliferator-activated receptors |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SCD1 | Stearoyl-CoA desaturase-1 |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SREBP | Sterol-regulatory element binding protein |
| T2DM | Type 2 diabetes mellitus |
| TCA cycle | Tricarboxylic acid cycle |
| TG | Triglycerides |
| TGF-β | Transforming growth factor-β |
| TLR-4 | Toll-like receptor-4 |
| TNF-α | Tumor necrosis factor-α |
| TSC | Tuberous sclerosis complex |
References
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.M.; Williams, C.D.; Harrison, S.A. Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2012, 10, 837–858. [Google Scholar] [CrossRef]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Yeoh, S.W.; Patrick, D.; Ket, S.; Marion, K.; Gow, P.; Angus, P.W. Characteristics of hepatocellular carcinoma in cirrhotic and non-cirrhotic non-alcoholic fatty liver disease. World J. Gastroenterol. 2015, 21, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef]
- Haga, Y.; Kanda, T.; Sasaki, R.; Nakamura, M.; Nakamoto, S.; Yokosuka, O. Nonalcoholic fatty liver disease and hepatic cirrhosis: Comparison with viral hepatitis-associated steatosis. World J. Gastroenterol. 2015, 21, 12989–12995. [Google Scholar] [CrossRef] [PubMed]
- Dzierlenga, A.L.; Clarke, J.D.; Hargraves, T.L.; Ainslie, G.R.; Vanderah, T.W.; Paine, M.F.; Cherrington, N.J. Mechanistic basis of altered morphine disposition in nonalcoholic steatohepatitis. J. Pharmacol. Exp. Ther. 2015, 352, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Moon, A.M.; Watkins, S.E.; Lok, A.S.; Firpi-Morell, R.J.; Trinh, H.N.; Kupec, J.T.; Schoen, C.; Neuschwander-Tetri, B.A.; Barritt, A.S. Opioid Use is More Common in Non-Alcoholic Fatty Liver Disease Patients with Cirrhosis, Higher Body Mass Index and Psychiatric Disease. Dig. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rogal, S.S.; Beste, L.A.; Youk, A.; Fine, M.J.; Ketterer, B.; Zhang, H.; Leipertz, S.; Chartier, M.; Good, C.B.; Kraemer, K.L.; et al. Characteristics of Opioid Prescriptions to Veterans With Cirrhosis. Clin. Gastroenterol. Hepatol. 2019, 17, 1165–1174.e3. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, D.; Sessa, G.; Salvatore, T.; Sasso, F.C.; Giugliano, D.; Lefebvre, P.J.; Torella, R. The involvement of the opioid system in human obesity: A study in normal weight relatives of obese people. J. Clin. Endocrinol. Metab. 1996, 81, 713–718. [Google Scholar]
- Haffner, S.M. Pre-diabetes, insulin resistance, inflammation and CVD risk. Diabetes Res. Clin. Pract. 2003, 61 (Suppl. 1), S9–S18. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabrò, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: The prospective AIRE Study. Cardiovasc. Diabetol. 2019, 18, 24. [Google Scholar] [CrossRef]
- Stols-Gonçalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef]
- Targher, G.; Lonardo, A.; Byrne, C.D. Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Cho, Y.K.; Kim, Y.; Sung, E.; Ahn, J.; Jung, H.S.; Yun, K.E.; Shin, H.; Ryu, S. Nonheavy Drinking and Worsening of Noninvasive Fibrosis Markers in Nonalcoholic Fatty Liver Disease: A Cohort Study. Hepatology 2019, 69, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus, P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Jonas, W.; Schürmann, A. Genetic and epigenetic factors determining NAFLD risk. Mol. Metab. 2020, 101111. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Teo, K.; Abeysekera, K.W.M.; Adams, L.; Aigner, E.; Anstee, Q.M.; Banales, J.M.; Banerjee, R.; Basu, P.; Berg, T.; Bhatnagar, P.; et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol. 2021, 74, 20–30. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [Green Version]
- Gijón, M.A.; Riekhof, W.R.; Zarini, S.; Murphy, R.C.; Voelker, D.R. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J. Biol. Chem. 2008, 283, 30235–30245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2020, 70, 940–950. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.L.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Greco, D.; Kotronen, A.; Westerbacka, J.; Puig, O.; Arkkila, P.; Kiviluoto, T.; Laitinen, S.; Kolak, M.; Fisher, R.M.; Hamsten, A.; et al. Gene expression in human NAFLD. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 294, G1281–G1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sánchez-Campos, S.; García-Mediavilla, M.V.; Fernández-Bermejo, M.; Lozano-Rodríguez, T.; Vargas-Castrillón, J.; Buqué, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, M.K.; Nellemann, B.; Stødkilde-Jørgensen, H.; Pedersen, S.B.; Grønbæk, H.; Nielsen, S. Impaired Insulin Suppression of VLDL-Triglyceride Kinetics in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2016, 101, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, M.; Sreekumar, R.; Rasmussen, D.; Lindor, K.; Nair, K.S. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 2002, 35, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Fujisawa, T.; Sugimoto, K.; Nojima, K.; Oze-Fukai, A.; Yoshikawa, Y.; Wang, X.; Yasuda, O.; Ikegami, H.; Rakugi, H. Involvement of microsomal triglyceride transfer protein in nonalcoholic steatohepatitis in novel spontaneous mouse model. J. Hepatol. 2010, 52, 903–912. [Google Scholar] [CrossRef]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Fujimoto, Y.; Fujitake, M.; Endo, H.; Takahashi, H.; Inamori, M.; Kobayashi, N.; et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 2009, 50, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Londos, C.; Honnor, R.C.; Dhillon, G.S. cAMP-dependent protein kinase and lipolysis in rat adipocytes. III. Multiple modes of insulin regulation of lipolysis and regulation of insulin responses by adenylate cyclase regulators. J. Biol. Chem. 1985, 260, 15139–15145. [Google Scholar] [CrossRef]
- Softic, S.; Boucher, J.; Solheim, M.H.; Fujisaka, S.; Haering, M.F.; Homan, E.P.; Winnay, J.; Perez-Atayde, A.R.; Kahn, C.R. Lipodystrophy Due to Adipose Tissue-Specific Insulin Receptor Knockout Results in Progressive NAFLD. Diabetes 2016, 65, 2187–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.K.; Garg, A. Congenital generalized lipodystrophy: Significance of triglyceride biosynthetic pathways. Trends Endocrinol. Metab. 2003, 14, 214–221. [Google Scholar] [CrossRef]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: A cross-sectional study. Sci. Rep. 2014, 4, 5832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 2005, 54, 3458–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef]
- Zhang, H.H.; Halbleib, M.; Ahmad, F.; Manganiello, V.C.; Greenberg, A.S. Tumor necrosis factor-alpha stimulates lipolysis in differentiated human adipocytes through activation of extracellular signal-related kinase and elevation of intracellular cAMP. Diabetes 2002, 51, 2929–2935. [Google Scholar] [CrossRef]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Lehrskov, L.L.; Christensen, R.H. The role of interleukin-6 in glucose homeostasis and lipid metabolism. Semin. Immunopathol. 2019, 41, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Karin, M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012, 56, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Dalamaga, M.; Chou, S.H.; Shields, K.; Papageorgiou, P.; Polyzos, S.A.; Mantzoros, C.S. Leptin at the intersection of neuroendocrinology and metabolism: Current evidence and therapeutic perspectives. Cell Metab. 2013, 18, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef]
- Honda, H.; Ikejima, K.; Hirose, M.; Yoshikawa, M.; Lang, T.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology 2002, 36, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, K.; Takei, Y.; Honda, H.; Hirose, M.; Yoshikawa, M.; Zhang, Y.J.; Lang, T.; Fukuda, T.; Yamashina, S.; Kitamura, T.; et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002, 122, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kadowaki, T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013, 17, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Bugianesi, E.; Pagotto, U.; Manini, R.; Vanni, E.; Gastaldelli, A.; de Iasio, R.; Gentilcore, E.; Natale, S.; Cassader, M.; Rizzetto, M.; et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J. Clin. Endocrinol. Metab. 2005, 90, 3498–3504. [Google Scholar] [CrossRef]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhao, Q.; Song, N.; Yan, Z.; Lin, R.; Wu, S.; Jiang, L.; Hong, S.; Xie, J.; Zhou, H.; et al. AdipoR1/AdipoR2 dual agonist recovers nonalcoholic steatohepatitis and related fibrosis via endoplasmic reticulum-mitochondria axis. Nat. Commun. 2020, 11, 5807. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L.; Yang, Z.; Yang, S.S. Roles of Adipokines in Digestive Diseases: Markers of Inflammation, Metabolic Alteration and Disease Progression. Int J. Mol. Sci. 2020, 21, 8308. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüning, J.C.; Michael, M.D.; Winnay, J.N.; Hayashi, T.; Hörsch, D.; Accili, D.; Goodyear, L.J.; Kahn, C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 1998, 2, 559–569. [Google Scholar] [CrossRef]
- Kim, J.K.; Michael, M.D.; Previs, S.F.; Peroni, O.D.; Mauvais-Jarvis, F.; Neschen, S.; Kahn, B.B.; Kahn, C.R.; Shulman, G.I. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Investig. 2000, 105, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannery, C.; Dufour, S.; Rabøl, R.; Shulman, G.I.; Petersen, K.F. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012, 61, 2711–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabøl, R.; Petersen, K.F.; Dufour, S.; Flannery, C.; Shulman, G.I. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 13705–13709. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; DeFronzo, R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 1999, 22, 1462–1470. [Google Scholar] [CrossRef]
- Kato, K.; Takeshita, Y.; Misu, H.; Zen, Y.; Kaneko, S.; Takamura, T. Liver steatosis is associated with insulin resistance in skeletal muscle rather than in the liver in Japanese patients with non-alcoholic fatty liver disease. J. Diabetes Investig. 2015, 6, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.C.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Relationship between sarcopenia and nonalcoholic fatty liver disease: The Korean Sarcopenic Obesity Study. Hepatology 2014, 59, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Kim, D.; Joo, S.K.; Kim, J.H.; Chang, M.S.; Kim, B.G.; Lee, K.L.; Kim, W. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J. Hepatol. 2017, 66, 123–131. [Google Scholar] [CrossRef]
- Bhanji, R.A.; Narayanan, P.; Allen, A.M.; Malhi, H.; Watt, K.D. Sarcopenia in hiding: The risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology 2017, 66, 2055–2065. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.C.; Liu, Y.Y.; Lin, C.C.; Wang, C.C.; Wu, Y.J.; Yong, C.C.; Chen, K.D.; Chuah, S.K.; Yao, C.C.; Huang, P.Y.; et al. Gut Microbiota Dysbiosis in Patients with Biopsy-Proven Nonalcoholic Fatty Liver Disease: A Cross-Sectional Study in Taiwan. Nutrients 2020, 12, 820. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [Green Version]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Faria Ghetti, F.; Oliveira, D.G.; de Oliveira, J.M.; de Castro Ferreira, L.; Cesar, D.E.; Moreira, A.P.B. Influence of gut microbiota on the development and progression of nonalcoholic steatohepatitis. Eur. J. Nutr. 2018, 57, 861–876. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Sartor, R.B.; Keku, J.; Schwab, J.H. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology 1990, 98, 414–423. [Google Scholar] [CrossRef]
- Sabaté, J.M.; Jouët, P.; Harnois, F.; Mechler, C.; Msika, S.; Grossin, M.; Coffin, B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes. Surg. 2008, 18, 371–377. [Google Scholar] [CrossRef]
- Wijarnpreecha, K.; Lou, S.; Watthanasuntorn, K.; Kroner, P.T.; Cheungpasitporn, W.; Lukens, F.J.; Pungpapong, S.; Keaveny, A.P.; Ungprasert, P. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 601–608. [Google Scholar] [CrossRef]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kadowaki, T. Imbalanced Insulin Actions in Obesity and Type 2 Diabetes: Key Mouse Models of Insulin Signaling Pathway. Cell Metab. 2017, 25, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001, 15, 1206–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Chu, Q.; Monks, B.R.; Birnbaum, M.J. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat. Commun. 2015, 6, 7078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.E.; Kokkotou, E.; Biddinger, S.B.; Kondo, T.; Gebhardt, R.; Kratzsch, J.; Mantzoros, C.S.; Kahn, C.R. High circulating leptin receptors with normal leptin sensitivity in liver-specific insulin receptor knock-out (LIRKO) mice. J. Biol. Chem. 2007, 282, 23672–23678. [Google Scholar] [CrossRef] [Green Version]
- Ling, A.V.; Gearing, M.E.; Semova, I.; Shin, D.J.; Clements, R.; Lai, Z.W.; Biddinger, S.B. FoxO1 Is Required for Most of the Metabolic and Hormonal Perturbations Produced by Hepatic Insulin Receptor Deletion in Male Mice. Endocrinology 2018, 159, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Alemán, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Haas, J.T.; Miao, J.; Chanda, D.; Wang, Y.; Zhao, E.; Haas, M.E.; Hirschey, M.; Vaitheesvaran, B.; Farese, R.V., Jr.; Kurland, I.J.; et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012, 15, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Kubota, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Mineyama, T.; Ogata, H.; Tokuyama, K.; Ohsugi, M.; et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab. 2008, 8, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Kubota, N.; Kubota, T.; Kajiwara, E.; Iwamura, T.; Kumagai, H.; Watanabe, T.; Inoue, M.; Takamoto, I.; Sasako, T.; Kumagai, K.; et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat. Commun. 2016, 7, 12977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, K.; Ogawa, W.; Matsumoto, M.; Nakamura, T.; Sakaue, H.; Kasuga, M. Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. J. Clin. Investig. 2002, 110, 1483–1491. [Google Scholar] [CrossRef]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, R.V.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, M.; Selinger, E.S.; Ballou, L.M.; Lin, R.Z. Ablation of PI3K p110-α prevents high-fat diet-induced liver steatosis. Diabetes 2011, 60, 1483–1492. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Cao, H.; Ye, C.; Chang, C.; Lu, M.; Jing, Y.; Zhang, D.; Yao, X.; Duan, Z.; Xia, H.; et al. Hepatic miR-378 targets p110α and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat. Commun. 2014, 5, 5684. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.; Lipina, C.; Tronche, F.; Sutherland, C.; Alessi, D.R. Deficiency of PDK1 in liver results in glucose intolerance, impairment of insulin-regulated gene expression and liver failure. Biochem. J. 2005, 385, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Ogawa, W.; Nishizawa, A.; Inoue, H.; Teshigawara, K.; Kinoshita, S.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; Sakaue, H.; et al. Restoration of glucokinase expression in the liver normalizes postprandial glucose disposal in mice with hepatic deficiency of PDK1. Diabetes 2007, 56, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- Leavens, K.F.; Easton, R.M.; Shulman, G.I.; Previs, S.F.; Birnbaum, M.J. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009, 10, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, B.; Wang, Y.; Stahl, A.; Bassilian, S.; Lee, W.P.; Kim, Y.J.; Sherwin, R.; Devaskar, S.; Lesche, R.; Magnuson, M.A.; et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 2082–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrou, M.; Bourgoin, L.; Poher, A.L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN deficiency improves muscle insulin sensitivity and decreases adiposity in mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends Endocrinol. Metab. 2010, 21, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ou, J.; Bashmakov, Y.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin inhibits transcription of IRS-2 gene in rat liver through an insulin response element (IRE) that resembles IREs of other insulin-repressed genes. Proc. Natl. Acad. Sci. USA 2001, 98, 3756–3761. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, Y.; Tsuruzoe, K.; Kodama, S.; Igata, M.; Toyonaga, T.; Ueki, K.; Kahn, C.R.; Araki, E. Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J. Endocrinol. 2003, 179, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Ide, T.; Shimano, H.; Yahagi, N.; Matsuzaka, T.; Nakakuki, M.; Yamamoto, T.; Nakagawa, Y.; Takahashi, A.; Suzuki, H.; Sone, H.; et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat. Cell Biol. 2004, 6, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Tsunekawa, S.; Demozay, D.; Briaud, I.; McCuaig, J.; Accili, D.; Stein, R.; Rhodes, C.J. FoxO feedback control of basal IRS-2 expression in pancreatic β-cells is distinct from that in hepatocytes. Diabetes 2011, 60, 2883–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, C.; Geißler, C.; Tackenberg, H.; El Gammal, A.T.; Wolter, S.; Spranger, J.; Mann, O.; Lehnert, H.; Kirchner, H. Multi-layered epigenetic regulation of IRS2 expression in the liver of obese individuals with type 2 diabetes. Diabetologia 2020, 63, 2182–2193. [Google Scholar] [CrossRef]
- Honma, M.; Sawada, S.; Ueno, Y.; Murakami, K.; Yamada, T.; Gao, J.; Kodama, S.; Izumi, T.; Takahashi, K.; Tsukita, S.; et al. Selective insulin resistance with differential expressions of IRS-1 and IRS-2 in human NAFLD livers. Int J. Obes. 2018, 42, 1544–1555. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; Zhang, D.; Abulizi, A.; Zhang, X.M.; et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Betters, J.L.; Lord, C.; Ma, Y.; Han, X.; Yang, K.; Alger, H.M.; Melchior, J.; Sawyer, J.; Shah, R.; et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J. Lipid Res. 2010, 51, 3306–3315. [Google Scholar] [CrossRef] [Green Version]
- Cantley, J.L.; Yoshimura, T.; Camporez, J.P.; Zhang, D.; Jornayvaz, F.R.; Kumashiro, N.; Guebre-Egziabher, F.; Jurczak, M.J.; Kahn, M.; Guigni, B.A.; et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 1869–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, K.; Zhang, Y.; Zhang, D.; Kahn, M.; Ter Horst, K.W.; Rodrigues, M.R.S.; Gaspar, R.C.; Hirabara, S.M.; Luukkonen, P.K.; Lee, S.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e5. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef]
- Wan, M.; Leavens, K.F.; Saleh, D.; Easton, R.M.; Guertin, D.A.; Peterson, T.R.; Kaestner, K.H.; Sabatini, D.M.; Birnbaum, M.J. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011, 14, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Kenerson, H.L.; Yeh, M.M.; Yeung, R.S. Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PLoS ONE 2011, 6, e18075. [Google Scholar]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado del Pozo, C.; Vesperinas-García, G.; Rubio, M.; Corripio-Sánchez, R.; Torres-García, A.J.; Obregon, M.J.; Calvo, R.M. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim. Biophys. Acta 2011, 1811, 1194–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [Green Version]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394. [Google Scholar] [CrossRef]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Horst, K.W.; Vatner, D.F.; Zhang, D.; Cline, G.W.; Ackermans, M.T.; Nederveen, A.J.; Verheij, J.; Demirkiran, A.; van Wagensveld, B.A.; Dallinga-Thie, G.M.; et al. Hepatic Insulin Resistance Is Not Pathway Selective in Humans With Nonalcoholic Fatty Liver Disease. Diabetes Care 2021, 44, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kubota, N.; Takamoto, I.; Obata, A.; Iwamoto, M.; Hayashi, T.; Aihara, M.; Kubota, T.; Nishihara, H.; Kadowaki, T. Role of insulin receptor substrates in the progression of hepatocellular carcinoma. Sci. Rep. 2017, 7, 5387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enooku, K.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Tsutsumi, T.; Nakagomi, R.; et al. Hepatic IRS1 and ß-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J. Gastroenterol. 2018, 53, 1261–1275. [Google Scholar] [CrossRef]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Sasako, T.; Ohsugi, M.; Kubota, N.; Itoh, S.; Okazaki, Y.; Terai, A.; Kubota, T.; Yamashita, S.; Nakatsukasa, K.; Kamura, T.; et al. Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat. Commun. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Summary of genetic variants related to the pathogenesis of NAFLD. Many genome-wide association studies have identified novel loci associated with disease severity phenotypes of NAFLD. The PNPLA3 (rs738409 C>G) variant, which causes impaired mobilization of TG from lipid droplets, predisposes to progression along the full spectrum of liver damage associated with fatty liver. The TM6SF2 (rs58542926 C>T) variant, a loss-of-function variant, is associated with hepatic steatosis via decreased VLDL-TG secretion in the liver. The MBOAT7 (rs641738 C>T) variant, resulting in lower MBOAT7 protein expression in the liver, is associated with increased hepatic fat content through inducing changes in the remodeling of phospholipids. In addition, other genetic variants known to be involved in the regulation of lipid metabolism (LYPLAL1, APOB, MTP, LPIN1, UCP2), glucose metabolism and lipogenesis (GCKR), innate immunity (IL28B, MERTK), insulin signaling (ENPP1, IRS1), oxidative stress (SOD2), and fibrogenesis (KLF6) have also been reported to be associated with the progression of NAFLD.
Figure 1.
Summary of genetic variants related to the pathogenesis of NAFLD. Many genome-wide association studies have identified novel loci associated with disease severity phenotypes of NAFLD. The PNPLA3 (rs738409 C>G) variant, which causes impaired mobilization of TG from lipid droplets, predisposes to progression along the full spectrum of liver damage associated with fatty liver. The TM6SF2 (rs58542926 C>T) variant, a loss-of-function variant, is associated with hepatic steatosis via decreased VLDL-TG secretion in the liver. The MBOAT7 (rs641738 C>T) variant, resulting in lower MBOAT7 protein expression in the liver, is associated with increased hepatic fat content through inducing changes in the remodeling of phospholipids. In addition, other genetic variants known to be involved in the regulation of lipid metabolism (LYPLAL1, APOB, MTP, LPIN1, UCP2), glucose metabolism and lipogenesis (GCKR), innate immunity (IL28B, MERTK), insulin signaling (ENPP1, IRS1), oxidative stress (SOD2), and fibrogenesis (KLF6) have also been reported to be associated with the progression of NAFLD.

Figure 2.
Summary of factors involved in the pathogenesis of MAFLD. Imbalance between energy intake and expenditure leads to fat accumulation in adipose tissue. When the accumulation exceeds the capacity for adipose tissue remodeling, increased release of FFAs and proinflammatory cytokines with dysregulated production of adipokines results in the development of insulin resistance in the skeletal muscle as well as adipose tissue. Glucose and FFAs not utilized in the skeletal muscle and adipose tissue become substrates for hepatic DNL. Increased uptake of FFAs and relatively decreased VLDL-TG secretion also promote hepatic fat accumulation. Compensatory increase in FFA oxidation is accompanied by enhanced oxidative stress and inflammation. Dysbiosis, defined as an imbalance in the microbiota composition, can disrupt the tight junctions in intestinal endothelial cells, leading to increased mucosal permeability and translocation of bacterial LPS from bacteria. Small intestinal bacterial overgrowth and gut-derived metabolites, such as endogenous ethanol, are also associated with the progression of MAFLD. In addition, people with specific genetic risk variants are also susceptible to the development and progression of MAFLD. Taken together, insulin resistance in adipose tissue and the skeletal muscle, dysbiosis, and genetic predisposition are synergistically involved in fat accumulation and inflammation in the liver, followed by subsequent fibrosis and carcinogenesis, in patients with MAFLD.
Figure 2.
Summary of factors involved in the pathogenesis of MAFLD. Imbalance between energy intake and expenditure leads to fat accumulation in adipose tissue. When the accumulation exceeds the capacity for adipose tissue remodeling, increased release of FFAs and proinflammatory cytokines with dysregulated production of adipokines results in the development of insulin resistance in the skeletal muscle as well as adipose tissue. Glucose and FFAs not utilized in the skeletal muscle and adipose tissue become substrates for hepatic DNL. Increased uptake of FFAs and relatively decreased VLDL-TG secretion also promote hepatic fat accumulation. Compensatory increase in FFA oxidation is accompanied by enhanced oxidative stress and inflammation. Dysbiosis, defined as an imbalance in the microbiota composition, can disrupt the tight junctions in intestinal endothelial cells, leading to increased mucosal permeability and translocation of bacterial LPS from bacteria. Small intestinal bacterial overgrowth and gut-derived metabolites, such as endogenous ethanol, are also associated with the progression of MAFLD. In addition, people with specific genetic risk variants are also susceptible to the development and progression of MAFLD. Taken together, insulin resistance in adipose tissue and the skeletal muscle, dysbiosis, and genetic predisposition are synergistically involved in fat accumulation and inflammation in the liver, followed by subsequent fibrosis and carcinogenesis, in patients with MAFLD.
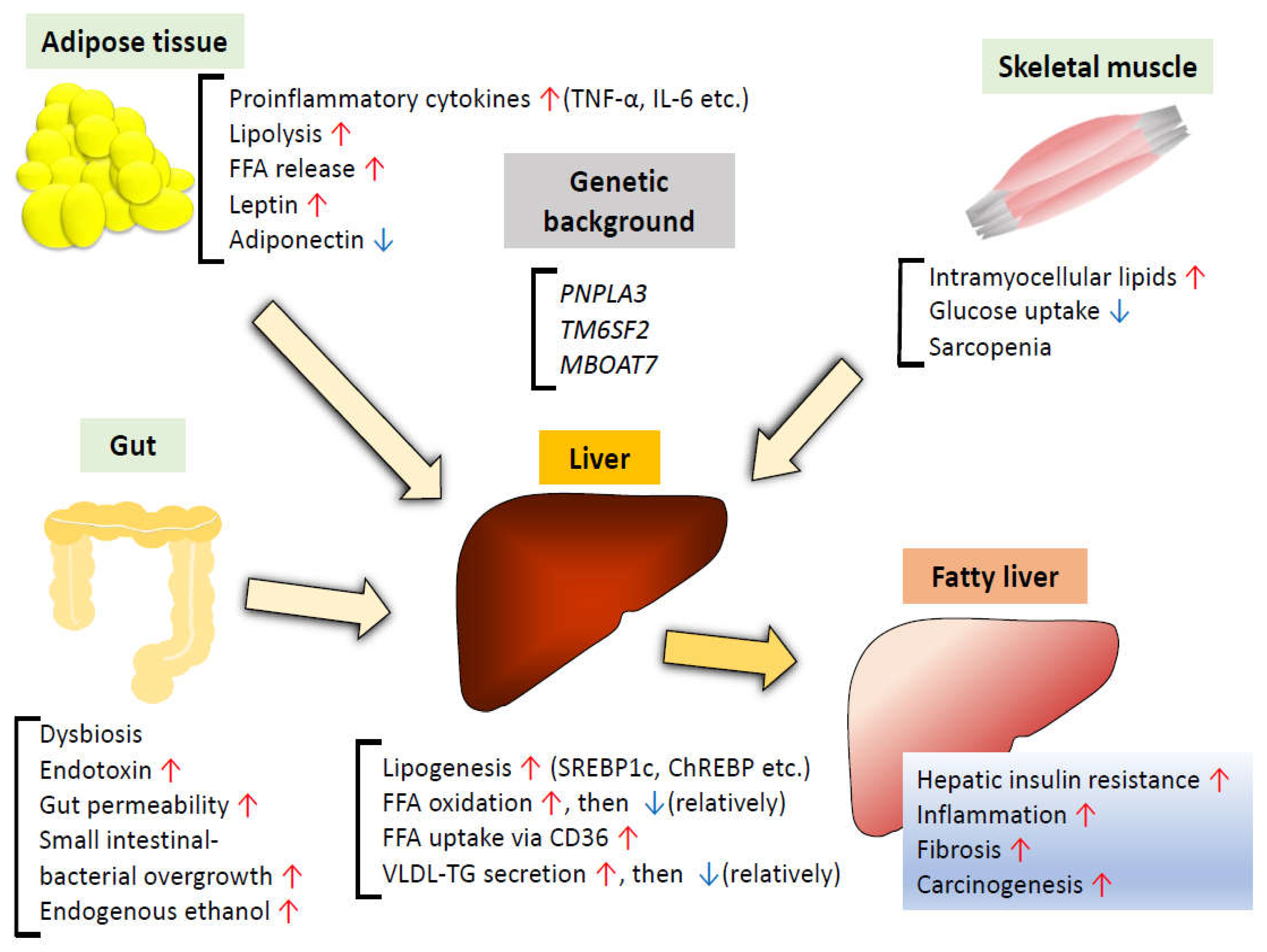
Figure 3.
After insulin binds to the insulin receptor, autophosphorylation of tyrosine residues in the intracellular subunit of each of the receptors causes docking and phosphorylation of Irs1/Irs2, followed by activation of the downstream kinase cascades, such as the PI3K/Akt pathway. For induction of SREBP1c, mTORC1 activation and concomitant inhibition of FoxO1 via the PI3K/Akt pathway are required. In addition to the DAG-PKCε pathway inhibiting physiological phosphorylation of the insulin receptor, hyperinsulinemia leads to downregulation of hepatic Irs2 expression, resulting in the progression of hepatic insulin resistance. GLUT2 is the major glucose transporter in hepatocytes. In response to an increase in the intracellular glucose concentration, ChREBP induces expression of various genes related to lipogenic enzymes, independent of insulin signaling. Due to elevated Irs1 expression in the perivenous (PV) zone, which is the primary site of lipogenesis, insulin signaling is rather enhanced in the presence of hyperinsulinemia despite the downregulation of Irs2, resulting in increased lipogenesis and development of steatosis. Independent of insulin signaling, ER stress also activates SREBP through increased cleavage of S1P by caspase-2.
Figure 3.
After insulin binds to the insulin receptor, autophosphorylation of tyrosine residues in the intracellular subunit of each of the receptors causes docking and phosphorylation of Irs1/Irs2, followed by activation of the downstream kinase cascades, such as the PI3K/Akt pathway. For induction of SREBP1c, mTORC1 activation and concomitant inhibition of FoxO1 via the PI3K/Akt pathway are required. In addition to the DAG-PKCε pathway inhibiting physiological phosphorylation of the insulin receptor, hyperinsulinemia leads to downregulation of hepatic Irs2 expression, resulting in the progression of hepatic insulin resistance. GLUT2 is the major glucose transporter in hepatocytes. In response to an increase in the intracellular glucose concentration, ChREBP induces expression of various genes related to lipogenic enzymes, independent of insulin signaling. Due to elevated Irs1 expression in the perivenous (PV) zone, which is the primary site of lipogenesis, insulin signaling is rather enhanced in the presence of hyperinsulinemia despite the downregulation of Irs2, resulting in increased lipogenesis and development of steatosis. Independent of insulin signaling, ER stress also activates SREBP through increased cleavage of S1P by caspase-2.
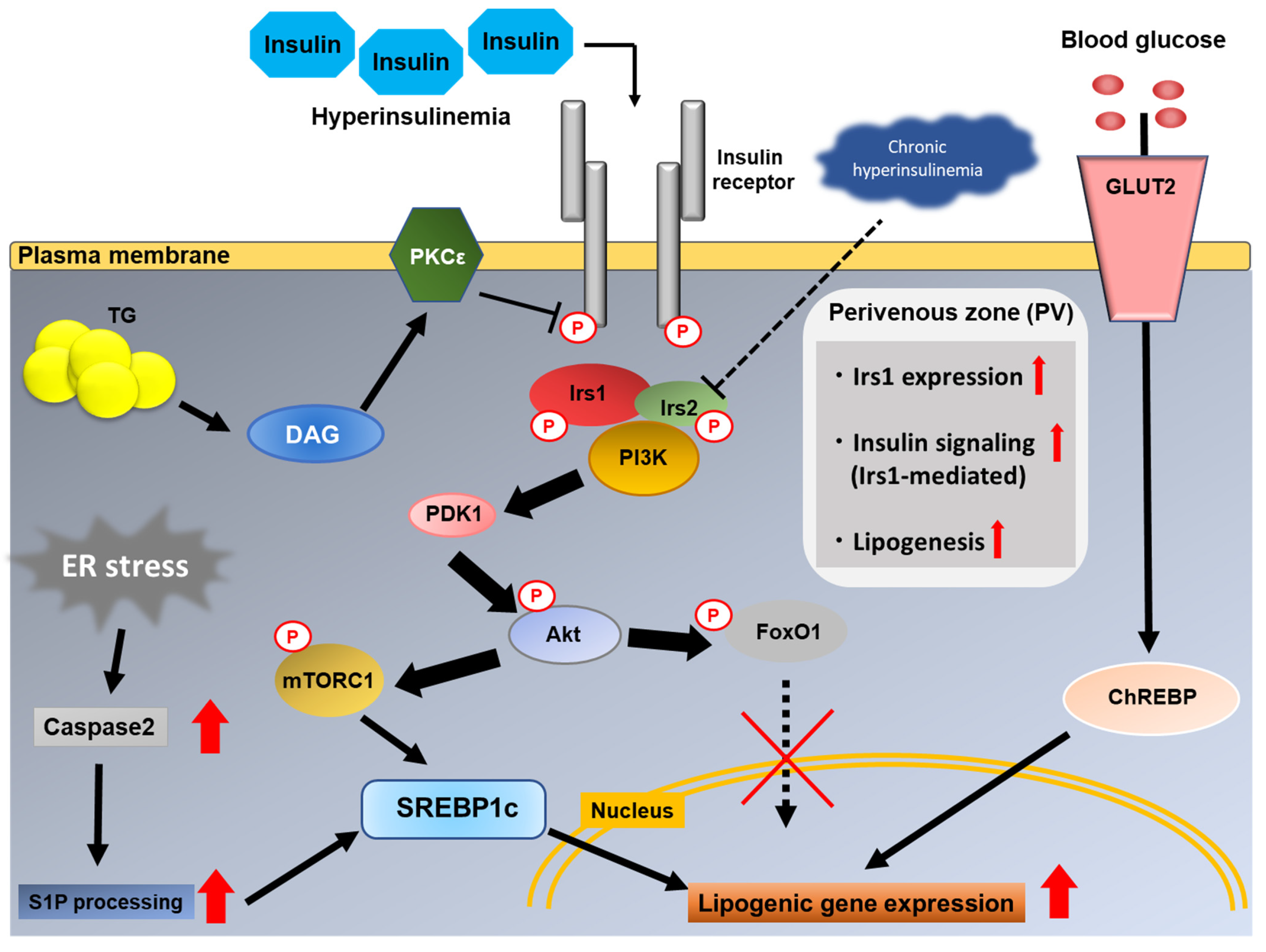

{kind=link}
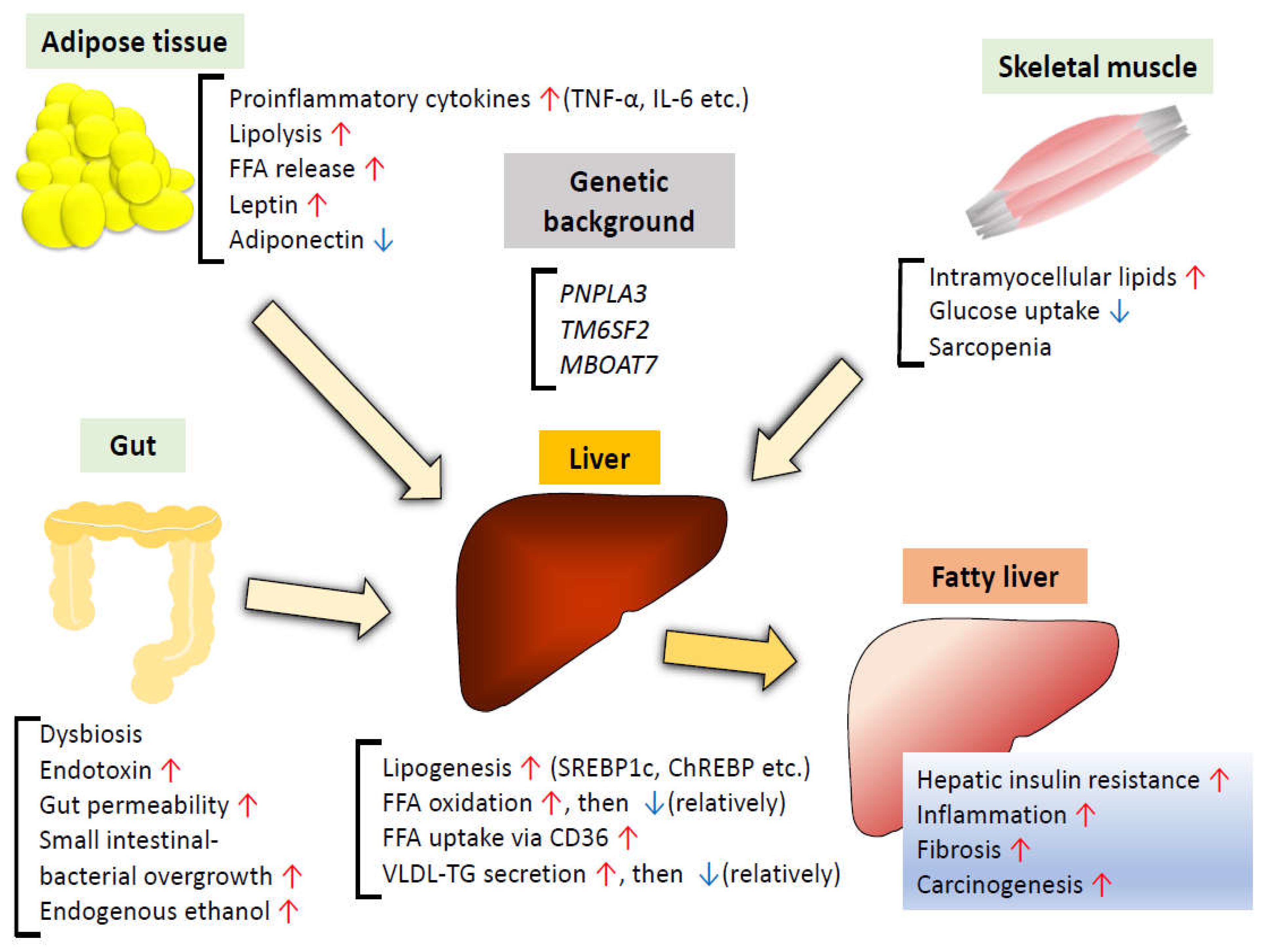{kind=link}
{kind=link}
Table 1.
Summary of genetically modified mouse models of insulin signaling. (N/A: not applicable).
| Type of Mice | Body Weight | Glucose Tolerance | Plasma Insulin | Hepatic Lipogenesis | Hepatic TG Content | Comments |
|---|---|---|---|---|---|---|
| LIRKO | Decreased, then catch up | Severely impaired | Increased | Decreased | Decreased in DIO, ob/ob mice | Metabolic phenotype becomes less severe with aging. |
| LIrs1KO | Normal | Mildly impaired (refeeding state) | Increased (re-feeding state) | Decreased (re-feeding state) | Decreased in DIO mice | N/A |
| LIrs2KO | Normal | Mildly impaired (fasting state) | Increased (fasting state) | Decreased (fasting state) | Similar to controls in DIO mice | N/A |
| LIrs1/2DKO | Normal | Impaired | Increased | Decreased | Decreased in DIO mice | N/A |
| LPI3K(p85)DKO | Normal | Impaired | Increased | Decreased | N/A | Small liver, liver dysfunction |
| LPI3K(p110α)KO | Increased, with increased fat mass | Impaired | Increased | Decreased | Decreased in DIO mice | Small liver, normal morphology |
| LPDK1KO | Normal | Impaired | Increased | Decreased | N/A | N/A |
| LAkt2KO | Normal | Normal | Normal | Normal | Decreased in DIO, ob/ob mice | N/A |
| LAkt1/2DKO | N/A | Impaired | Increased | Decreased | N/A | N/A |
| LPTENKO | Normal | Improved | Decreased | Increased | Increased | Development of HCC with aging |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084156
AMA Style
Sakurai Y, Kubota N, Yamauchi T, Kadowaki T. Role of Insulin Resistance in MAFLD. International Journal of Molecular Sciences. 2021; 22(8):4156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084156
Chicago/Turabian StyleSakurai, Yoshitaka, Naoto Kubota, Toshimasa Yamauchi, and Takashi Kadowaki. 2021. "Role of Insulin Resistance in MAFLD" International Journal of Molecular Sciences 22, no. 8: 4156. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084156
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.