
Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management?

 and
and
Abstract
:1. Introduction
2. Monogenic vs. Polygenic Determination

{kind=link}
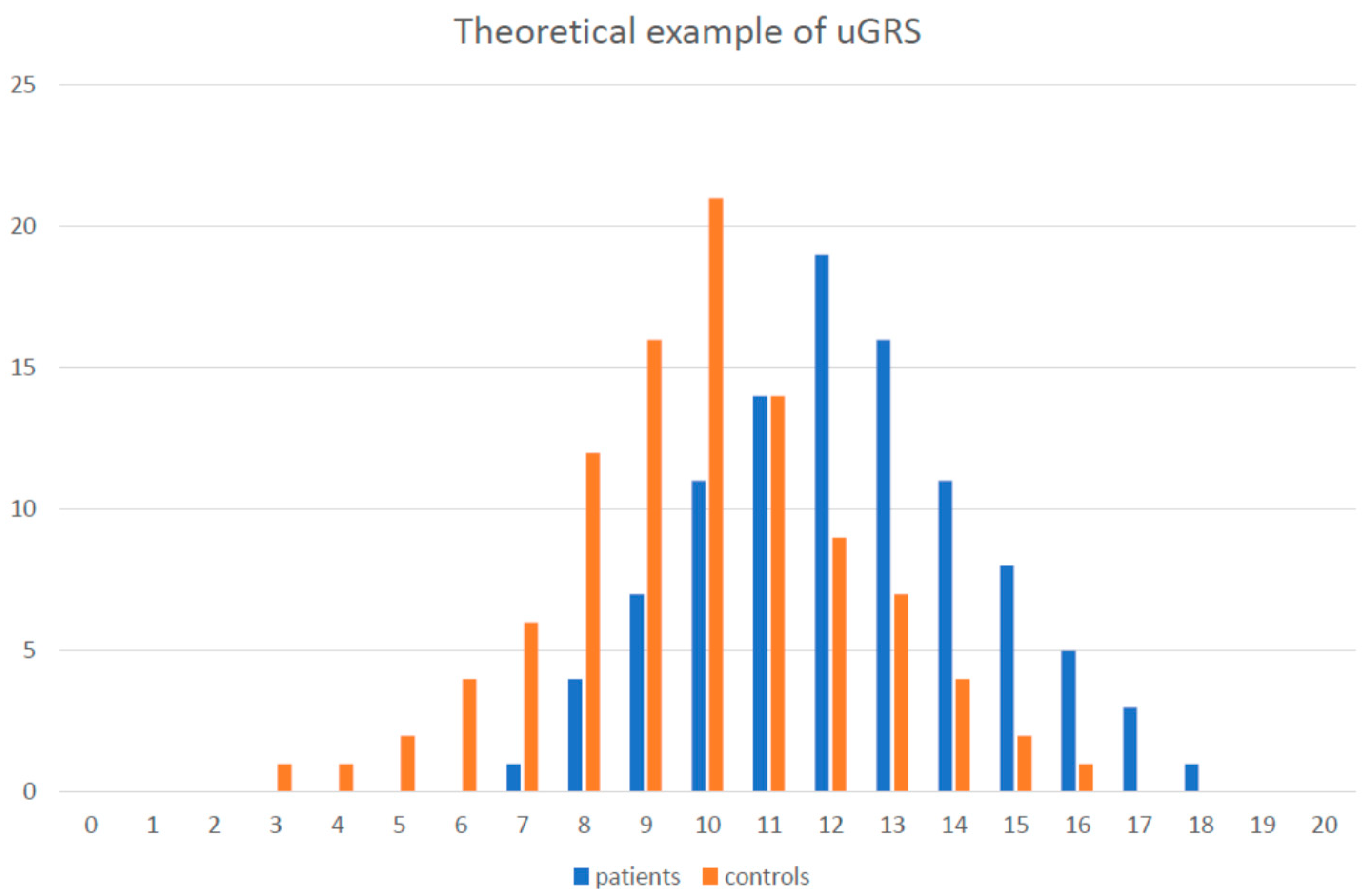{kind=link}
| Gene(s) | CVD | Manifestation | Frequency |
|---|---|---|---|
| LDLR, APOB, PCSK9 | Familial hypercholesterolemia | High concentrations of LDL and total cholesterol; xanthomas; arcus lipoides cornae; xanthalesmas; coronary heart disease | 1:200–250 |
| ABCG5, ABCG8 | Sitosterolemia | High plasma sitosterol, campesterol; hypercholesterolemia; premature coronary heart disease; xanthomas | 1:2000 |
| MYH7, MYBPC3, TNNT2, TPM1, MYL2, MYL3, PLN, | Hypertrophic cardiomyopathy | Hypertrophy of left ventricle, shortness of breath, diastolic dysfunction, left ventricular outflow ischemia | 1:500 |
| PKP2, DSP, DSG2, JUP, TMEM43 | Arrhythmogenic right ventricular cardiomyopathy | Ventricular arrhythmias, right ventricular cardiomyopathy | 1:5000 |
| MYH7, MYBPC3, TNNT2, MYH6, MYPN, ANKRD1, RAF1, DES, DMD | Familial dilated cardiomyopathy | Diastolic dysfunction, left ventricular hypertrophy, atrial fibrillation, congestive heart failure | 1:2500 |
| FBN1, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, SKI | Marfan’s syndrome | Aortic aneurysm or dissection, valvular heart disease, enlargement of the proximal pulmonary artery, congestive heart failure, arrhythmias | 1:5000 |
| ACTA2, FBN1, MYH11, TGFBR1/2, LOX, COL3A1, TGFB2/3 | Thoracic aortic aneurysm and dissection | Chest pain, renal cysts, thumb-palm sign, temporal arteritis, bicuspid aortic valve, abdominal aneurysm, intracranial aneurysm, | unknown |
| BMPR2, BMPR1B, CAV1, KCNK3, SMAD9, ACVRL1, ENG, EIF2AK4 | Pulmonary arterial hypertension | Right ventricular failure, impaired brachial artery flow-mediated dilation, increased pulmonary vascular resistance | 15:1,000,000 |
| KCNQ1/H2/E1/J2, SCN5A, CAV3, CALM1/2 | Long QT syndrome | Malignant arrhythmia, palpitations, syncope, anoxic seizures secondary to ventricular arrhythmia | 1:2000 |
| KCNH2 | Short QT syndrome | Abbreviated QTc interval on the ECG, propensity for atrial and ventricular arrhythmias, | unknown |
| SCN5A | Brugada syndrome | Elevation of the ST, ventricular fibrillation, syncope, arrhythmia | 1:2000 |
3. Association Studies
4. Genome Wide Association Studies (GWAS)
| Gene | Effect on | Ref |
|---|---|---|
| ANRIL | Risk of myocardial infarction | [24] |
| SORT1 | Plasma cholesterol levels | [21,28] |
| APOA5 | Plasma triglyceride levels | [16,49] |
| FTO | BMI values, risk of T2DM and myocardial infarction | [34,35,36,38,39] |
| TCF7L2 | Risk of T2DM | [50] |
| APOE | Plasma cholesterol levels | [15] |
| MC4R | BMI values | [51] |
| CHRNA5-A3-B4 | Smoking addiction | [52] |
| UMOD | Hypertension | [53] |
5. Gene Score
5.1. Polygenic Predisposition
5.2. Unweighted and Weighted Genetic Risk Score
5.3. GRS Examples
6. Nutrigenetics
7. Pharmacogenetic of CVD Treatment
8. Epigenetics
8.1. Regulatory Non-Coding miRNA
| miRNA | Function |
|---|---|
| miR-15 family, miR-34 family, miR-499, miR-320, miR-24, miR-1, miR-16, miR-21, miR-92a, miR-375, miR-103/107, miR-133a/b, miR-214 | Differently regulated in heart tissue in response to myocardial infarction |
| miR-34a, miR-217, miR-146a | Endothelial cell senescence |
| miR-126, miR-31, miR-17-3p | Vascular inflammation |
| miR-21, miR-221, miR-222, miR-143/145 cluster, miR-1, miR-10a | SMC (smooth muscle cell) differentiation, survival, proliferation, and dedifferentiation |
| miR-155, miR-125a-5p | Monocytes/macrophages lipid uptake and inflammatory responses |
| miR-146a, miR-128, miR-365, miR-503 | Effect on migration of macrophages |
| miR-33, miR-302a, miR-122, miR-370, miR-335, miR-378, miR-27, miR-125a-5p, miR-33a/b, miR-144, miR-223, miR-148a, miR-128-1 | Cholesterol homeostasis and fatty acid oxidation |
8.2. DNA Methylation
9. Telomeres
10. Geographical and Ethnical Differences
11. Heritability of CVD risk Modifiers: Less Understood Piece of the Puzzle
12. Conclusions and Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular disease statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, E.; Wilson, L.; Wickramasinghe, K.; Bhatnagar, P.; Leal, J.; Luengo-Fernandez, R.; Burns, R.; Rayner, M.; Townsend, N. European Cardiovascular Disease Statistics 2017; European Heart Network: Brussels, Belgium, 2017. [Google Scholar]
- Hartley, A.; Marshall, D.C.; Salciccioli, J.D.; Sikkel, M.B.; Maruthappu, M.; Shalhoub, J. Trends in mortality from ischemic heart disease and cerebrovascular disease in Europe: 1980 to 2009. Circulation 2016, 133, 1916–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, e783–e831. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Kathiresan, S. Genetics of common, complex coronary artery disease. Cell 2019, 177, 132–145. [Google Scholar] [CrossRef]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of familial hypercholesterolemia: New insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef]
- Mytilinaiou, M.; Kyrou, I.; Khan, M.; Grammatopoulos, D.K.; Randeva, H.S. Familial hypercholesterolemia: New horizons for diagnosis and effective management. Front. Pharmacol. 2018, 9, 707. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.; Futema, M.; Nair, D.; Humphries, S.E. Genetic architecture of familial hypercholesterolaemia. Curr. Cardiol. Rep. 2017, 19, 44. [Google Scholar] [CrossRef]
- Brandts, J.; Dharmayat, K.I.; Ray, K.K.; Vallejo-Vaz, A.J. Familial hypercholesterolemia: Is it time to separate monogenic from polygenic familial hypercholesterolemia? Curr. Opin. Lipidol. 2020, 31, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.B.; Hui, R.T.; Song, L. The era of clinical application of gene diagnosis in cardiovascular diseases is coming. Chronic Dis. Transl. Med. 2020, 5, 214–220. [Google Scholar] [CrossRef]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Lee, M.H.; Hazard, S.; Brooks-Wilson, A.; Hidaka, H.; Kojima, H.; Ose, L.; Stalenhoef, A.F.; Mietinnen, T.; Bjorkhem, I.; et al. Two genes that map to the STSL locus cause sitosterolemia: Genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am. J. Hum. Genet. 2001, 69, 278–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 591, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pennacchio, L.A.; Olivier, M.; Hubacek, J.A.; Cohen, J.C.; Cox, D.R.; Fruchart, J.C.; Krauss, R.M.; Rubin, E.M. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 2001, 294, 169–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, R.J. The genetic epidemiology of melanocortin 4 receptor variants. Eur. J. Pharmacol. 2011, 660, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Gianfagna, F.; Cugino, D.; Santimone, I.; Iacoviello, L. From candidate gene to genome-wide association studies in cardiovascular disease. Thromb. Res. 2012, 129, 320–324. [Google Scholar] [CrossRef]
- Uitterlinden, A.G. An introduction to genome-wide association studies: GWAS for dummies. Semin. Reprod. Med. 2016, 34, 196–204. [Google Scholar] [CrossRef]
- Larson, M.G.; Atwood, L.D.; Benjamin, E.J.; Cupples, L.A.; D’Agostino, R.B., Sr.; Fox, C.S.; Govindaraju, D.R.; Guo, C.Y.; Heard-Costa, N.L.; Hwang, S.J.; et al. Framingham Heart Study 100K project: Genome-wide associations for cardiovascular disease outcomes. BMC Med. Genet. 2007, 8 (Suppl. 1), S5. [Google Scholar] [CrossRef]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdmann, J.; Kessler, T.; Munoz Venegas, L.; Schunkert, H. A decade of genome-wide association studies for coronary artery disease: The challenges ahead. Cardiovasc. Res. 2018, 114, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Palomaki, G.E.; Melillo, S.; Bradley, L.A. Association between 9p21 genomic markers and heart disease: A meta-analysis. JAMA 2010, 303, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Pfeiffer, R.M.; Hyland, P.L.; Shi, J.; Gu, F.; Wang, Z.; Bhattacharjee, S.; Luo, J.; Xiong, X.; Yeager, M.; et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis 2014, 35, 2698–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggs, J.L.; Yaspan, B.L.; Hauser, M.A.; Kang, J.H.; Allingham, R.R.; Olson, L.M.; Abdrabou, W.; Fan, B.J.; Wang, D.Y.; Brodeur, W.; et al. Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet. 2012, 8, e1002654. [Google Scholar] [CrossRef] [Green Version]
- Safa, A.; Noroozi, R.; Taheri, M.; Ghafouri-Fard, S. Association analysis of ANRIL polymorphisms and haplotypes with autism spectrum disorders. J. Mol. Neurosci. 2021, 71, 187–192. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Strong, A.; Rader, D.J. Sortilin as a regulator of lipoprotein metabolism. Curr. Atheroscler. Rep. 2012, 14, 211–218. [Google Scholar] [CrossRef]
- Zhong, L.Y.; Cayabyab, F.S.; Tang, C.K.; Zheng, X.L.; Peng, T.H.; Lv, Y.C. Sortilin: A novel regulator in lipid metabolism and atherogenesis. Clin. Chim. Acta 2016, 460, 11–17. [Google Scholar] [CrossRef]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Ruda, V.M.; et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef]
- Kjolby, M.; Andersen, O.M.; Breiderhoff, T.; Fjorback, A.W.; Pedersen, K.M.; Madsen, P.; Jansen, P.; Heeren, J.; Willnow, T.E.; Nykjaer, A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tveten, K.; Strøm, T.B.; Cameron, J.; Berge, K.E.; Leren, T.P. Mutations in the SORT1 gene are unlikely to cause autosomal dominant hypercholesterolemia. Atherosclerosis 2012, 225, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Dina, C.; Meyre, D.; Gallina, S.; Durand, E.; Körner, A.; Jacobson, P.; Carlsson, L.M.; Kiess, W.; Vatin, V.; Lecoeur, C.; et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 2007, 39, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT; Lund University; Novartis Institutes of BioMedical Research; Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [PubMed]
- Loos, R.J.; Yeo, G.S. The bigger picture of FTO: The first GWAS-identified obesity gene. Nat. Rev. Endocrinol. 2014, 10, 51–61. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Stanek, V.; Gebauerová, M.; Pilipcincová, A.; Dlouhá, D.; Poledne, R.; Aschermann, M.; Skalická, H.; Matousková, J.; Kruger, A.; et al. A FTO variant and risk of acute coronary syndrome. Clin. Chim. Acta 2010, 411, 1069–1072. [Google Scholar] [CrossRef]
- Doney, A.S.; Dannfald, J.; Kimber, C.H.; Donnelly, L.A.; Pearson, E.; Morris, A.D.; Palmer, C.N. The FTO gene is associated with an atherogenic lipid profile and myocardial infarction in patients with type 2 diabetes: A Genetics of Diabetes Audit and Research Study in Tayside Scotland (Go-DARTS) study. Circ. Cardiovasc Genet. 2009, 2, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Hubacek, J.A.; Viklicky, O.; Dlouha, D.; Bloudickova, S.; Kubinova, R.; Peasey, A.; Pikhart, H.; Adamkova, V.; Brabcova, I.; Pokorna, E.; et al. The FTO gene polymorphism is associated with end-stage renal disease: Two large independent case-control studies in a general population. Nephrol. Dial. Transplant. 2012, 27, 1030–1035. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Tosto, G.; Mayeux, R.; Luchsinger, J.A.; NIA-LOAD/NCRAD Family Study Group; Alzheimer’s Disease Neuroimaging Initiative. Genetic variants in the Fat and Obesity Associated (FTO) gene and risk of Alzheimer’s disease. PLoS ONE 2012, 7, e50354. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Dlouha, D.; Klementova, M.; Lanska, V.; Neskudla, T.; Pelikanova, T. The FTO variant is associated with chronic complications of diabetes mellitus in Czech population. Gene 2018, 642, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, E.; Kring, S.I.; Berentzen, T.L.; Holst, C.; Pers, T.H.; Hansen, T.; Pedersen, O.; Sørensen, T.I.; Jess, T. Fatness-associated FTO gene variant increases mortality independent of fatness—In cohorts of Danish men. PLoS ONE 2009, 4, e4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, Y.C.L.; Yeo, G.S.H.; O’Rahilly, S.; Coll, A.P. Obesity and FTO: Changing focus at a complex locus. Cell Metab. 2014, 20, 710–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerken, T.; Girard, C.A.; Tung, Y.C.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dlouha, D.; Pitha, J.; Lanska, V.; Hubacek, J.A. Association between FTO 1st intron tagging variant and telomere length in middle aged females. 3PMFs study. Clin. Chim. Acta 2012, 413, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hambly, B.D.; McLachlan, C.S. FTO associations with obesity and telomere length. J. Biomed. Sci. 2017, 24, 65. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Saunders, R.A.; Szkudlarek-Mikho, M.; Serna Ide, L.; Chin, K.V. The obesity-associated Fto gene is a transcriptional coactivator. Biochem. Biophys. Res. Commun. 2010, 401, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Nadkarni, G.N.; Gignoux, C.R.; Sorokin, E.P.; Daya, M.; Rahman, R.; Barnes, K.C.; Wassel, C.L.; Kenny, E.E. Worldwide frequencies of APOL1 renal risk variants. N. Engl. J. Med. 2018, 379, 2571–2572. [Google Scholar] [CrossRef]
- Grant, S.F.A. The TCF7L2 locus: A genetic window into the pathogenesis of type 1 and type 2 diabetes. Diabetes Care 2019, 42, 1624–1629. [Google Scholar] [CrossRef] [Green Version]
- Xi, B.; Chandak, G.R.; Shen, Y.; Wang, Q.; Zhou, D. Association between common polymorphism near the MC4R gene and obesity risk: A systematic review and meta-analysis. PLoS ONE 2012, 7, e45731. [Google Scholar] [CrossRef] [PubMed]
- Saccone, N.L.; Saccone, S.F.; Hinrichs, A.L.; Stitzel, J.A.; Duan, W.; Pergadia, M.L.; Agrawal, A.; Breslau, N.; Grucza, R.A.; Hatsukami, D.; et al. Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Lip, S.; Padmanabhan, S. Genomics of blood pressure and hypertension: Extending the mosaic theory toward stratification. Can. J. Cardiol. 2020, 36, 694–705. [Google Scholar] [CrossRef]
- Lambert, S.A.; Abraham, G.; Inouye, M. Towards clinical utility of polygenic risk scores. Hum. Mol. Genet. 2019, 28, R133–R142. [Google Scholar] [CrossRef]
- Rao, A.S.; Knowles, J.W. Polygenic risk scores in coronary artery disease. Curr. Opin. Cardiol. 2019, 34, 435–440. [Google Scholar] [CrossRef]
- Igo, R.P., Jr.; Kinzy, T.G.; Cooke Bailey, J.N. Genetic risk scores. Curr. Protoc. Hum. Genet. 2019, 104, e95. [Google Scholar] [CrossRef] [PubMed]
- Kooner, J.S.; Chambers, J.C.; Aguilar-Salinas, C.A.; Hinds, D.A.; Hyde, C.L.; Warnes, G.R.; Gómez Pérez, F.J.; Frazer, K.A.; Elliott, P.; Scott, J.; et al. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 2008, 40, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Ceska, R.; Adamkova, V.; Peasey, A.; Pikhart, H.; Kubinova, R.; Marmot, M.; Bobak, M.; Hubacek, J.A. MLXIPL variant in individuals with low and high triglyceridemia in white population in Central Europe. Hum. Genet. 2008, 124, 553–555. [Google Scholar] [CrossRef]
- Rašlová, K.; Dobiášová, M.; Hubáček, J.A.; Bencová, D.; Siváková, D.; Danková, Z.; Franeková, J.; Jabor, A.; Gašparovič, J.; Vohnout, B. Association of metabolic and genetic factors with cholesterol esterification rate in HDL plasma and atherogenic index of plasma in a 40 years old Slovak population. Physiol. Res. 2011, 60, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kawashiri, M.A.; Nomura, A.; Teramoto, R.; Hosomichi, K.; Nohara, A.; Inazu, A.; Mabuchi, H.; Tajima, A.; Yamagishi, M. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J. Clin. Lipidol. 2018, 12, 1436–1444. [Google Scholar] [CrossRef]
- Poledne, R.; Hubácek, J.; Písa, Z.; Pistulková, H.; Valenta, Z. Genetic markers in hypercholesterolemic and normocholesterolemic Czech children. Clin. Genet. 1994, 46, 88–91. [Google Scholar] [CrossRef]
- Pedersen, J.C.; Berg, K. Gene-gene interaction between the low density lipoprotein receptor and apolipoprotein E loci affects lipid levels. Clin. Genet. 1990, 38, 287–294. [Google Scholar] [PubMed]
- Shabana; Shahid, S.U.; Hasnain, S. Use of a gene score of multiple low-modest effect size variants can predict the risk of obesity better than the individual SNPs. Lipids Health Dis. 2018, 17, 155. [Google Scholar] [CrossRef] [Green Version]
- Hubacek, J.A.; Dlouha, D.; Adamkova, V.; Schwarzova, L.; Lanska, V.; Ceska, R.; Satny, M.; Vrablik, M. The gene score for predicting hypertriglyceridemia: New insights from a Czech case-control study. Mol. Diagn. Ther. 2019, 23, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.T.; Wang, J.; Lanktree, M.B.; McIntyre, A.D.; Ban, M.R.; Martins, R.A.; Kennedy, B.A.; Hassell, R.G.; Visser, M.E.; Schwartz, S.M.; et al. An increased burden of common and rare lipid-associated risk alleles contributes to the phenotypic spectrum of hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1916–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Rouby, N.; McDonough, C.W.; Gong, Y.; McClure, L.A.; Mitchell, B.D.; Horenstein, R.B.; Talbert, R.L.; Crawford, D.C.; eMERGE Network; Gitzendanner, M.A.; et al. Genome-wide association analysis of common genetic variants of resistant hypertension. Pharmacogenom. J. 2019, 19, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Cooper, J.A.; Morris, R.W.; Dudbridge, F.; Shah, T.; Engmann, J.; Dale, C.; White, J.; McLachlan, S.; Zabaneh, D.; et al. Sixty-five common genetic variants and prediction of type 2 diabetes. Diabetes 2015, 64, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Läll, K.; Mägi, R.; Morris, A.; Metspalu, A.; Fischer, K. Personalized risk prediction for type 2 diabetes: The potential of genetic risk scores. Genet. Med. 2017, 19, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.W.; Cooper, J.A.; Shah, T.; Wong, A.; Drenos, F.; Engmann, J.; McLachlan, S.; Jefferis, B.; Dale, C.; Hardy, R.; et al. Marginal role for 53 common genetic variants in cardiovascular disease prediction. Heart 2016, 102, 1640–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinder, M.; Li, X.; DeCastro, M.L.; Cermakova, L.; Sadananda, S.; Jackson, L.M.; Azizi, H.; Mancini, G.B.J.; Francis, G.A.; Frohlich, J.; et al. Risk of premature atherosclerotic disease in patients with monogenic versus polygenic familial hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 512–522. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Zekavat, S.M.; Collins, R.L.; Roselli, C.; Natarajan, P.; Lichtman, J.H.; D’Onofrio, G.; Matera, J.; Dreyer, R.; et al. Whole-genome sequencing to characterize monogenic and polygenic contributions in patients hospitalized with early-onset myocardial infarction. Circulation 2019, 139, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef] [Green Version]
- Hubacek, J.A.; Bobkova, D. Role of cholesterol 7alpha-hydroxylase (CYP7A1) in nutrigenetics and pharmacogenetics of cholesterol lowering. Mol. Diagn. Ther. 2006, 10, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hubacek, J.A.; Pitha, J.; Skodová, Z.; Poledne, R.; Lánská, V.; Waterworth, D.M.; Humphries, S.E.; Talmud, P.J. Czech MONICA Study. Polymorphisms in CYP-7A1, not APOE, influence the change in plasma lipids in response to population dietary change in an 8 year follow-up; results from the Czech MONICA study. Clin. Biochem. 2003, 36, 263–267. [Google Scholar] [CrossRef]
- Kovár, J.; Suchánek, P.; Hubácek, J.A.; Poledne, R. The A-204C polymorphism in the cholesterol 7alpha-hydroxylase (CYP7A1) gene determines the cholesterolemia responsiveness to a high-fat diet. Physiol. Res. 2004, 53, 565–568. [Google Scholar] [PubMed]
- MacKay, D.S.; Eck, P.K.; Gebauer, S.K.; Baer, D.J.; Jones, P.J. CYP7A1-rs3808607 and APOE isoform associate with LDL cholesterol lowering after plant sterol consumption in a randomized clinical trial. Am. J. Clin. Nutr. 2015, 102, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Hofman, M.K.; Weggemans, R.M.; Zock, P.L.; Schouten, E.G.; Katan, M.B.; Princen, H.M. CYP7A1 A-278C polymorphism affects the response of plasma lipids after dietary cholesterol or cafestol interventions in humans. J. Nutr. 2004, 134, 2200–2204. [Google Scholar] [CrossRef] [Green Version]
- Merritt, D.C.; Jamnik, J.; El-Sohemy, A. FTO genotype, dietary protein intake, and body weight in a multiethnic population of young adults: A cross-sectional study. Genes Nutr. 2018, 13, 4. [Google Scholar] [CrossRef]
- Qi, Q.; Downer, M.K.; Kilpeläinen, T.O.; Taal, H.R.; Barton, S.J.; Ntalla, I.; Standl, M.; Boraska, V.; Huikari, V.; Kiefte-de Jong, J.C.; et al. Dietary intake, FTO genetic variants, and adiposity: A combined analysis of over 16,000 children and adolescents. Diabetes 2015, 64, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Kim, J. Are genes destiny? Exploring the role of intrauterine environment in moderating genetic influences on body mass. Am. J. Hum. Biol. 2020, 32, e23354. [Google Scholar] [CrossRef]
- Holmes, M.V.; Newcombe, P.; Hubacek, J.A.; Sofat, R.; Ricketts, S.L.; Cooper, J.; Breteler, M.M.; Bautista, L.E.; Sharma, P.; Whittaker, J.C.; et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: A meta-analysis of genetic studies and randomised trials. Lancet 2011, 378, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, Y.; Huang, T.; Cheng, H.L.; Campos, H.; Qi, L. Sugar-sweetened beverage intake, chromosome 9p21 variants, and risk of myocardial infarction in Hispanics. Am. J. Clin. Nutr. 2016, 103, 1179–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Romero, A.C.; Navas-Carrillo, D.; Marín, F.; Orenes-Piñero, E. The future of nutrition: Nutrigenomics and nutrigenetics in obesity and cardiovascular diseases. Crit. Rev. Food Sci. Nutr. 2018, 58, 3030–3041. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Annunziata, G.; Bordoni, L.; Muscogiuri, G.; Colao, A.; Savastano, S.; Obesity Programs of Nutrition, Education, Research and Assessment (OPERA) Group. Nutrigenetics-personalized nutrition in obesity and cardiovascular diseases. Int. J. Obes. Suppl. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mullins, V.A.; Bresette, W.; Johnstone, L.; Hallmark, B.; Chilton, F.H. Genomics in personalized nutrition: Can you “Eat for your genes”? Nutrients 2020, 12, 3118. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Pikhart, H.; Peasey, A.; Malyutina, S.; Pajak, A.; Tamosiunas, A.; Voevoda, M.; Holmes, M.V.; Bobak, M. The association between the FTO gene variant and alcohol consumption and binge and problem drinking in different gene-environment background: The HAPIEE study. Gene 2019, 707, 30–35. [Google Scholar] [CrossRef]
- Johnson, J.A.; Cavallari, L.H. Pharmacogenetics and cardiovascular disease—Implications for personalized medicine. Pharmacol. Rev. 2013, 65, 987–1009. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez Vicente, A.E.; Herrero Cervera, M.J.; Bernal, M.L.; Rojas, L.; Peiró, A.M. Personalized medicine into health national services: Barriers and potentialities. Drug Metab. Pers. Ther. 2018, 33, 159–163. [Google Scholar] [CrossRef]
- Vrablik, M.; Zlatohlavek, L.; Stulc, T.; Adamkova, V.; Prusikova, M.; Schwarzova, L.; Hubacek, J.A.; Ceska, R. Statin-associated myopathy: From genetic predisposition to clinical management. Physiol. Res. 2014, 63 (Suppl. 3), S327–S334. [Google Scholar] [CrossRef]
- Canestaro, W.J.; Austin, M.A.; Thummel, K.E. Genetic factors affecting statin concentrations and subsequent myopathy: A HuGENet systematic review. Genet. Med. 2014, 16, 810–819. [Google Scholar] [CrossRef] [Green Version]
- Neřoldová, M.; Stránecký, V.; Hodaňová, K.; Hartmannová, H.; Piherová, L.; Přistoupilová, A.; Mrázová, L.; Vrablík, M.; Adámková, V.; Hubáček, J.A.; et al. Rare variants in known and novel candidate genes predisposing to statin-associated myopathy. Pharmacogenomics 2016, 17, 1405–1414. [Google Scholar] [CrossRef]
- SEARCH Collaborative Group; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar]
- Voora, D.; Shah, S.H.; Spasojevic, I.; Ali, S.; Reed, C.R.; Salisbury, B.A.; Ginsburg, G.S. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 2009, 54, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunham, L.R.; Lansberg, P.J.; Zhang, L.; Miao, F.; Carter, C.; Hovingh, G.K.; Visscher, H.; Jukema, J.W.; Stalenhoef, A.F.; Ross, C.J.; et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenom. J. 2012, 12, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Hubáček, J.A.; Dlouhá, D.; Adámková, V.; Zlatohlavek, L.; Viklický, O.; Hrubá, P.; Češka, R.; Vrablík, M. SLCO1B1 polymorphism is not associated with risk of statin-induced myalgia/myopathy in a Czech population. Med. Sci. Monit. 2015, 21, 1454–1459. [Google Scholar] [PubMed] [Green Version]
- Swen, J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.; Mulder, H.; Rongen, G.; van Schaik, R.; Schalekamp, T.; Touw, D.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Abi Khalil, C. The emerging role of epigenetics in cardiovascular disease. Ther. Adv. Chronic. Dis. 2014, 5, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Fang, F. Histone methylation and transcriptional regulation in cardiovascular disease. Cardiovasc Hematol. Disord. Drug Targets 2014, 14, 89–97. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Obsteter, J.; Dovc, P.; Kunej, T. Genetic variability of microRNA regulome in human. Mol. Genet. Genomic Med. 2015, 3, 30–39. [Google Scholar] [CrossRef]
- Dlouhá, D.; Hubáček, J.A. Regulatory RNAs and cardiovascular disease—With a special focus on circulating microRNAs. Physiol. Res. 2017, 66 (Suppl. 1), S21–S38. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, X.; Lu, Y.; Yang, B. MiRNAs at the heart of the matter. J. Mol. Med. 2008, 86, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Laffont, B.; Rayner, K.J. Micrornas in the pathobiology and therapy of atherosclerosis. Can. J. Cardiol. 2017, 33, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gonzalo-Calvo, D.; Iglesias-Gutiérrez, E.; Llorente-Cortés, V. Epigenetic biomarkers and cardiovascular disease: Circulating microRnas. Rev. Esp. Cardiol. 2017, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Dong, Y.H.; Du, W.; Shi, C.Y.; Wang, K.; Tariq, M.A.; Wang, J.X.; Li, P.F. The role of microRnas in myocardial infarction: From molecular mechanism to clinical application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Economou, E.K.; Oikonomou, E.; Siasos, G.; Papageorgiou, N.; Tsalamandris, S.; Mourouzis, K.; Papaioanou, S.; Tousoulis, D. The role of micrornas in coronary artery disease: From pathophysiology to diagnosis and treatment. Atherosclerosis 2015, 241, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Wang, H.; Jia, C.; Zhu, S.; Chu, X.; Ma, Q.; Wei, J.; Chen, E.; Zhu, W.; Macon, C.J.; et al. MicroRna-146a induces lineage-negative bone marrow cell apoptosis and senescence by targeting polo-like kinase 2 expression. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRna 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, Y.; Wang, C.; Manes, T.D.; Pober, J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of e-selectin and intercellular adhesion molecule-1 on human endothelial cells: Feedback control of inflammation. J. Immunol. 2010, 184, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Asgeirsdottir, S.A.; Van Solingen, C.; Murniati, N.F.; Zwiers, P.J.; Heeringa, P.; Van Meurs, M.; Satchell, S.C.; Saleem, M.A.; Mathieson, P.W.; Banas, B.; et al. MicroRna-126 contributes to renal macrovascular heterogeneity of vcam-1 protein expression in acute inflammation. Am. J. Physiol. Renal. Physiol. 2012, 302, F1630–F1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Williams, D.; Sur, S.; Wang, J.Y.; Jo, H. Role of flow-sensitive microRNAs and long noncoding RNAs in vascular dysfunction and atherosclerosis. Vascul. Pharmacol. 2019, 114, 76–92. [Google Scholar] [CrossRef]
- Zampetaki, A.; Dudek, K.; Mayr, M. Oxidative Stress in Atherosclerosis: The role of microRnas in arterial remodeling. Free Radic. Biol. Med. 2013, 64, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Thavarajah, T.; Gu, W.; Cai, J.; Xu, Q. Impact of miRna in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, E159–E170. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.S.; Hu, G.Q.; Lin, B.; Lin, Z.Y.; Sun, C.C. MicroRNA-155 silencing enhances inflammatory response and lipid uptake in oxidized low-density lipoprotein-stimulated human THP-1 macrophages. J. Investig. Med. 2010, 58, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Huang, Z.; Wang, L.; Wang, Y.; Wu, F.; Meng, S.; Wang, C. MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc. Res. 2009, 83, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Hernando, C.; Suárez, Y.; Rayner, K.J.; Moore, K.J. MicroRnas in lipid metabolism. Curr. Opin. Lipidol. 2011, 22, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Rotllan, N.; Price, N.; Pati, P.; Goedeke, L.; Fernández-Hernando, C. MicroRNAs in lipoprotein metabolism and cardiometabolic disorders. Atherosclerosis 2016, 246, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Dlouha, D.; Blaha, M.; Blaha, V.; Fatorova, I.; Hubacek, J.A.; Stavek, P.; Lanska, V.; Parikova, A.; Pitha, J. Analysis of circulating miRNAs in patients with familial hypercholesterolaemia treated by LDL/Lp(a) apheresis. Atheroscler. Suppl. 2017, 30, 128–134. [Google Scholar] [CrossRef]
- Zampetaki, A.; Willeit, P.; Drozdov, I.; Kiechl, S.; Mayr, M. Profiling of circulating microRNAs: From single biomarkers to re-wired networks. Cardiovasc Res. 2012, 93, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The impact of epigenetics on cardiovascular disease. Biochem. Cell. Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol. 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [Green Version]
- Sae-Lee, C.; Corsi, S.; Barrow, T.M.; Kuhnle, G.G.C.; Bollati, V.; Mathers, J.C.; Byun, H.M. Dietary intervention modifies DNA methylation age assessed by the epigenetic clock. Mol. Nutr. Food Res. 2018, 62, e1800092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, G.; Mendelson, M.M.; Ward-Caviness, C.K.; Joehanes, R.; Huan, T.; Gondalia, R.; Salfati, E.; Brody, J.A.; Fiorito, G.; Bressler, J.; et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation 2019, 140, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Talens, R.P.; Jukema, J.W.; Trompet, S.; Kremer, D.; Westendorp, R.G.; Lumey, L.H.; Sattar, N.; Putter, H.; Slagboom, P.E.; Heijmans, B.T.; et al. Hypermethylation at loci sensitive to the prenatal environment is associated with increased incidence of myocardial infarction. Int. J. Epidemiol. 2012, 41, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, L.; Wahl, S.; Pilling, L.C.; Reischl, E.; Sandling, J.K.; Kunze, S.; Holdt, L.M.; Kretschmer, A.; Schramm, K.; Adamski, J.; et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ. Cardiovasc. Genet. 2015, 8, 334–342. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, I.; Kleber, M.; Delgado, G.; Herrmann, W.; März, W.; Herrmann, M. Telomere length and mortality in the Ludwigshafen Risk and Cardiovascular Health study. PLoS ONE 2018, 13, e0198373. [Google Scholar] [CrossRef] [Green Version]
- Arbeev, K.G.; Verhulst, S.; Steenstrup, T.; Kark, J.D.; Bagley, O.; Kooperberg, C.; Reiner, A.P.; Hwang, S.J.; Levy, D.; Fitzpatrick, A.L.; et al. Association of leukocyte telomere length with mortality among adult participants in 3 longitudinal studies. JAMA Netw. Open 2020, 3, e200023. [Google Scholar] [CrossRef] [Green Version]
- Mundstock, E.; Sarria, E.E.; Zatti, H.; Mattos Louzada, F.; Kich Grun, L.; Herbert Jones, M.; Guma, F.T.; Mazzola, I.M.J.; Epifanio, M.; Stein, R.T.; et al. Effect of obesity on telomere length: Systematic review and meta-analysis. Obesity 2015, 23, 2165–2174. [Google Scholar] [CrossRef]
- Tellechea, M.L.; Pirola, C.J. The impact of hypertension on leukocyte telomere length: A systematic review and meta-analysis of human studies. J. Hum. Hypertens. 2017, 31, 99–105. [Google Scholar] [CrossRef]
- Koriath, M.; Müller, C.; Pfeiffer, N.; Nickels, S.; Beutel, M.; Schmidtmann, I.; Rapp, S.; Münzel, T.; Westermann, D.; Karakas, M.; et al. Relative telomere length and cardiovascular risk factors. Biomolecules 2019, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhang, M.; Niu, W.; Yang, R.; Zhang, Y.; Qiu, Z.; Sun, B.; Zhao, Z. Analysis of DNA methylation in various swine tissues. PLoS ONE 2011, 6, e16229. [Google Scholar] [CrossRef]
- Dlouha, D.; Maluskova, J.; Kralova Lesna, I.; Lanska, V.; Hubacek, J.A. Comparison of the relative telomere length measured in leukocytes and eleven different human tissues. Physiol. Res. 2014, 63 (Suppl. 3), S343–S350. [Google Scholar] [CrossRef]
- Jiang, R.; Jones, M.J.; Chen, E.; Neumann, S.M.; Fraser, H.B.; Miller, G.E.; Kobor, M.S. Discordance of DNA methylation variance between two accessible human tissues. Sci. Rep. 2015, 5, 8257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrella, M.M.; Parekh, R.S. The expanding role of APOL1 risk in chronic kidney disease and cardiovascular disease. Semin. Nephrol. 2017, 37, 520–529. [Google Scholar] [CrossRef]
- Vanhamme, L.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Nolan, D.P.; Lins, L.; Van Den Abbeele, J.; Pays, A.; Tebabi, P.; Van Xong, H.; Jacquet, A.; et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 2003, 422, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Hubacek, J.A. Apolipoprotein A5 fifteen years anniversary: Lessons from genetic epidemiology. Gene 2016, 592, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Hubáček, J.A.; Šedová, L.; Olišarová, V.; Adámková, V.; Tóthová, V. Different prevalence of T2DM risk alleles in Roma population in comparison with the majority Czech population. Mol. Genet. Genom. Med. 2020, 8, e1361. [Google Scholar] [CrossRef]
- Werissa, N.A.; Piko, P.; Fiatal, S.; Kosa, Z.; Sandor, J.; Adany, R. SNP-based genetic risk score modeling suggests no increased genetic susceptibility of the Roma population to type 2 diabetes mellitus. Genes 2019, 10, 942. [Google Scholar] [CrossRef] [Green Version]
- Dick, D.M.; Bierut, L.J. The genetics of alcohol dependence. Curr. Psychiatry Rep. 2006, 8, 151–157. [Google Scholar] [CrossRef]
- Borinskaya, S.; Kal’ina, N.; Marusin, A.; Faskhutdinova, G.; Morozova, I.; Kutuev, I.; Koshechkin, V.; Khusnutdinova, E.; Stepanov, V.; Puzyrev, V.; et al. Distribution of the alcohol dehydrogenase ADH1B*47His allele in Eurasia. Am. J. Hum. Genet. 2009, 84, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.N.; Williams, R.R. A survey of 246 suggested coronary risk factors. Atherosclerosis 1981, 40, 1–52. [Google Scholar] [CrossRef]
- Authors/Task Force Members; ESC Committee for Practice Guidelines (CPG); ESC National Cardiac Societies. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205, Erratum in: Atherosclerosis 2020, 292, 160–162. Erratum in: Atherosclerosis 2020, 294, 80–82. [Google Scholar]
- Graziano, F.; Biino, G.; Bonati, M.T.; Neale, B.M.; Do, R.; Concas, M.P.; Vaccargiu, S.; Pirastu, M.; Terradura-Vagnarelli, O.; Cirillo, M.; et al. Estimation of metabolic syndrome heritability in three large populations including full pedigree and genomic information. Hum. Genet. 2019, 138, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Taron, M.; Llerena, A.; Manolopoulos, V.G.; Rodriguez-Antona, C.; Stankovic, S.; van Schaik, R.H.N. The need of the clinical implementation of pharmacogenetics in European health services for routine drug prescription. What’s next? An urgent clinical unmet need for patients. Drug Metab. Pers. Ther. 2020, in press. [Google Scholar] [CrossRef]
- Merched, A.J.; Chan, L. Nutrigenetics and nutrigenomics of atherosclerosis. Curr. Atheroscler. Rep. 2013, 15, 328. [Google Scholar] [CrossRef] [Green Version]
- Lovegrove, J.A.; Gitau, R. Personalized nutrition for the prevention of cardiovascular disease: A future perspective. J. Hum. Nutr. Diet. 2008, 21, 306–316. [Google Scholar] [CrossRef]
- Di Renzo, L.; Gualtieri, P.; Romano, L.; Marrone, G.; Noce, A.; Pujia, A.; Perrone, M.A.; Aiello, V.; Colica, C.; De Lorenzo, A. Role of personalized nutrition in chronic-degenerative diseases. Nutrients 2019, 11, 1707. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrablik, M.; Dlouha, D.; Todorovova, V.; Stefler, D.; Hubacek, J.A. Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management? Int. J. Mol. Sci. 2021, 22, 4182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084182
Vrablik M, Dlouha D, Todorovova V, Stefler D, Hubacek JA. Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management? International Journal of Molecular Sciences. 2021; 22(8):4182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084182
Chicago/Turabian StyleVrablik, Michal, Dana Dlouha, Veronika Todorovova, Denes Stefler, and Jaroslav A. Hubacek. 2021. "Genetics of Cardiovascular Disease: How Far Are We from Personalized CVD Risk Prediction and Management?" International Journal of Molecular Sciences 22, no. 8: 4182. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084182