
Harnessing the Endogenous Plasticity of Pancreatic Islets: A Feasible Regenerative Medicine Therapy for Diabetes?

 ,
, 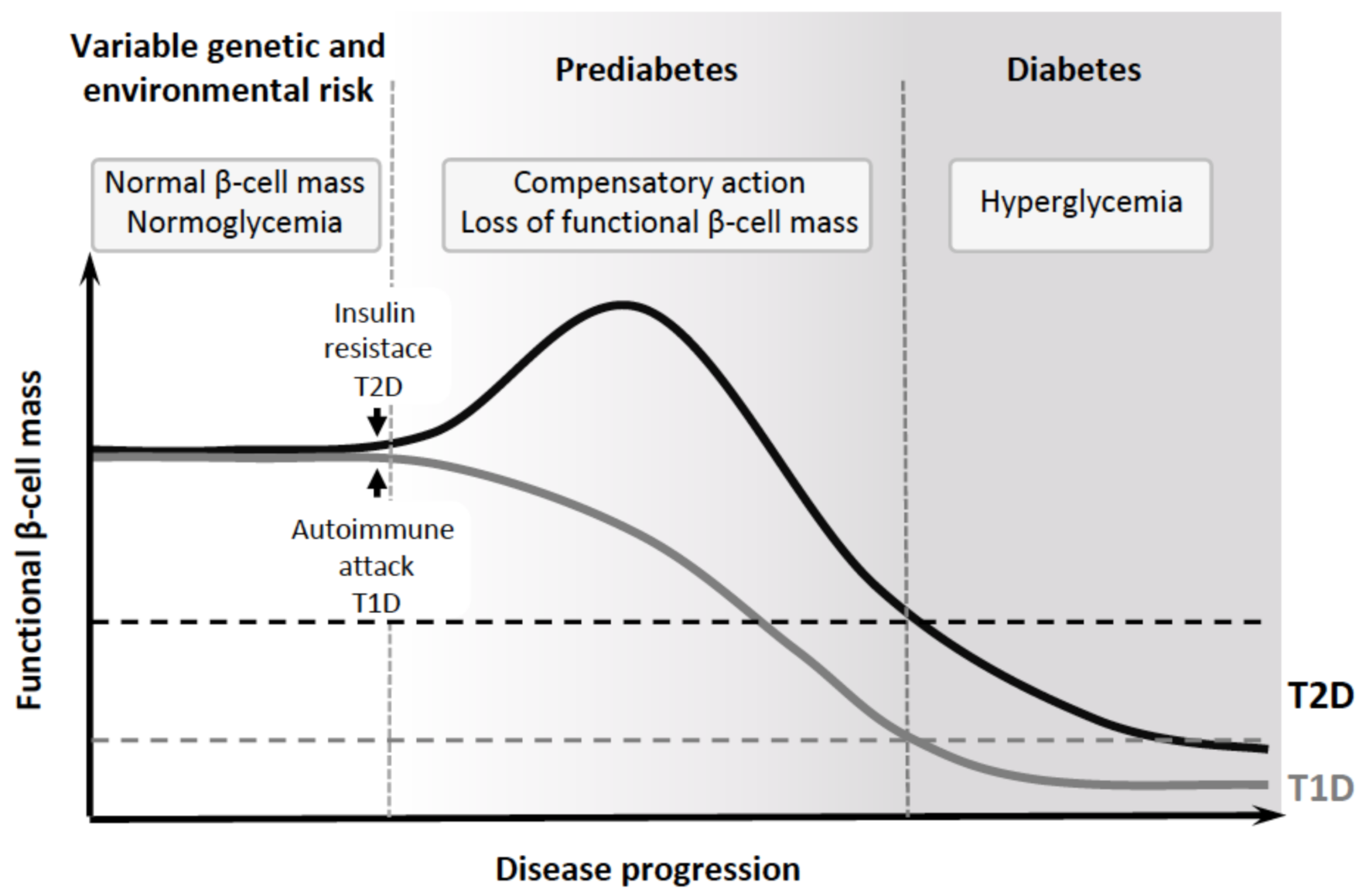{kind=link}
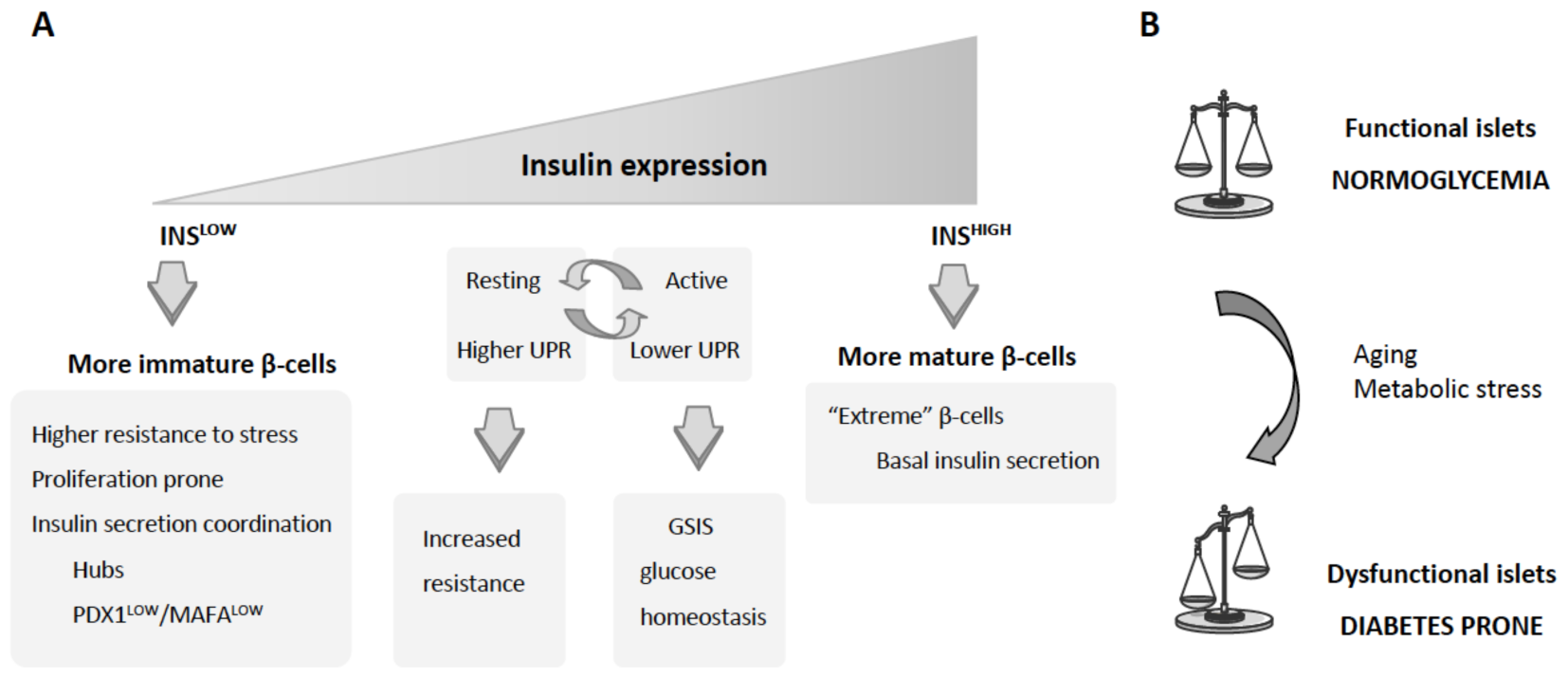{kind=link}
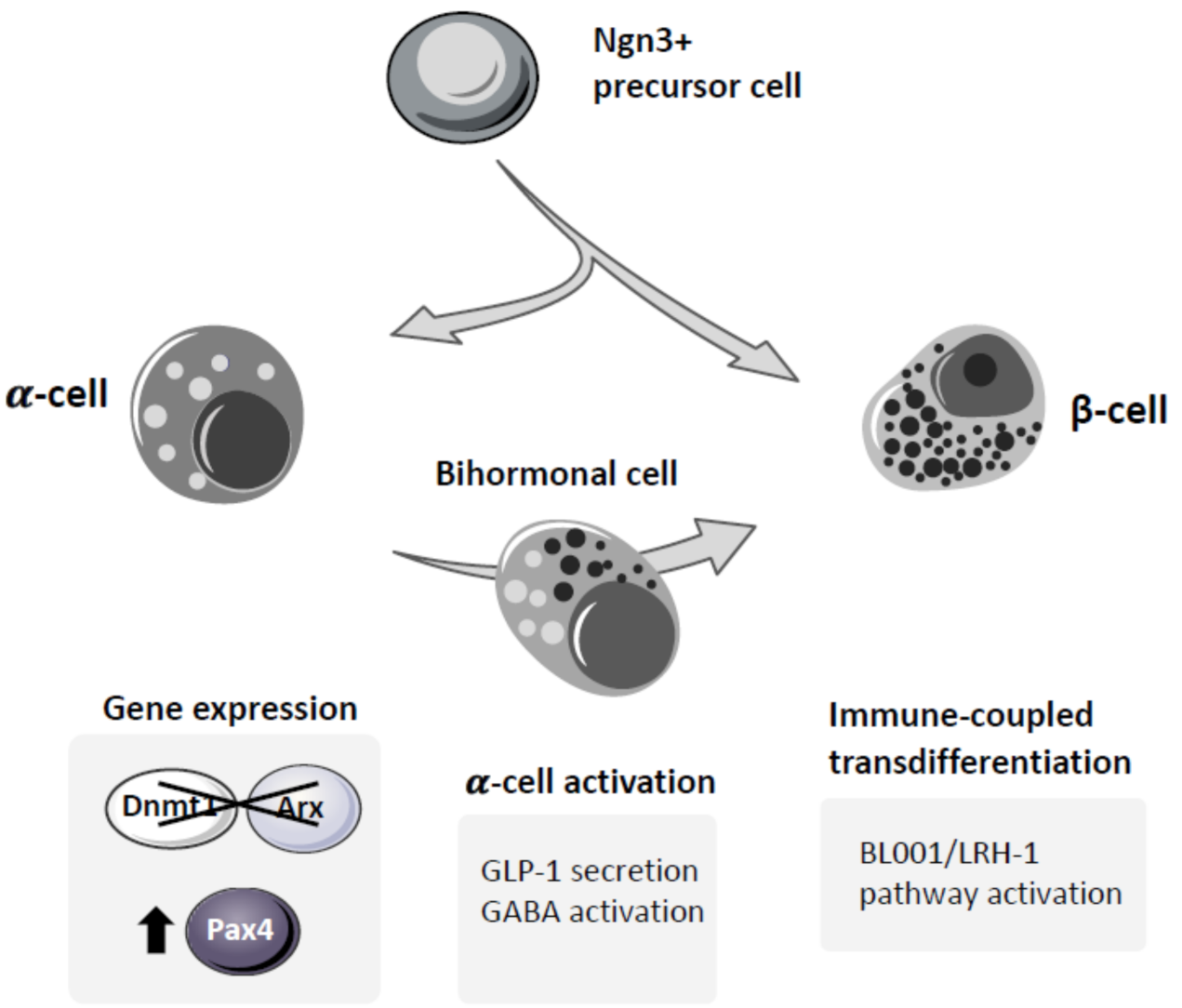{kind=link}
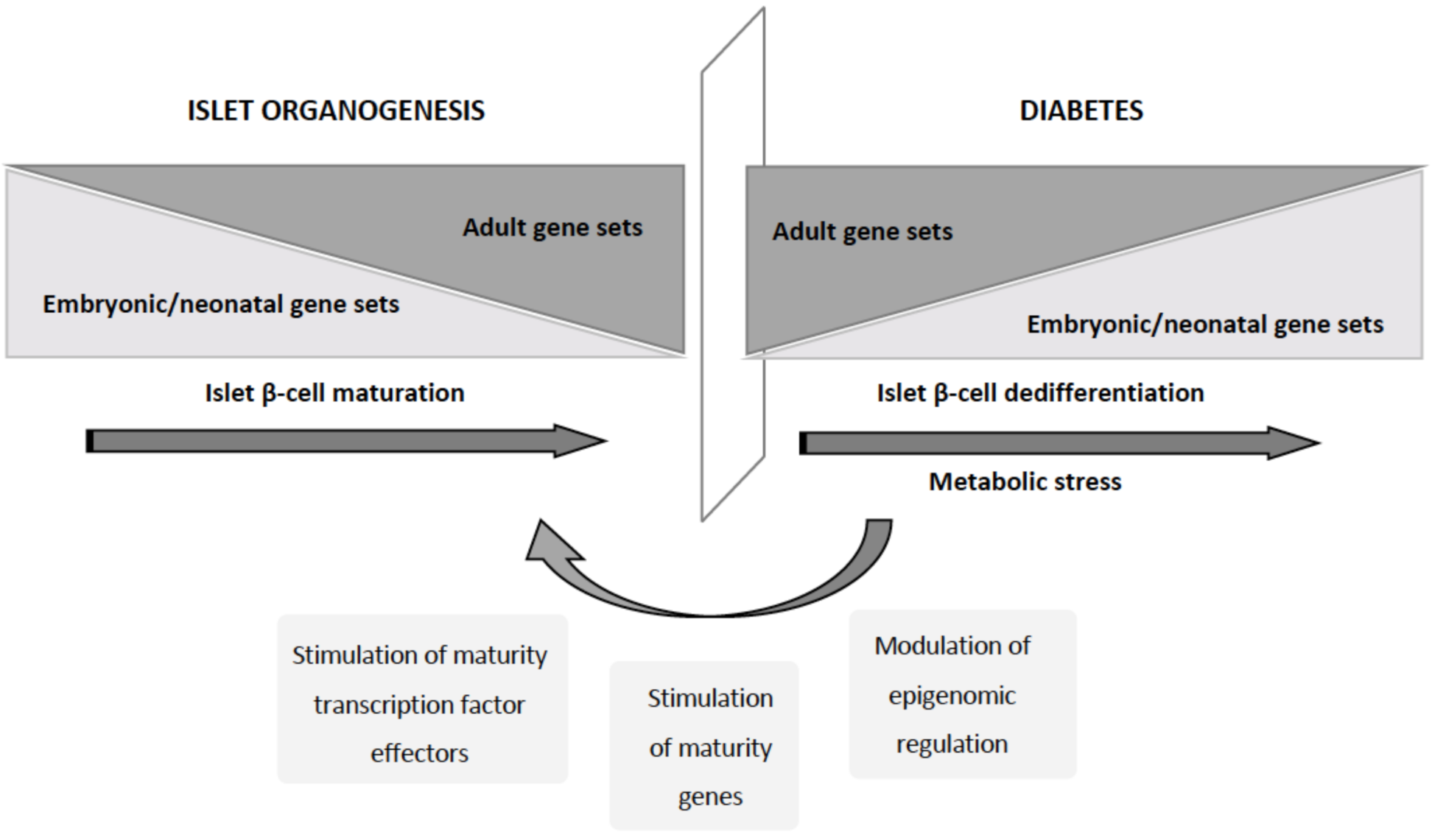{kind=link}
Abstract
:1. Introduction
The Pancreatic Islet
2. Heterogeneity within β-Cell Population of the Islets Can Modulate Islet Plasticity
2.1. β-Cell Heterogeneity in Adult Islets
2.2. Functional Implications of β-Cell Heterogeneity
2.3. Maintenance of β-Cell Heterogeneity
2.4. PAX4 in β-Cell Heterogeneity
2.5. Future Directions in β-Cell Heterogeneity Studies
3. Transdifferentiation of Pancreatic α-Cells into β-Cells
3.1. ⍺-Cells as Source for New β-Cells
3.2. ⍺- to β-Cell Transdifferentiation
3.2.1. GLP-1 Stimulates ⍺- to β-Cell Transdifferentiation
3.2.2. Can GABA Stimulate ⍺- to β-Cell Transdifferentiation?
3.2.3. Immune-Coupled ⍺- to β-Cell Transdifferentiation: Role of LRH-1
4. Redifferentiation of β-Cells
4.1. Dedifferentiation of β-Cells during Diabetes
4.2. Redifferentiation of Dedifferentiatiated β-Cells: Mimicking Organogenesis
4.3. HMG20A Role in Functional Maturation of β-Cells
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matveyenko, A.V.; Butler, P.C. Relationship between beta-cell mass and diabetes onset. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 23–31. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.J.; Breuer, T.G.; Bonadonna, R.C.; Tannapfel, A.; Uhl, W.; Schmidt, W.E.; Schrader, H.; Menge, B.A. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia 2012, 55, 1346–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 (Suppl. 3), S16–S21. [Google Scholar] [CrossRef] [Green Version]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas organogenesis: From lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef] [Green Version]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Li, C.; Zhang, J.; Schug, J.; Avrahami, R.; Rao, S.; Stadler, M.B.; Burger, L.; Schubeler, D.; Glaser, B.; et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab. 2015, 22, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Arda, H.E.; Li, L.; Tsai, J.; Torre, E.A.; Rosli, Y.; Peiris, H.; Spitale, R.C.; Dai, C.; Gu, X.; Qu, K.; et al. Age-Dependent pancreatic gene regulation reveals mechanisms governing human beta cell function. Cell Metab. 2016, 23, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of beta cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Van Haaren, M.; Mruk, M.; Lee, T.B., Jr.; Crawford, C.; Hollister-Lock, J.; Sullivan, B.A.; Johnson, J.W.; Ebrahimi, A.; Dreyfuss, J.M.; et al. β cell aging markers have heterogeneous distribution and are induced by insulin resistance. Cell Metab. 2017, 25, 898–910.e895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, P.I.; Fuente-Martin, E.; Brun, T.; Cobo-Vuilleumier, N.; Jimenez-Moreno, C.M.; Herrera Gomez, I.G.; Lopez Noriega, L.; Mellado-Gil, J.M.; Martin-Montalvo, A.; Soria, B.; et al. PAX4 defines an expandable beta-cell subpopulation in the adult pancreatic islet. Sci. Rep. 2015, 5, 15672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef]
- Roscioni, S.S.; Migliorini, A.; Gegg, M.; Lickert, H. Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016, 12, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Katsuta, H.; Akashi, T.; Katsuta, R.; Nagaya, M.; Kim, D.; Arinobu, Y.; Hara, M.; Bonner-Weir, S.; Sharma, A.J.; Akashi, K.; et al. Single pancreatic beta cells co-express multiple islet hormone genes in mice. Diabetologia 2010, 53, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuta, H.; Aguayo-Mazzucato, C.; Katsuta, R.; Akashi, T.; Hollister-Lock, J.; Sharma, A.J.; Bonner-Weir, S.; Weir, G.C. Subpopulations of GFP-marked mouse pancreatic β-cells differ in size, granularity, and insulin secretion. Endocrinology 2012, 153, 5180–5187. [Google Scholar] [CrossRef] [Green Version]
- Szabat, M.; Luciani, D.S.; Piret, J.M.; Johnson, J.D. Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology 2009, 150, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Szabat, M.; Lynn, F.C.; Hoffman, B.G.; Kieffer, T.J.; Allan, D.W.; Johnson, J.D. Maintenance of β-cell maturity and plasticity in the adult pancreas: Developmental biology concepts in adult physiology. Diabetes 2012, 61, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Van Der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dolleman, S.; Liu, S.; Ackermann, A.M.; Caceres, E.; Hunter, A.E.; et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metab. 2017, 25, 911–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Zhang, J.; Saravanakumar, S.; Flisher, M.F.; Grimm, D.R.; Van Der Meulen, T.; Huising, M.O. Virgin beta cells at the neogenic niche proliferate normally and mature slowly. Diabetes 2021, db200679. [Google Scholar] [CrossRef]
- Camunas-Soler, J.; Dai, X.Q.; Hang, Y.; Bautista, A.; Lyon, J.; Suzuki, K.; Kim, S.K.; Quake, S.R.; MacDonald, P.E. Patch-Seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab. 2020, 31, 1017–1031. [Google Scholar] [CrossRef]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Mousavy Gharavy, S.N.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader β-cells coordinate Ca2+ dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef] [Green Version]
- Nasteska, D.; Fine, N.H.F.; Ashford, F.B.; Cuozzo, F.; Viloria, K.; Smith, G.; Dahir, A.; Dawson, P.W.J.; Lai, Y.C.; Bastidas-Ponce, A.; et al. PDX1(LOW) MAFA(LOW) β-cells contribute to islet function and insulin release. Nat. Commun. 2021, 12, 674. [Google Scholar] [CrossRef]
- Farack, L.; Golan, M.; Egozi, A.; Dezorella, N.; Bahar Halpern, K.; Ben-Moshe, S.; Garzilli, I.; Toth, B.; Roitman, L.; Krizhanovsky, V.; et al. Transcriptional heterogeneity of beta cells in the intact pancreas. Dev. Cell 2019, 48, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Y.; Dominguez Gutierrez, G.; Okamoto, H.; Kim, J.; Lee, A.H.; Adler, C.; Ni, M.; Yancopoulos, G.D.; Murphy, A.J.; Gromada, J. Pseudotime ordering of single human β-cells reveals states of insulin production and unfolded protein response. Diabetes 2018, 67, 1783–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.B.; O’Donnell, A.C.; Stamateris, R.E.; Ha, B.; McCloskey, K.M.; Reynolds, P.R.; Arvan, P.; Alonso, L.C. Insulin demand regulates β cell number via the unfolded protein response. J. Clin. Investig. 2015, 125, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Qiu, W.L.; Yu, X.X.; Zhang, Y.; He, M.Y.; Li, L.C.; Yang, L.; Zhang, W.; Franti, M.; Ye, J.; et al. Characterizing pancreatic β-cell heterogeneity in the streptozotocin model by single-cell transcriptomic analysis. Mol. Metab. 2020, 37, 100982. [Google Scholar] [CrossRef]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef]
- Brun, T.; Franklin, I.; St-Onge, L.; Biason-Lauber, A.; Schoenle, E.; Wollheim, C.B.; Gauthier, B.R. The diabetes-linked transcription factor Pax4 promotes β-cell proliferation and survival in rat and human islets. J. Cell Biol. 2004, 167, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Hecksher-Sorensen, J.; Broccoli, V.; Krull, J.; Ponte, I.; Mundiger, T.; Smith, J.; Gruss, P.; Serup, P.; Mansouri, A. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the ⍺-and β-cell lineages in the mouse endocrine pancreas. Development 2005, 132, 2969–2980. [Google Scholar] [CrossRef] [Green Version]
- Brun, T.; Duhamel, D.L.; Hu He, K.H.; Wollheim, C.B.; Gauthier, B.R. The transcription factor Pax4 acts as a survival gene in the insulinoma INS1E cells. Oncogene 2007, 26, 4261–4271. [Google Scholar] [CrossRef]
- Hu He, K.; Lorenzo, P.I.; Brun, T.; Jimenez Moreno, C.M.; Aeberhard, D.; Ortega, J.V.; Cornu, M.; Thorel, F.; Gjinovci, A.; Thorens, B.; et al. In vivo conditional Pax4 overexpression in mature islet β-cells prevents stress-induced hyperglycemia in mice. Diabetes 2011, 60, 1705–1715. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montalvo, A.; Lorenzo, P.I.; Lopez-Noriega, L.; Gauthier, B.R. Targeting pancreatic expressed PAX genes for the treatment of diabetes mellitus and pancreatic neuroendocrine tumors. Expert Opin. Ther. Targets 2017, 21, 77–89. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Juarez-Vicente, F.; Cobo-Vuilleumier, N.; Garcia-Dominguez, M.; Gauthier, B.R. The diabetes-linked transcription factor PAX4: From gene to functional consequences. Genes 2017, 8, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Elghazi, L.; Parker, S.E.; Kizilocak, H.; Asano, M.; Sussel, L.; Sosa-Pineda, B. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev. Biol. 2004, 266, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, A.L.; Li, S.; Jones, K.; Melton, D.A. Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech. Dev. 2007, 124, 97–107. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, W.; Jiang, W.; Sun, X.; Han, Y.; Ding, M.; Shi, Y.; Deng, H. P21cip-Overexpression in the mouse beta cells leads to the improved recovery from streptozotocin-induced diabetes. PLoS ONE 2009, 4, e8344. [Google Scholar] [CrossRef]
- Mellado-Gil, J.M.; Jimenez-Moreno, C.M.; Martin-Montalvo, A.; Alvarez-Mercado, A.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Lorenzo, P.I.; Bru-Tari, E.; De Gracia Herrera-Gomez, I.; Lopez-Noriega, L.; et al. PAX4 preserves endoplasmic reticulum integrity preventing beta cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia 2016, 59, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, P.I.; Cobo-Vuilleumier, N.; Gauthier, B. Therapeutic potential of pancreatic PAX4-regulated pathways in treating diabetes mellitus. Curr. Opin. Pharmacol. 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pipeleers, D.; In’t Veld, P.I.; Maes, E.; Van De Winkel, M. Glucose-Induced insulin release depends on functional cooperation between islet cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7322–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtusciszyn, A.; Armanet, M.; Morel, P.; Berney, T.; Bosco, D. Insulin secretion from human beta cells is heterogeneous and dependent on cell-to-cell contacts. Diabetologia 2008, 51, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, D.C.; Brissova, M.; Phillips, N.; Shrestha, S.; Walker, J.T.; Aramandla, R.; Poffenberger, G.; Flaherty, D.K.; Weller, K.P.; Pelletier, J.; et al. Ectonucleoside triphosphate diphosphohydrolase-3 antibody targets adult human pancreatic beta cells for in vitro and in vivo analysis. Cell Metab. 2019, 29, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthault, C.; Staels, W.; Scharfmann, R. Purification of pancreatic endocrine subsets reveals increased iron metabolism in beta cells. Mol. Metab. 2020, 42, 101060. [Google Scholar] [CrossRef]
- Nasteska, D.; Hodson, D.J. The role of beta cell heterogeneity in islet function and insulin release. J. Mol. Endocrinol. 2018, 61, R43–R60. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Martin, M.; Spenle, C.; Poulet, M.; Collin, C.; Gradwohl, G. Competence of failed endocrine progenitors to give rise to acinar but not ductal cells is restricted to early pancreas development. Dev. Biol. 2012, 361, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Lee, C.; Choung, J.S.; Jung, H.S.; Jun, H.S. Glucagon-Like peptide 1 increases β-cell regeneration by promoting ⍺-to β-cell transdifferentiation. Diabetes 2018, 67, 2601–2614. [Google Scholar] [CrossRef] [Green Version]
- Haedersdal, S.; Lund, A.; Knop, F.K.; Vilsboll, T. The role of glucagon in the pathophysiology and treatment of type 2 diabetes. Mayo Clin. Proc. 2018, 93, 217–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, K.; Chera, S.; Van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Pineros, A.R.; Gao, H.; Wu, W.; Liu, Y.; Tersey, S.A.; Mirmira, R.G. Single-Cell transcriptional profiling of mouse islets following short-term obesogenic dietary intervention. Metabolites 2020, 10, 513. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Q.; Camunas-Soler, J.; Briant, L.J.B.; Dos Santos, T.; Spigelman, A.F.; Walker, E.M.; Arrojo e Drigo, R.; Bautista, A.; Jones, R.C.; Lyon, J.; et al. Heterogenous impairment of α-cell function in type 2 diabetes is linked to cell maturation state. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ghazvini Zadeh, E.H.; Huang, Z.; Xia, J.; Li, D.; Davidson, H.W.; Li, W.H. ZIGIR, a granule-specific Zn(2+) indicator, reveals human islet ⍺ cell heterogeneity. Cell Rep. 2020, 32, 107904. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Rupnik, M.; Gaisano, H.Y. Unperturbed islet α-cell function examined in mouse pancreas tissue slices. J. Physiol. 2011, 589, 395–408. [Google Scholar] [CrossRef]
- Collombat, P.; Mansouri, A.; Hecksher-Sorensen, J.; Serup, P.; Krull, J.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and subsequently β cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Al-Hasani, K.; Pfeifer, A.; Courtney, M.; Ben-Othman, N.; Gjernes, E.; Vieira, A.; Druelle, N.; Avolio, F.; Ravassard, P.; Leuckx, G.; et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 2013, 26, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Druelle, N.; Vieira, A.; Shabro, A.; Courtney, M.; Mondin, M.; Rekima, S.; Napolitano, T.; Silvano, S.; Navarro-Sanz, S.; Hadzic, B.; et al. Ectopic expression of Pax4 in pancreatic δ cells results in β-like cell neogenesis. J. Cell. Biol. 2017, 216, 4299–4311. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting adult pancreatic islet α cells into β cells by targeting both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Hao, E.; Piran, R.; Keinan, E.; Levine, F. Pancreatic β-cell neogenesis by direct conversion from mature α-cells. Stem Cells 2010, 28, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Mezza, T.; Cinti, F.; Cefalo, C.M.A.; Pontecorvi, A.; Kulkarni, R.N.; Giaccari, A. β-cell fate in human insulin resistance and type 2 diabetes: A perspective on islet plasticity. Diabetes 2019, 68, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.H.; Tang, J.; Yadev, A.K.; Saghafi, S.T.; Kibbe, C.R.; Linnemann, A.K.; Merrins, M.J.; Davis, D.B. Intra-Islet GLP-1, but not CCK, is necessary for β-cell function in mouse and human islets. Sci. Rep. 2020, 10, 2823. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hu, Y.; Xu, N.; Zhou, W.; Yang, L.; Chen, R.; Yang, R.; Sun, J.; Chen, H. A new way for beta cell neogenesis: Transdifferentiation from alpha cells induced by glucagon-like peptide 1. J. Diabetes Res. 2019, 2019, 2583047. [Google Scholar] [CrossRef] [PubMed]
- Sarnobat, D.; Moffett, C.R.; Tanday, N.; Reimann, F.; Gribble, F.M.; Flatt, P.R.; Tarasov, A.I. Antidiabetic drug therapy alleviates type 1 diabetes in mice by promoting pancreatic α-cell transdifferentiation. Biochem. Pharmacol. 2020, 182, 114216. [Google Scholar] [CrossRef]
- Villalba, A.; Rodriguez-Fernandez, S.; Perna-Barrull, D.; Ampudia, R.M.; Gomez-Munoz, L.; Pujol-Autonell, I.; Aguilera, E.; Coma, M.; Cano-Sarabia, M.; Vazquez, F.; et al. Repurposed analog of GLP-1 ameliorates hyperglycemia in type 1 diabetic mice through pancreatic cell reprogramming. Front. Endocrinol. 2020, 11, 258. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.K.; Neuman, J.C.; Battiola, T.J.; Wisinski, J.A.; Kimple, M.E.; Davis, D.B. Glucagon-Like peptide-1 regulates cholecystokinin production in β-cells to protect from apoptosis. Mol. Endocrinol. 2015, 29, 978–987. [Google Scholar] [CrossRef] [Green Version]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Clemente, G.; Hu, J.; Pontecorvi, A.; Holst, J.J.; Giaccari, A.; Kulkarni, R.N. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014, 63, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilimnik, G.; Kim, A.; Steiner, D.F.; Friedman, T.C.; Hara, M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of β-cell regeneration. Islets 2010, 2, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Brun, T.; He, K.H.; Lupi, R.; Boehm, B.; Wojtusciszyn, A.; Sauter, N.; Donath, M.; Marchetti, P.; Maedler, K.; Gauthier, B.R. The diabetes-linked transcription factor Pax4 is expressed in human pancreatic islets and is activated by mitogens and GLP-1. Hum. Mol. Genet. 2008, 17, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Franklin, I.K.; Wollheim, C.B. GABA in the endocrine pancreas: Its putative role as an islet cell paracrine-signalling molecule. J. Gen. Physiol. 2004, 123, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Kumar, M.; Zhang, Y.; Gyulkhandanyan, A.; Xiang, Y.Y.; Ye, B.; Perrella, J.; Hyder, A.; Zhang, N.; Wheeler, M.; et al. Gamma-Aminobutyric acid up-and downregulates insulin secretion from beta cells in concert with changes in glucose concentration. Diabetologia 2006, 49, 697–705. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ren, L.; Wan, Y.; Prud’homme, G.J. GABAergic regulation of pancreatic islet cells: Physiology and antidiabetic effects. J. Cell Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-Term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell 2017, 168, 73–85.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins target GABAA receptor signaling and impair α cell identity. Cell 2017, 168, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meulen, T.; Lee, S.; Noordeloos, E.; Donaldson, C.J.; Adams, M.W.; Noguchi, G.M.; Mawla, A.M.; Huising, M.O. Artemether does not turn α cells into β cells. Cell Metab. 2018, 27, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, A.M.; Moss, N.G.; Kaestner, K.H. GABA and artesunate do not induce pancreatic α-to- β cell transdifferentiation in vivo. Cell Metab. 2018, 28, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Gurzov, E.N. Can GABA turn pancreatic α-cells into β-cells? Nat. Rev. Endocrinol. 2018, 14, 629–630. [Google Scholar] [CrossRef]
- St-Onge, L.; Baquie, M.; Wollheim, C.B.; Gauthier, B. Novel Methods for Preventing Diabetes. International Patent No. WO/2011/144725, 24 November 2011. [Google Scholar]
- Cobo-Vuilleumier, N.; Lorenzo, P.I.; Rodriguez, N.G.; Herrera Gomez, I.G.; Fuente-Martin, E.; Lopez-Noriega, L.; Mellado-Gil, J.M.; Romero-Zerbo, S.Y.; Baquie, M.; Lachaud, C.C.; et al. LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat. Commun. 2018, 9, 1488. [Google Scholar] [CrossRef]
- Liu, X.; Turban, S.; Carter, R.N.; Ahmad, S.; Ramage, L.; Webster, S.P.; Walker, B.R.; Seckl, J.R.; Morton, N.M. β-cell-specific glucocorticoid reactivation attenuates inflammatory β-cell destruction. Front. Endocrinol. 2014, 5, 165. [Google Scholar] [CrossRef] [Green Version]
- Cobo-Vuilleumier, N.; Gauthier, B.R. Time for a paradigm shift in treating type 1 diabetes mellitus: Coupling inflammation to islet regeneration. Metabolism 2020, 104, 154137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Zhang, M.; Zeng, S.; Huang, Y.; Qin, M.; Nasri, U.; Santamaria, P.; Riggs, A.D.; Jin, L.; Zeng, D. Reversal of autoimmunity by mixed chimerism enables reactivation of β cells and transdifferentiation of α cells in diabetic NOD mice. Proc. Natl. Acad. Sci. USA 2020, 117, 31219–31230. [Google Scholar] [CrossRef]
- Cobo-Vuilleumier, N.; Lorenzo, P.I.; Gauthier, B.R. Targeting LRH-1/NR5A2 to treat type 1 diabetes mellitus. Cell Stress 2018, 2, 141–143. [Google Scholar] [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Wang, M.; Wang, Q.A.; Spurgin, S.B.; Wang, Z.V.; Sun, K.; Scherer, P.E. Autonomous interconversion between adult pancreatic α-cells and β-cells after differential metabolic challenges. Mol. Metab. 2016, 5, 437–448. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Wang, Y.J.; Schug, J.; Feleke, E.; Gao, L.; Liu, C.; Consortium, H.; Naji, A.; Glaser, B.; Kaestner, K.H. Single-Cell transcriptomics of human islet ontogeny defines the molecular basis of β-cell dedifferentiation in T2D. Mol. Metab. 2020, 42, 101057. [Google Scholar] [CrossRef] [PubMed]
- Marselli, L.; Suleiman, M.; Masini, M.; Campani, D.; Bugliani, M.; Syed, F.; Martino, L.; Focosi, D.; Scatena, F.; Olimpico, F.; et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 2014, 57, 362–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amo-Shiinoki, K.; Tanabe, K.; Hoshii, Y.; Matsui, H.; Harano, R.; Fukuda, T.; Takeuchi, T.; Bouchi, R.; Takagi, T.; Hatanaka, M.; et al. Islet cell dedifferentiation is a pathologic mechanism of long-standing progression of type 2 diabetes. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Christensen, A.A.; Gannon, M. The beta cell in type 2 diabetes. Curr. Diabet. Rep. 2019, 19, 81. [Google Scholar] [CrossRef]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.J.; Chatterjee, A.; Shen, E.; Cox, A.R.; Kushner, J.A. Low-Level insulin content within abundant non-β islet endocrine cells in long-standing type 1 diabetes. Diabetes 2019, 68, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershengorn, M.C.; Hardikar, A.A.; Wei, C.; Geras-Raaka, E.; Marcus-Samuels, B.; Raaka, B.M. Epithelial-To-Mesenchymal transition generates proliferative human islet precursor cells. Science 2004, 306, 2261–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, A.; Nolan, A.L.; Blacken, R.A.; Habener, J.F. Redifferentiation of insulin-secreting cells after in vitro expansion of adult human pancreatic islet tissue. Biochem. Biophys. Res. Commun. 2005, 327, 581–588. [Google Scholar] [CrossRef]
- Tabak, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimaki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [Green Version]
- White, M.G.; Shaw, J.A.; Taylor, R. Type 2 diabetes: The pathologic basis of reversible β-cell dysfunction. Diabetes Care 2016, 39, 2080–2088. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.J.; Pennartz, C.; Schenker, N.; Menge, B.A.; Schmidt, W.E.; Heise, T.; Kapitza, C.; Veldhuis, J.D. Hyperglycaemia is associated with impaired pulsatile insulin secretion: Effect of basal insulin therapy. Diabetes Obes. Metab. 2013, 15, 258–263. [Google Scholar] [CrossRef]
- Balakrishnan, S.; Dhavamani, S.; Prahalathan, C. β-Cell specific transcription factors in the context of diabetes mellitus and β-cell regeneration. Mech. Dev. 2020, 163, 103634. [Google Scholar] [CrossRef]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an alpha cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.L.; Liu, F.F.; Sander, M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013, 4, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, W.; Takahashi, S.; Yasuda, K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia 2015, 58, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Swisa, A.; Avrahami, D.; Eden, N.; Zhang, J.; Feleke, E.; Dahan, T.; Cohen-Tayar, Y.; Stolovich-Rain, M.; Kaestner, K.H.; Glaser, B.; et al. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J. Clin. Investig. 2017, 127, 230–243. [Google Scholar] [CrossRef]
- Gutierrez, G.D.; Bender, A.S.; Cirulli, V.; Mastracci, T.L.; Kelly, S.M.; Tsirigos, A.; Kaestner, K.H.; Sussel, L. Pancreatic beta cell identity requires continual repression of non-beta cell programs. J. Clin. Investig. 2017, 127, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Noriega, L.; Callingham, R.; Martinez-Sánchez, A.; Pizza, G.; Haberman, N.; Cvetesic, N.; Nguyen-Tu, M.S.; Lenhard, B.; Marchetti, P.; Piemonti, L.; et al. The long non-coding RNA Pax6os1/PAX6-AS1 modulates pancreatic β-cell identity and function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, F.F.; Liu, Y.H.; Wang, D.W.; Liu, T.S.; Yang, Y.; Guo, J.M.; Pan, Y.; Zhang, Y.F.; Du, H.; Li, L.; et al. Obesity-Induced reduced expression of the lncRNA ROIT impairs insulin transcription by downregulation of Nkx6.1 methylation. Diabetologia 2020, 63, 811–824. [Google Scholar] [CrossRef] [Green Version]
- Arnes, L.; Akerman, I.; Balderes, D.A.; Ferrer, J.; Sussel, L. βlinc1 encodes a long noncoding RNA that regulates islet β-cell formation and function. Genes Dev. 2016, 30, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Noriega, L.; Rutter, G.A. Long non-coding rnas as key modulators of pancreatic β-cell mass and function. Front. Endocrinol. 2020, 11, 610213. [Google Scholar] [CrossRef]
- Pullen, T.J.; Khan, A.M.; Barton, G.; Butcher, S.A.; Sun, G.; Rutter, G.A. Identification of genes selectively disallowed in the pancreatic islet. Islets 2010, 2, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Manteiga, J.M.; D’Alessandro, R.; Meldolesi, J. News about the role of the transcription factor REST in neurons: From physiology to pathology. Int. J. Mol. Sci. 2019, 21, 235. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Grapin-Botton, A. The importance of REST for development and function of beta cells. Front. Cell Dev. Biol. 2017, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Kim, Y.H.; Sever, D.; Mao, C.A.; Haefliger, J.A.; Grapin-Botton, A. REST represses a subset of the pancreatic endocrine differentiation program. Dev. Biol. 2015, 405, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Allagnat, F.; Chaffard, G.; Caille, D.; Fukuda, M.; Regazzi, R.; Abderrahmani, A.; Waeber, G.; Meda, P.; Maechler, P.; et al. Functional significance of repressor element 1 silencing transcription factor (REST) target genes in pancreatic beta cells. Diabetologia 2008, 51, 1429–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuente-Martin, E.; Mellado-Gil, J.M.; Cobo-Vuilleumier, N.; Martin-Montalvo, A.; Romero-Zerbo, S.Y.; Diaz Contreras, I.; Hmadcha, A.; Soria, B.; Martin Bermudo, F.; Reyes, J.C.; et al. Dissecting the brain/islet axis in metabesity. Genes 2019, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, P.I.; Martin-Montalvo, A.; Cobo Vuilleumier, N.; Gauthier, B.R. Molecular modelling of islet β-cell adaptation to inflammation in pregnancy and gestational diabetes mellitus. Int. J. Mol. Sci. 2019, 20, 6171. [Google Scholar] [CrossRef] [Green Version]
- Ceballos-Chavez, M.; Rivero, S.; Garcia-Gutierrez, P.; Rodriguez-Paredes, M.; Garcia-Dominguez, M.; Bhattacharya, S.; Reyes, J.C. Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc. Natl. Acad. Sci. USA 2012, 109, 8085–8090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado-Gil, J.M.; Fuente-Martin, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Lopez-Noriega, L.; Martin-Montalvo, A.; Gomez, I.G.H.; Ceballos-Chavez, M.; Gomez-Jaramillo, L.; Campos-Caro, A.; et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet beta cell functional maturity. Cell Death Dis. 2018, 9, 279. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, B.R.; Lorenzo, P.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Mellado-Gil, J.M.; Bermudez-Silva, F.J.; Rojo Martúnez, G.; Reyes, J.C. LSD1 Inhibitors for Use in the Treatment of Type 2 Diabetes. Patent Application No. EP19382051, 24 January 2019. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorenzo, P.I.; Cobo-Vuilleumier, N.; Martín-Vázquez, E.; López-Noriega, L.; Gauthier, B.R. Harnessing the Endogenous Plasticity of Pancreatic Islets: A Feasible Regenerative Medicine Therapy for Diabetes? Int. J. Mol. Sci. 2021, 22, 4239. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084239
Lorenzo PI, Cobo-Vuilleumier N, Martín-Vázquez E, López-Noriega L, Gauthier BR. Harnessing the Endogenous Plasticity of Pancreatic Islets: A Feasible Regenerative Medicine Therapy for Diabetes? International Journal of Molecular Sciences. 2021; 22(8):4239. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084239
Chicago/Turabian StyleLorenzo, Petra I., Nadia Cobo-Vuilleumier, Eugenia Martín-Vázquez, Livia López-Noriega, and Benoit R. Gauthier. 2021. "Harnessing the Endogenous Plasticity of Pancreatic Islets: A Feasible Regenerative Medicine Therapy for Diabetes?" International Journal of Molecular Sciences 22, no. 8: 4239. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084239