
Ampelopsin Inhibits Cell Proliferation and Induces Apoptosis in HL60 and K562 Leukemia Cells by Downregulating AKT and NF-κB Signaling Pathways



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
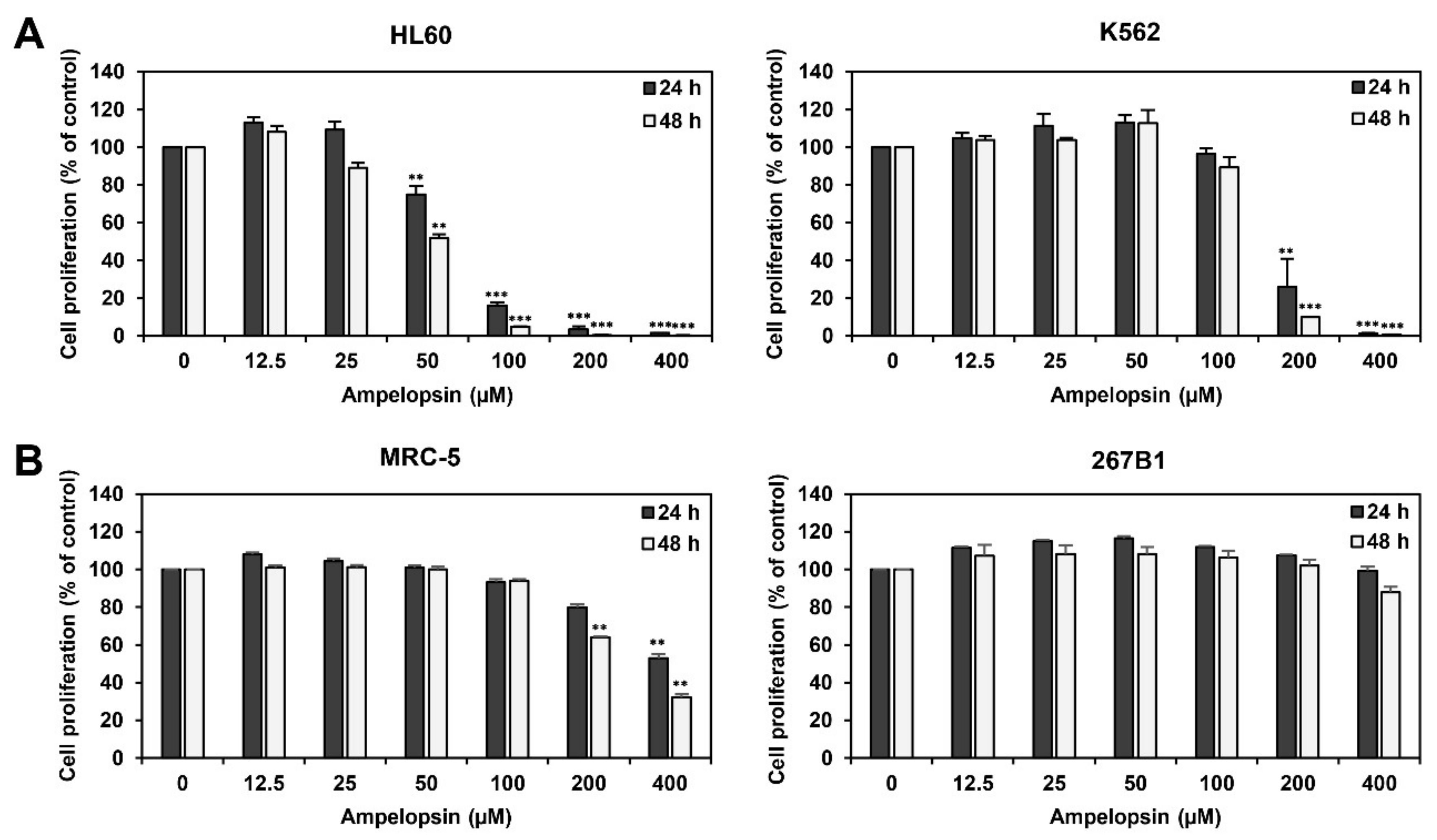
2.1. Ampelopsin Inhibits Proliferation of Leukemia Cells
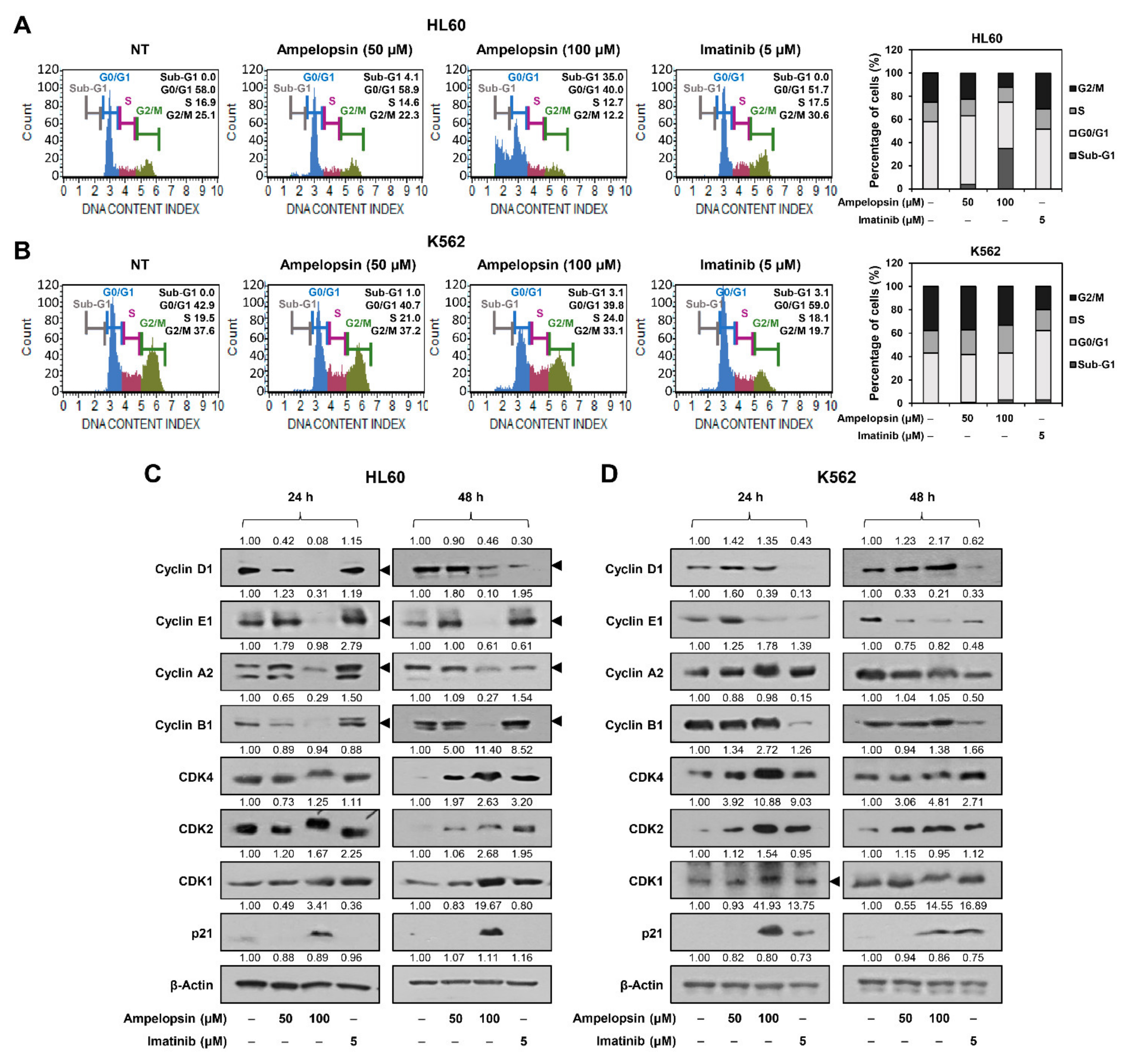
2.2. Ampelopsin Induces Cell Cycle Arrest of Leukemia Cells
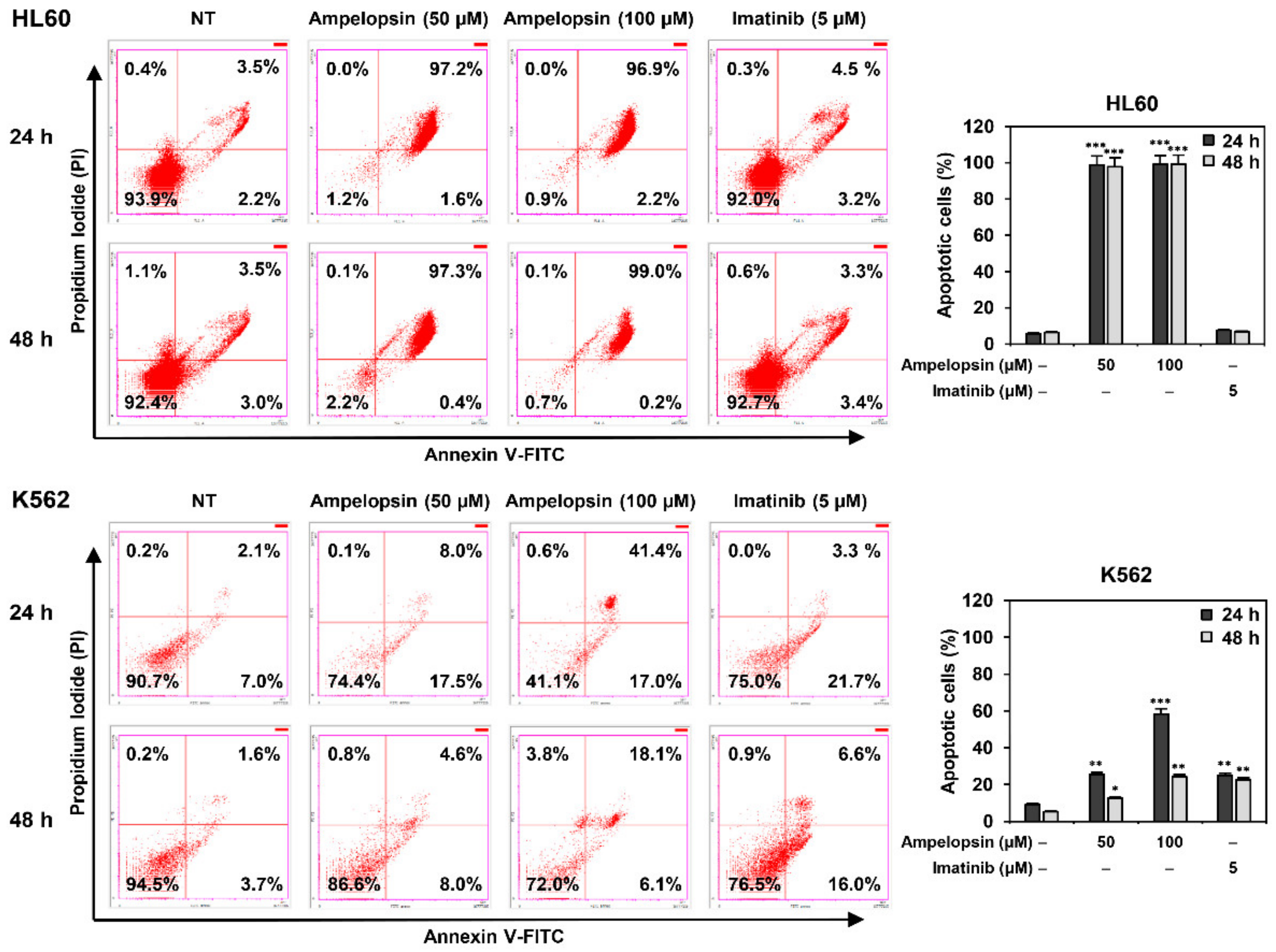
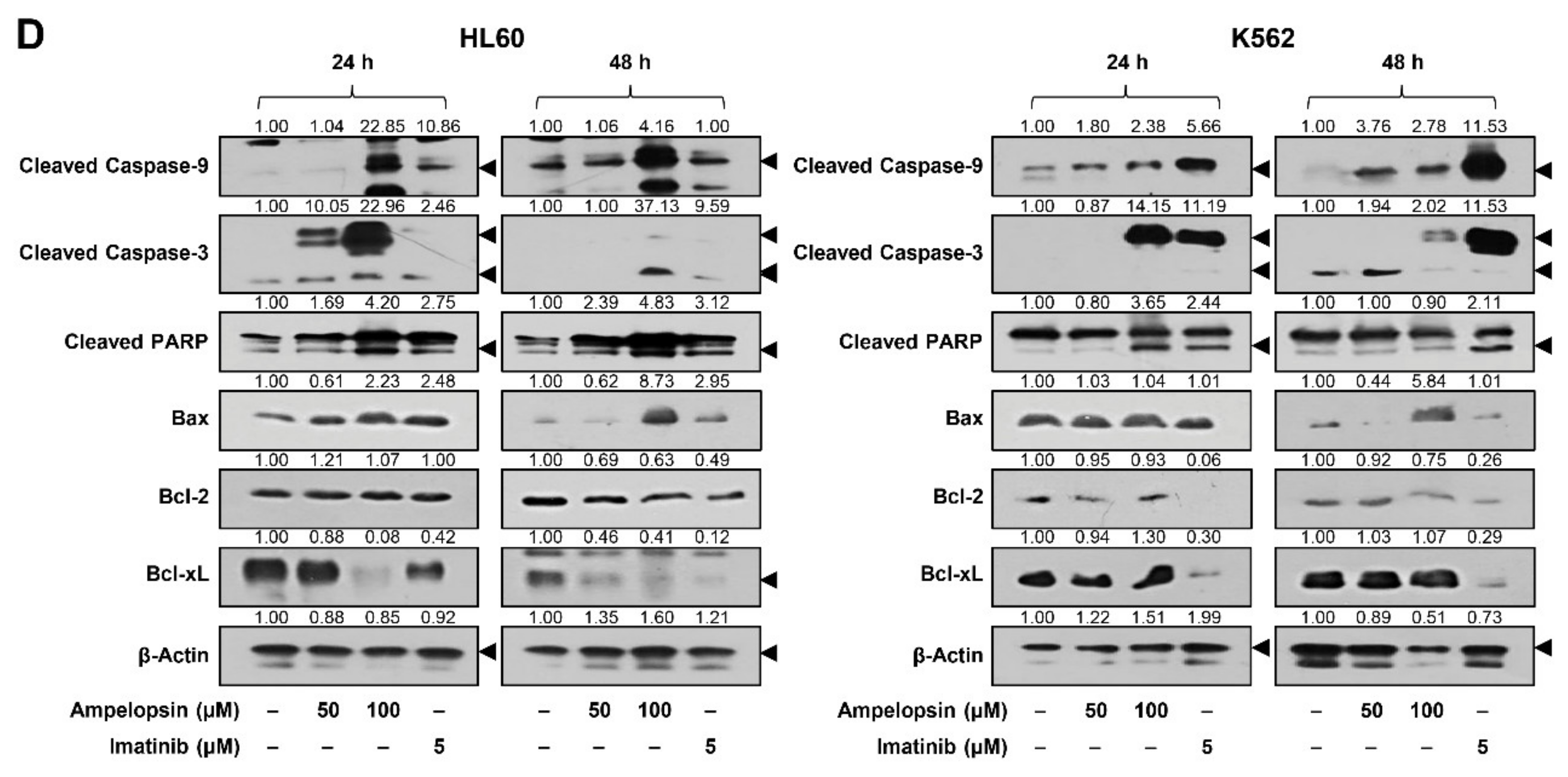
2.3. Ampelopsin Induces Apoptosis in Leukemia Cells
2.4. Ampelopsin Downregulates AKT and NF-κB Signaling in Leukemia Cells
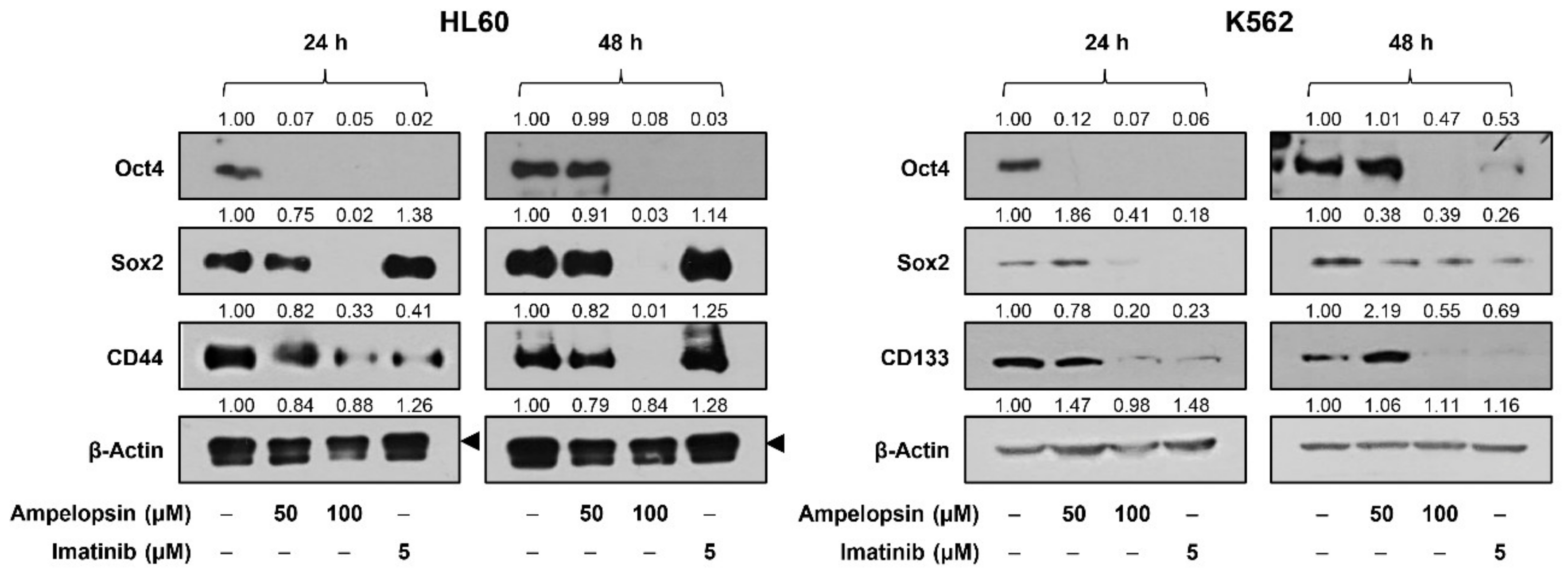
2.5. Ampelopsin Inhibits the Expression of Stemness Markers in Leukemia Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Cell Cycle Analysis
4.5. Western Blot Analysis
4.6. Apoptosis Analysis
4.7. DAPI Staining
4.8. Mitochondrial Membrane Potential (MMP) Measurement
4.9. Intracellular Reactive Oxygen Species (ROS) Measurement
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Juliusson, G.; Hough, R. Leukemia. Prog. Tumor Res. 2016, 43, 87–100. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Rea, D.; Mahon, F.X. How I manage relapse of chronic myeloid leukaemia after stopping tyrosine kinase inhibitor therapy. Br. J. Haematol. 2018, 180, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute promyelocytic leukemia: A constellation of molecular events around a single PML-RARA fusion gene. Cancers 2020, 12, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Fu, L.W. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit. Rev. Oncol. Hematol. 2015, 93, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Sutamtewagul, G.; Vigil, C.E. Clinical use of FLT3 inhibitors in acute myeloid leukemia. OncoTargets Ther. 2018, 11, 7041–7052. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutny, M.A.; Alonzo, T.A.; Gerbing, R.B.; Wang, Y.C.; Raimondi, S.C.; Hirsch, B.A.; Fu, C.H.; Meshinchi, S.; Gamis, A.S.; Feusner, J.H.; et al. Arsenic trioxide consolidation allows anthracycline dose reduction for pediatric patients with acute promyelocytic leukemia: Report from the children’s oncology group phase III historically controlled trial AAML0631. J. Clin. Oncol. 2017, 35, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Sabino Pinho de Oliveira, B.; Putti, S.; Naro, F.; Pellegrini, M. Bone marrow transplantation as therapy for ataxia-telangiectasia: A systematic review. Cancers 2020, 12, 3207. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Gaidano, G. Messengers of cell death: Apoptotic signaling in health and disease. Haematologica 2003, 88, 212–218. [Google Scholar] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Riccioni, R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica 2007, 92, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Liu, G.J.; Song, J.Y.; Chen, L.; Wang, A.H.; Gao, X.X.; Wang, Z.J. Preliminary results indicate resveratrol affects proliferation and apoptosis of leukemia cells by regulating PTEN/PI3K/AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4285–4292. [Google Scholar] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Oliveira, P.; Otero, P.; Pereira, A.G.; Chamorro, F.; Carpena, M.; Echave, J.; Fraga-Corral, M.; Simal-Gandara, J.; Prieto, M.A. Status and challenges of plant-anticancer compounds in cancer treatment. Pharmaceuticals 2021, 14, 157. [Google Scholar] [CrossRef]
- Hodgson, J.M.; Croft, K.D.; Puddey, I.B.; Mori, T.A.; Beilin, L.J. Soybean isoflavonoids and their metabolic products inhibit in vitro lipoprotein oxidation in serum. J. Nutr. Biochem. 1996, 7, 664–669. [Google Scholar] [CrossRef]
- Sultana, N. Clinically useful anticancer, antitumor, and antiwrinkle agent, ursolic acid and related derivatives as medicinally important natural product. J. Enzyme Inhib. Med. Chem. 2011, 26, 616–642. [Google Scholar] [CrossRef]
- Zhang, H.W.; Hu, J.J.; Fu, R.Q.; Liu, X.; Zhang, Y.H.; Li, J.; Liu, L.; Li, Y.N.; Deng, Q.; Luo, Q.S.; et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of PI3Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in cancer and apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kim, I.S.; Rehman, S.U.; Na, C.S.; Yoo, H.H. HPLC determination of bioactive flavonoids in Hovenia dulcis fruit extracts. J. Chromatogr. Sci. 2016, 54, 130–135. [Google Scholar] [PubMed]
- Weng, L.; Zhang, H.; Li, X.; Zhan, H.; Chen, F.; Han, L.; Xu, Y.; Cao, X. Ampelopsin attenuates lipopolysaccharide-induced inflammatory response through the inhibition of the NF-κB and JAK2/STAT3 signaling pathways in microglia. Int. Immunopharmacol. 2017, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zhang, J.; Ahmad, H.; Zhang, H.; Xu, Z.; Wang, T. Evaluation of antioxidant activities of ampelopsin and its protective effect in lipopolysaccharide-induced oxidative stress piglets. PLoS ONE 2014, 9, e1083142014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.M.; Lim, H.N.; Jung, H.J. Hovenia dulcis Thunb. and its active compound ampelopsin inhibit angiogenesis through suppression of VEGFR2 signaling and HIF-1α expression. Oncol. Rep. 2017, 38, 3430–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.N.; Wang, F.; Yuan, Y.T.; Liu, J.; Liu, Y.Z.; Yi, X. Antibacterial activity and mode of action of dihydromyricetin from Ampelopsis grossedentata leaves against food-borne bacteria. Molecules 2019, 24, 2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, X.; Chang, H.; Shu, F.; Wu, Y.; Zhang, T.; Fu, Y.; Zhang, Q.; Zhu, J.D.; Mi, M. Ampelopsin-induced autophagy protects breast cancer cells from apoptosis through Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci. 2014, 105, 1279–1287. [Google Scholar] [CrossRef]
- Kao, S.J.; Lee, W.J.; Chang, J.H.; Chow, J.M.; Chung, C.L.; Hung, W.Y.; Chien, M.H. Suppression of reactive oxygen species-mediated ERK and JNK activation sensitizes dihydromyricetin-induced mitochondrial apoptosis in human non-small cell lung cancer. Environ. Toxicol. 2017, 32, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yin, J.Q.; Wu, M.S.; Song, G.; Xie, X.B.; Zou, C.; Tang, Q.; Wu, Y.; Lu, J.; Wang, Y.; et al. Dihydromyricetin activates AMP-activated protein kinase and P38(MAPK) exerting antitumor potential in osteosarcoma. Cancer Prev. Res. 2014, 7, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, S.; Chan, H.F.; Lu, H.; Lin, Z.; He, C.; Chen, M. Dihydromyricetin induces apoptosis and reverses drug resistance in ovarian cancer cells by p53-mediated downregulation of survivin. Sci. Rep. 2017, 7, 46060. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Li, R.; Zeng, G.F.; Liu, B.; Liu, J.; Shu, Y.; Liu, Z.K.; Qiu, Z.D.; Wang, D.J.; Miao, H.L.; et al. Dihydromyricetin inhibits migration and invasion of hepatoma cells through regulation of MMP-9 expression. World J. Gastroenterol. 2014, 20, 10082–10093. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.J.; Tian, X.F.; Liu, X.W.; Fu, L.B.; Wu, Y.Y.; Fang, X.D.; Jin, H.Y. Dihydromyricetin induces cell apoptosis via a p53-related pathway in AGS human gastric cancer cells. Genet. Mol. Res. 2015, 14, 15564–15571. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Guozhang, H.; Wang, H.; Li, Z.; Liu, N. Ampelopsin inhibits human glioma through inducing apoptosis and autophagy dependent on ROS generation and JNK pathway. Biomed. Pharmacother. 2019, 116, 108524. [Google Scholar] [CrossRef]
- Zhou, D.Z.; Sun, H.Y.; Yue, J.Q.; Peng, Y.; Chen, Y.M.; Zhong, Z.J. Dihydromyricetin induces apoptosis and cytoprotective autophagy through ROS-NF-κB signalling in human melanoma cells. Free Radic. Res. 2017, 51, 517–528. [Google Scholar]
- Gamas, P.; Marchetti, S.; Puissant, A.; Grosso, S.; Jacquel, A.; Colosetti, P.; Pasquet, J.M.; Mahon, F.X.; Cassuto, J.P.; Auberger, P. Inhibition of imatinib-mediated apoptosis by the caspase-cleaved form of the tyrosine kinase Lyn in chronic myelogenous leukemia cells. Leukemia 2009, 23, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milella, M.; Kornblau, S.M.; Estrov, Z.; Carter, B.Z.; Lapillonne, H.; Harris, D.; Konopleva, M.; Zhao, S.; Estey, E.; Andreeff, M. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J. Clin. Investig. 2001, 108, 851–859. [Google Scholar] [CrossRef]
- Styczynski, J.; Drewa, T. Leukemic stem cells: From metabolic pathways and signaling to a new concept of drug resistance targeting. Acta. Biochim. Pol. 2007, 54, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, D.M.; Still, P.C.; Pérez, L.B.; Grever, M.R.; Kinghorn, A.D. Potential of plant-derived natural products in the treatment of leukemia and lymphoma. Curr. Drug Targets 2010, 11, 812–822. [Google Scholar] [CrossRef]
- Saraei, R.; Marofi, F.; Naimi, A.; Talebi, M.; Ghaebi, M.; Javan, N.; Salimi, O.; Hassanzadeh, A. Leukemia therapy by flavonoids: Future and involved mechanisms. J. Cell. Physiol. 2019, 234, 8203–8220. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Li, W.; Miao, H.; Zhang, H.; Zhou, Y.; Li, Z.; You, Q.; Zhao, L.; Guo, Q. Wogonin reverses multi-drug resistance of human myelogenous leukemia K562/A02 cells via downregulation of MRP1 expression by inhibiting Nrf2/ARE signaling pathway. Biochem. Pharmacol. 2014, 92, 220–234. [Google Scholar]
- Sun, Y.; Liu, W.; Wang, C.; Meng, Q.; Liu, Z.; Huo, X.; Yang, X.; Sun, P.; Sun, H.; Ma, X.; et al. Combination of dihydromyricetin and ondansetron strengthens antiproliferative efficiency of adriamycin in K562/ADR through downregulation of SORCIN: A new strategy of inhibiting P-glycoprotein. J. Cell. Physiol. 2019, 234, 3685–3696. [Google Scholar]
- Zhu, H.; Luo, P.; Fu, Y.; Wang, J.; Dai, J.; Shao, J.; Yang, X.; Chang, L.; Weng, Q.; Yang, B.; et al. Dihydromyricetin prevents cardiotoxicity and enhances anticancer activity induced by Adriamycin. Oncotarget 2015, 6, 3254–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.H.; Zhang, Q.; Shu, G.; Lin, J.C.; Zhao, L.; Liang, X.X.; Yin, L.; Shi, F.; Fu, H.L.; Yuan, Z.X. Dihydromyricetin sensitizes human acute myeloid leukemia cells to retinoic acid-induced myeloid differentiation by activating STAT1. Biochem. Biophys. Res. Commun. 2018, 495, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Sak, K.; Everaus, H. Established human cell lines as models to study anti-leukemic effects of flavonoids. Curr. Genomics 2017, 18, 3–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.M.; Kim, H.L.; Jung, H.J. Ampelopsin Inhibits Cell Proliferation and Induces Apoptosis in HL60 and K562 Leukemia Cells by Downregulating AKT and NF-κB Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084265
Han JM, Kim HL, Jung HJ. Ampelopsin Inhibits Cell Proliferation and Induces Apoptosis in HL60 and K562 Leukemia Cells by Downregulating AKT and NF-κB Signaling Pathways. International Journal of Molecular Sciences. 2021; 22(8):4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084265
Chicago/Turabian StyleHan, Jang Mi, Hong Lae Kim, and Hye Jin Jung. 2021. "Ampelopsin Inhibits Cell Proliferation and Induces Apoptosis in HL60 and K562 Leukemia Cells by Downregulating AKT and NF-κB Signaling Pathways" International Journal of Molecular Sciences 22, no. 8: 4265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084265